
ABSTRACT
The Ataxia-telangiectasia mutated (ATM) kinase and the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) are activated by DNA double-strand breaks (DSBs). These DSBs occur in the context of chromatin but how chromatin influences the activation of these kinases is not known. Here we show that loss of the replication-dependent chromatin assembly factors ASF1A/B or CAF-1 compromises ATM activation, while augmenting DNA-PKcs activation, in response to DNA DSBs. Cells deficient in ASF1A/B or CAF-1 exhibit reduced histone H4 lysine 16 acetylation (H4K16ac), a histone mark known to promote ATM activation. ASF1A interacts with the histone acetyl transferase, hMOF that mediates H4K16ac. ASF1A depletion leads to increased recruitment of DNA-PKcs to DSBs. We propose normal chromatin assembly and H4K16ac during DNA replication is required to regulate ATM and DNA-PKcs activity in response to the subsequent induction of DNA DSBs.
KEYWORDS:
Introduction
DNA double-strand breaks (DSBs) are the most perilous form of DNA lesion, as failure to repair them may lead to cell death, genomic instability or tumorigenesis [Citation1]. There are two main DSB repair pathways in eukaryotic cells, non-homologous end joining (NHEJ) and homologous recombination (HR) [Citation2]. DSBs also activate a complex signalling network, called the DNA damage response (DDR), which coordinates repair of DSBs and cell-cycle checkpoint activation [Citation3].
ATM and DNA-PKcs are Phosphoinositide 3 kinase-like (PIKKs) serine/threonine kinases [Citation4,Citation5]. DNA-PKcs and ATM are activated early in the response to DNA DSBs and orchestrate DSB repair. DNA-PKcs is recruited and activated at DSBs by the Ku70 and Ku80 heterodimer [Citation5]. ATM is recruited and activated at DSBs by the Mre11-Rad50-Nbs1 (MRN) complex [Citation4]. Once activated, ATM promotes NHEJ in G1 phase of the cell cycle while promoting HR in S/G2 [Citation4]. In contrast, DNA-PKcs primarily functions in DSB repair by NHEJ. ATM and DNA-PKcs are recruited to the same DNA ends [Citation6], where they regulate each other. ATM-mediated phosphorylation of DNA-PKcs, displaces DNA-PKcs from DNA ends [Citation7], while DNA-PKcs-mediated phosphorylation of ATM suppresses the catalytic activity of ATM [Citation6]. How the activities of ATM and DNA-PKcs, and their negative influence on each other, are controlled is unclear.
Within our cells, DSB repair occurs in the context of chromatin, where each fundamental repeating unit of chromatin, a nucleosome, comprises 147bp of DNA wrapped around the outside of an octamer of core histones [Citation8]. Histone post-translational modifications help recruit proteins involved in the DDR to DSBs [Citation9]. Furthermore, H4 K16 acetylation, by the histone acetyl transferase hMOF, has been shown to promote the efficient activation of ATM in response to DSBs (Gupta et al., 2005). Histones also impose a physical block to the DNA repair machinery such that histones are removed from around DSBs to enable DNA repair, while histones are restored to the DNA after DSB repair is complete [Citation10–Citation13]. Intriguingly, the assembly of histones after DNA synthesis via the “replication-dependent” chromatin assembly pathway, by the Asf1 and CAF-1 histone chaperones, after DSB repair is required for turning off the DDR in yeast [Citation10,Citation14,Citation15]. However, whether chromatin assembly plays a role in inactivation of the DDR after DNA repair in metazoans is unknown.
In this work, we set out to determine if the role of chromatin assembly in inactivation of the DDR after DSB repair was conserved from yeast to humans. Unexpectedly, we find reduced activation of ATM upon depletion of human ASF1 or CAF-1. Depletion of ASF1 or CAF-1 also led to reduced H4 K16 acetylation globally and during DNA repair, likely mediated via a novel interaction between hMOF and ASF1A. ASF1-depletion enhanced activation of DNA-PKcs and enhanced localization of active DNA-PKcs to induced DSBs. We propose that chromatin structure restricts precocious access of DNA-PKcs to DNA lesions and regulates the balance of ATM vs. DNA-PKcs activation in human cells.
Results
Replication-dependent chromatin assembly promotes ATM activation in response to DSBs
To determine if chromatin assembly was required for inactivating the DNA damage response after DNA repair is complete in human cells, we knocked down ASF1A/B, induced DNA damage with the radiomimetic bleomycin, and examined checkpoint activation at increasing times post bleomycin removal. In response to DNA DSBs, the histone H2AX is phosphorylated by ATM and DNA-PKcs forming γH2AX in chromatin flanking the DSB. Phosphorylation of H2AX was slightly more persistent after ASF1A/B depletion compared to the scrambled shRNA control (ShControl) after inducing DSBs (). However, we consistently observed reduced and shorter-lived ATM-dependent phosphorylation of Chk2 at Thr68 (Chk2T68p) upon depletion of human ASF1A/B (, S1A, S2, S3). Both ASF1A and ASF1B contribute to the efficient activation of Chk2 after DSB induction (Figures S2, S3). Reduced Chk2 phosphorylation in the absence of ASF1 also occurred following treatment with neocarzinostatin (NCS), another radiomimetic drug (data not shown). These data demonstrate that ASF1 promotes efficient activation of Chk2 after DSB induction.
Figure 1. Replication-dependent chromatin assembly promotes efficient activation of Chk2 but has minimal effect on γH2AX. (a) HeLa cells transduced with lentiviruses expressing scrambed shRNA or shRNAs against both ASF1A and ASF1B for 72 hours were treated with 40 μg/ml bleomycin for 2 hours, following by washing out the bleomycin at time 0. Samples were taken at the indicated time points after washing out the bleomycin and analyzed by western blotting with the indicated antibodies. Numbers indicate the normalized changes in γH2AX, normalized to GAPDH, and to 1 at time = 0 for each set of lanes within the brackets. (b) As in A, but with analysis by immunofluorescence of γH2AX. (c) As in A, with analysis of Chk2 phosphorylation. Numbers indicate the normalized changes in Chk2p, normalized to Chk2, and to 1 at time = 4. (d) As in B, but with knockdown of CAF-1 p150. (e) As in C, but with knockdown of HIRA. Numbers indicate the normalized changes, as described in D. (f). As in A, but with ASF1A/B and CAF-1 depletion and analysis of KAP-1 phosphorylation.
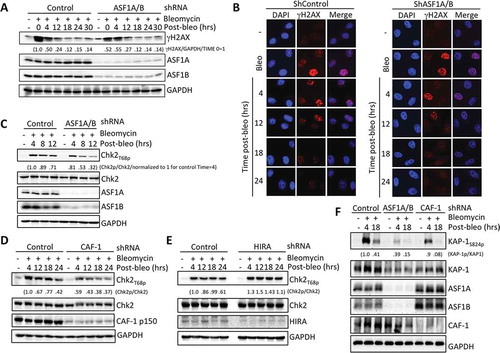
ASF1 is the upstream histone chaperone for replication-independent chromatin assembly of histones H3.3/H4 by the HIRA histone chaperone, and for replication-dependent chromatin assembly of histones H3.1/H4 by CAF-1 [Citation16]. To determine whether either of these chromatin assembly pathways is required for efficient activation of Chk2 after DSB induction, we knocked down HIRA and CAF-1 individually. Knockdown of the large subunit of CAF-1, CAF-1 p150, but not HIRA, led to reduced Chk2 phosphorylation following DSB induction (, S1B, S1C). These data indicate that only replication-dependent chromatin assembly, but not the replication-independent chromatin assembly, promotes Chk2 activation after DSB induction in humans. In agreement, phosphorylation of another ATM substrate, KAP-1, was greatly reduced upon ASF1A/B or CAF-1 depletion after DSB induction (). These differences do not appear to be due to cell cycle changes, because we do not see consistent cell cycle changes during the time frame of ASF1A/B and CAF-1 knockdown used in our experiments (Figure S4).
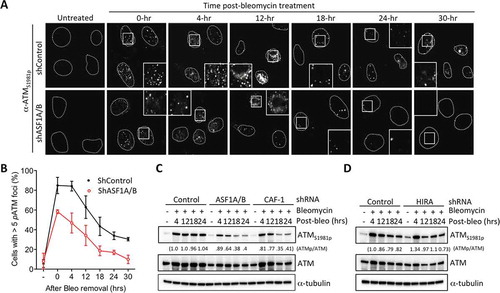
Reduced Chk2 and KAP-1 phosphorylation suggested that ATM is not efficiently activated after DSB induction upon ASF1A/B and CAF-1 depletion (). Indeed, inhibition of ATR had no effect on Chk2 phosphorylation with or without ASF1A/B knockdown (Figure S3). Therefore, we asked whether the levels of active ATM were reduced upon ASF1A/B and CAF-1 depletion, by measuring autophosphorylation of ATM on Ser1981 (ATMS1981p), which is a marker of ATM activity [Citation17]. We saw reduced activation of ATM after bleomycin treatment by immunofluorescence analysis upon ASF1A/B depletion (), S3). By western blot analysis, ATMS1981p was reduced following ASF1A/B or CAF-1 p150 depletion, but not upon HIRA depletion following DSB induction (). Taken together, these data demonstrate that ATM activation is compromised in response to DSBs when the replication-dependent chromatin assembly pathway is inactivated.
Figure 2. Replication-dependent chromatin assembly promotes efficient activation of ATM. (a) ASF1A/B promotes DSB induced ATMS1981p foci formation. Cells were treated as in , followed by staining for ATMS1981p. Squares indicate areas shown enlarged in the insets. (b) Quantitation of (a). Over 100 cells per sample per experiment were randomly picked to determine the number of foci. Average and SD for 3 independent experiments are shown. (c) Experiment was performed as in , followed by western blotting with the indicated antibodies. (d) As in C, but for HIRA depletion.
Replication-dependent chromatin assembly promotes H4 K16 acetylation
In yeast, ASF1 and CAF-1 promote H4K16ac [Citation18] and in metazoan cells, reduced basal levels of H4K16ac reduce ATM activation [Citation19]. We found that depletion of both ASF1A/B greatly reduced the global basal level of H4 K16ac with shRNAs or siRNA in HeLa cells (, S5). Both ASF1A and ASF1B contribute to the global H4 K16 acetylation, because knockdown of either ASF1A or ASF1B partially reduced the H4 K16 acetylation levels (). This is not due to off target effects as re-introduction of an shRNA resistant ASF1A restored the H4 K16 acetylation levels (). Interestingly, knockdown of ASF1A also reduced levels of ASF1B, while rescue of ASF1A restored ASF1B levels (). This phenomena makes it difficult to conclude exactly how much ASF1A contributes to H4 K16Ac directly. We conclude that ASF1A/B promote global H4 K16 acetylation in human cells.
Figure 3. Replication-dependent chromatin assembly promotes H4 K16 acetylation and ASF1A binds hMOF. (a) ASF1A/B were depleted from HeLa cells with shRNAs or siRNAs, followed by western blotting with the indicated antibodies. (b) shRNA depletion of ASF1A or ASF1B alone, as indicated. In the case of ASF1A depletion, either an empty vector or a plasmid expressing H1-ASF1A was reintroduced. (c) hMOF interacts with ASF1A. Immunoprecipitation of hMOF via a FLAG epitope. * refers to a non-specific band running slightly faster than FLAG-hMOF, most likely an antibody chain. Some of the HA-ASF1A in the IP appears to have undergone degradation to a smaller band. (d) Experiment was performed as in with the indicated shRNA depletions followed by western blotting with the indicated antibodies. (e) As in D upon HIRA depletion.
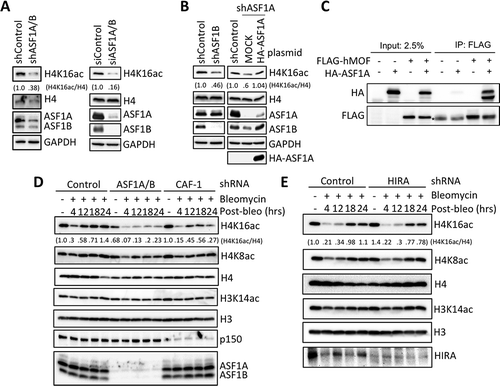
Asf1 promotes other histone acetylation events through the binding of HATs, such as Rtt109 [Citation20], and CBP [Citation21]. Therefore, we asked whether ASF1A binds to the human H4K16 HAT hMOF, as a potential means to enhance the basal level of H4 K16 acetylation. By immunprecipitation analysis, we found that hMOF effectively pulled down ASF1A (). These data suggest that ASF1A and hMOF interact in human cells, consistent with the ASF1A-hMOF interaction serving to deliver histones to hMOF for their acetylation on H4 lysine 16 by hMOF.
The levels of H4 K16ac are known to initially decrease in response to DSBs, and subsequently increase over time during DSB repair, mediated by another HAT, KAT5/TIP60 [Citation19,Citation22]. We asked whether ASF1A/B and CAF-1 influence the increase in H4 K16 acetylation that occurs during DNA repair. We find that ASF1A/B depletion obliterated the increase of H4 K16 acetylation during repair (). CAF-1 depletion had a similar, although slightly reduced effect, while HIRA depletion had no influence on either the global levels of H4 K16ac or the acetylation of H4 K16 during DNA repair (). Taken together, these data indicate that replication-dependent chromatin assembly factors promote H4 K16 acetylation globally and after DNA damage in human cells, which could contribute to reduced ATM activation in the absence of replication-dependent chromatin assembly.
ASF1A/B depletion leads to enhanced persistent DNA-PKcs activation
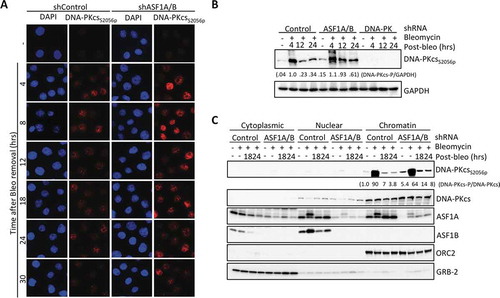
Depletion of ASF1A/B or CAF-1 leads to reduced ATM activation (, , S3), yet there was no striking differences in γH2AX levels after induction of DSBs upon ASF1A/B depletion () or CAF-1 depletion [Citation23]. As both ATM and DNA-PKcs contribute to γH2AX formation [Citation24] we asked if DNA-PKcs activity is enhanced upon ASF1A/B depletion. Upon treatment of ASF1-deficienct cells with bleomycin, DNA-PKcs autophosphorylated on Serine 2056 (DNA-PKcsS2056p), a mark of DNA-PKcs activation, accumulated at higher levels and with more persistence in the nucleus as compared to control cells (, S6). In addition, in ASF1-depleted cells we also observed more persistent DNA-PKcsS2056p by western blotting after DNA damage (, S7). Furthermore, there is DNA-PKcsS2056p apparent in cells depleted of ASF1A/B even without induced DNA damage (, S6). Chromatin fractionation demonstrated that the activated DNA-PKcsS2056p remained on the chromatin in cells depleted of ASF1A/B (). We also observed persistent phosphorylation of DNA-PKcs on threonine T2609 upon ASF1A/B depletion (data not shown), which is a substrate of not only ATM, but also ATR [Citation25]. These data indicate that there is enhanced persistent activation of DNA-PKcs upon depletion of ASF1A/B.
Figure 4. Enhanced DNA-PKcs activation in response to DSBs upon ASF1A/B depletion. (a) Immunofluorescence staining of active DNA-PKcs at the indicated time after washing out 50 μg/ml Bleomcyin upon ASF1A/B depletion by shRNA. (b) The experiment was performed as in , with the indicated shRNA depletions followed by western blotting with the indicated antibodies. (c) The experiment was performed as in , with the indicated shRNA depletions, and fractionation into cytoplasmic, nuclear and chromatin fragments, followed by western blotting with the indicated antibodies.
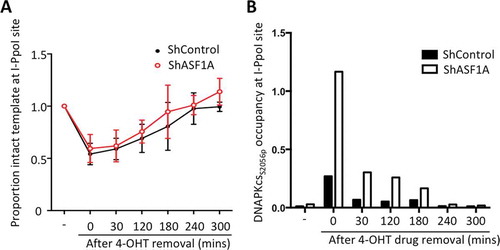
In order to ask whether the increased levels of DNA-PKcsS2056p in human cells depleted of ASF1A/B reflects its localization to DNA breaks, we used the inducible I-PpoI endonuclease system which cuts at known locations in the human genome [Citation26]. The nuclear localization of the I-PpoI endonuclease fused to ER nuclear localization sequence was induced by tamoxifen (4-OHT) treatment and addition of Shield-1 to stabilize the enzyme in HCT116 cells [Citation12]. Repair was enabled by washing away the 4-OHT and Shield-1 to destabilize and export the I-PpoI to the cytoplasm. Using quantitative PCR analysis over the I-PpoI site within the human SLCO5A gene [Citation13], we find that the extent and kinetics of DNA cutting and repair were unchanged upon ASF1A knockdown (). We had previously extensively characterized repair at this particular site to be via NHEJ and not HR, depending on Ku but not Rad51 [Citation13]. To examine DNA-PKcsS2056p recruitment during NHEJ, we performed chromatin immunoprecipitation (ChIP) analysis on samples from the same repair time course, using phospho-DNA-PKcsS2056 antibodies. In the control cells, DNA-PKcsS2056p levels were highest at the time of most DNA cutting, and gradually decreased during repair (). However, depletion of ASF1A significantly elevated levels of active DNA-PKcs at each time point (). Taken together, these results indicate that depletion of ASF1 leads to increased DNA-PKcs association with chromatin at DSB sites and augmented DNA-PKcs activation.
Figure 5. Depletion of ASF1A leads to enhanced levels of DNA-PKcs to DSBs during NHEJ. (a) Cutting and NHEJ repair of the inducible I-PpoI site within the SLCO5A gene is unaffected by ASF1A depletion. Real-time PCR analysis over the break was used to measure the intactness of the I-PpoI site and values were normalized to GAPDH in each sample. Average and SEM are plotted for three individual experiments. (b) ChIP analysis of DNA-PKcsS2056p adjacent to the I-PpoI lesion determined from the same time course shown in A. Representative results are shown.
Discussion
In response to DSBs, the activities of the ATM and DNA-PKcs kinases are balanced to regulate the DDR, but how this balance is regulated is poorly understood. Our work reveals an unexpected role for the replication-dependent chromatin assembly pathway in regulating the subsequent activities of ATM and DNA-PKcs in response to DSBs. Specifically, depletion of the histone chaperones CAF-1 and ASF1A/B that mediate chromatin assembly following DNA synthesis reduced activation of ATM and enhanced activation of DNA-PKcs. This work provides novel insight into how chromatin structure regulates the activities of these key DNA damage response kinases.
Cells transiently depleted for the replication-dependent chromatin assembly factors ASF1A/B and CAF-1 have reduced ATM activation (, , S3). Depletion of ASF1 or CAF-1 leads to a more open chromatin structure, due to reduced ability to assemble histones following DNA replication. This is apparent in the greater accessibility of the human genome to micrococcal nuclease upon depletion of ASF1A/B [Citation27]. However, not all chromatin perturbations that make the genome less tightly packaged lead to reduced ATM activation. For example, it has been previously shown that agents that perturb chromatin structure without necessarily causing DNA damage activate ATM [Citation17]. Conversely, forced local chromatin condensation is sufficient to activate ATM without DNA damage [Citation28]. In either scenario, the molecular mechanisms by which changes in chromatin states activate ATM are unclear.
Histone acetylation also influences ATM activation in response to DSBs. Specifically, reduced H4 K16 acetylation in cells lacking the HAT hMOF, have reduced ATM activation [Citation19,Citation29]. A potential explanation for the reduced activation of ATM in cells depleted of ASF1A/B or CAF-1, could be due to the reduced acetylation of H4 K16Ac (, S5). The yeast enzyme, Sas2, which acetylates H4 K16 exists in the SAS-I complex. SAS-I binds to yeast CAF-1 [Citation30] and Asf1 in yeast [Citation31]. Precedent for how ASF1 may function to promote acetylation comes from its ability to present histones to the HAT Rtt109 [Citation32] and CBP in human cells [Citation21,Citation33] which enhances their HAT activities towards histones. Indeed, ASF1A physically interacts with hMOF (), potentially explaining a mechanism by which it promotes H4 K16 acetylation. Moreover, the incorporation of acetylated H4 K16 into chromatin occurs mainly during DNA replication in a chromatin assembly-dependent manner [Citation34]. As such, the reduced replication-dependent chromatin assembly that occurs upon ASF1A/B or CAF-1 depletion would be sufficient to account for the global reduction in H4 K16Ac. Furthermore, the fact that the damage-dependent increase in H4 K16Ac depended on ASF1 and CAF-1 () indicates that this increase on H4 K16Ac is occurring on histones that are being newly-assembled onto DNA during DSB repair. It will be interesting to determine exactly how H4 K16Ac promotes ATM activation. H4 K16 acetylation opens up the chromatin structure [Citation35], which potentially could increase recruitment of MRN to DNA ends. ASF1A has recently been proposed to contribute to NHEJ via potentiating the interaction between Mdc1 and ATM [Citation36]. Given that both H4 K16ac and H2AX phosphorylation recruit Mdc1 [Citation37], the role of ASF1A in promoting H4 K16ac could explain the contribution of ASF1A to NHEJ [Citation36].
It is possible that the increased DNA-PKcs activity in cells depleted for ASF1A/B is a consequence of the more open chromatin structure due to histone loss, promoting access of Ku to the DNA ends, leading to more DNA-PKcs activation. Alternatively, it is possible that the reduced activation of ATM in cells depleted for ASF1A/B (, , S1-S3) may promote increased DNA-PKcs activation and localization at DSBs (, , S6, S7), due to the reduced amount of ATM-dependent DNA-PKcs phosphorylation, which has been reported to promote dissociation of DNA-PKcs from DNA ends [Citation7,Citation38]. This may be unlikely though because it has been proposed that if a lesion fails to be repaired in a timely manner by a DNA-PKcs-dependent end-joining mechanism, ATM-mediated DNA-PKcs phosphorylation would induce dissociation of DNA-PKcs from DNA ends, but clearly there is no defect in NHEJ in our hands upon depletion of ASF1A/B nor CAF-1 () [Citation13].
Conversely, it is also possible that the elevated DNA-PKcs activity could be responsible for the reduced amount of ATM activation in cells depleted for ASF1A/B. DNA-PKcs phosphorylates ATM at many residues to reduce its activity [Citation6]. Indeed, a phosphomimetic allele of ATM that mimics the DNA-PKcs-mediated ATM phosphorylation has reduced levels of ATM autophosphorylation (ATMS1981p) indicating reduced ATM activity [Citation6]. In agreement with the idea that elevated DNA-PKcs activity in cells depleted of ASF1A/B may reduce ATM activation is the fact that we find background levels of DNA-PKcs activation even in the absence of induced DSBs upon ASF1A/B depletion (, S7). It will be interesting to determine exactly why DNA-PKcs is active in cells lacking ASF1A/B in the absence of induced DSBs. Perhaps the reduced histone occupancy on the genome leads to increased accessibility of Ku to the DNA, in the absence of DNA ends. Nevertheless, our work establishes a role for the chromatin assembly machinery in the balance of activity of ATM and DNA-PKcs in response to DNA DSBs.
Materials and methods
Cell culture, antibodies, DNA damaging agent treatments
HeLa and HCT116 were grown in complete DMEM with 10% FBS and 1% penicillin/streptomycin. Human osteosarcoma U-2 OS cells were grown in McCoy’s 5a Medium with 10% FBS and 1% penicillin/streptomycin. All the above cells were grown at 37°C with 5% CO2. For transient transfection, 2 × 106 cells were seeded per 60mm dish for 24 hours prior to the delivery of plasmid DNA using Lipofectamine 2000 (Invitrogen). shRNA viral particles freshly produced from 293T cells were added to desired cells for 2 days to efficiently knockdown gene expressions. To induce DNA damage, cells were generally treated with 40 μg/ml Bleomycin (#9041–93-4, Cayman Chemical Company) followed by harvesting at indicated time points after washing with PBS. The antibodies used in this study were listed in .
Table 1. A list of antibodies used in this study.
SiRNA and shRNA knockdowns
100 nM of siASF1A and siASF1B were mixed with INTERFERin siRNA transfection reagent (#409–10, Polyplus transfection) according to the manufacturer’s protocol to perform ASF1A/B knockdowns for 48 hours. The siRNA control was a random scrambled RNA sequence. shRNA lentiviral plasmids against ASF1A, ASF1B, HIRA, CAF-1, and the scrambled control were obtained from the shRNA and ORFeome Core Facility at the University of Texas MD Anderson Cancer Center (Houston, TX). To generate shRNA knockdown cells, cells were incubated with shRNA lentiviral supernatant for 24 hours followed by 1 μg/ml Puromycin for 48 hours. The knockdown efficiency of each target protein was determined by western blotting. Each experiment was repeated at least 3 times and representative results are shown. All siRNA and shRNA target sequences used in this study are listed in .
Table 2. A list of siRNAs, shRNAs and gRNA used in this study.
Flow cytometry analysis
To analyze DNA content of cells, harvested cells were washed in PBS with 1% FBS followed by fixation in 70% ethanol overnight at 4°C. Fixed cells were spun down to aspirate ethanol and then were washed in PBS once. Cell pellets were resuspended in PBS with 1% FBS containing 10 μg/ml propidium iodide (PI) (#81,845, Sigma-Aldrich) and 0.5 μg/ml RNase A (#10,109,142,001, Sigma-Aldrich) and kept in the dark at room temperature for 1 hour followed by flow cytometry analysis using BD LSRII flow cytometer.
Immunofluorescence analysis
Cells were plated on poly-L-lysine-coated coverslips for 24 hours. After bleomycin treatment, coverslips were washed with PBS. For DNA damage foci analysis, 0.1% Triton-X-100 in cytoskeletal (CSK) buffer (100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 10 mM PIPES (pH 6.8)) was added to permeabilize cells for 3 minutes at room temperature. Cells were washed once with TBS-T (0.05% Tween-20) and fixed with 4% paraformaldehyde for 10 minutes. Fixed cells were blocked with 3% BSA for 30 minutes. Primary antibodies diluted in 3% BSA blocking buffer were subsequently incubated with coverslips overnight at 4°C. Cells were washed 3 times with TBS-T before incubating with fluorescence conjugated-secondary antibodies for 1 hour at room temperature in the dark. After washing in PBS, cells were mounted in prolong gold antifade mountant with DAPI and kept at 4°C in the dark. Cells were imaged using a 3i confocal microscope system and images were analyzed via ImageJ software.
Protein extraction and immunoblotting
After washing once in PBS, cells were resuspended in pre-chilled RIPA buffer (50 mM Tris-HCl, pH7.4, 1 mM EDTA, 0.5 mM EGTA, 1% Triton-X-100, 0.1% Nadeoxycholate, 0.1% SDS, 150 mM NaCl, protease inhibitors (complete Mini EDTA-free, #4,693,159,001, Roche) and phosphatase inhibitors (PhosSTOP, #4,906,837,001, Roche) followed by sonication. Cell debris was removed by centrifugation at 14,000 r.p.m., at 4°C for 10 minutes. Supernatants were collected and protein concentrations quantitated by Bradford protein assay (#500–0006, Bio-Rad). Equivalent protein amounts were loaded on SDS-PAGE or Criterion 4–12% precast gels (#5,671,085, Bio-Rad). After electrophoresis, proteins were transferred to Nitrocellulose membrane (#10,600,048, GE Healthcare Life Sciences) for 90 minutes at 4°C and membranes were incubated with 5% non-fat milk for 1 hour at room temperature before overnight incubation with diluted primary antibodies at 4°C, followed by incubation with secondary antibodies in 5% non-fat milk for 1 hour at room temperature. Enhanced chemiluminescence (ECL) Western blotting substrate (# RPN 3243, Amersham ECL, GE Healthcare Life Sciences) were used for detection, using a protein simple analysis machine.
Immunoprecipitation
2 x 106 HeLa cells were seeded into 60mm dishes and incubated overnight. HA-tagged ASF1A plasmid was transfected into cells for 24 hours before harvesting. Cells were then lysed with pre-chilled modified RIPA buffer (50 mM Tris-HCl, pH7.4, 1 mM EDTA, 0.5 mM EGTA, 0.5% NP-40, 150 mM NaCl, protease inhibitors (complete Mini EDTA-free, #4,693,159,001, Roche) for 15 minutes on ice. To collect supernatant, lysates were centrifuged at 14,000 r.p.m. at 4°C for 10 minutes. Cell lysates were precleared with Dynabeads Protein A (#10001D, ThermoFisher Scientific) for 1 hour followed by incubation with anti-FLAG-Agarose (#F2426, Sigma-Aldrich) for 3 hours at 4°C. After centrifugation, the HA-Agarose protein complexes were washed in RIPA buffer 3 times for 15 minutes and boiled with SDS loading buffer.
Chromatin fractionation analysis
This analysis was performed as described previously with slight modification [Citation39]. Whole cell extract (WCE) samples were taken from the same batch of samples used for chromatin fractionation and prepared separately. Cells harvested from 6-well plates were re-suspended in buffer A (10 mM HEPES, pH7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, complete Mini (EDTA-free) protease and PhosSTOP phosphatase inhibitors) before adding an equal volume of buffer A with 0.2% Triton X-100 and were incubated on ice for 4.5 minutes. Cytoplasmic protein fractions were collected after centrifuge at 1,300 x g, for 4 minutes at 4°C. The insoluble pellet, including chromatin, was washed with buffer A and then the supernatant was aspirated. The pellet was resuspended in buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, complete Mini protease and PhosSTOP phosphatase inhibitors) for 15 minutes, followed by centrifugation at 1,700 x g, 4 minutes at 4°C. Nuclear protein fractions were obtained from the supernatant. The pellet was washed pellet once in buffer B and S7 nuclease with RIPA buffer and 5 mM CaCl2 added to each sample for 10 minutes on ice, followed by sonication to solubilize. This yields the “chromatin” fraction.
I-PpoI-based inducible DSB system
The pBABE-dd-I-PpoI (Addgene #49052) plasmid developed by Michael Kastan lab was introduced into HCT116 cells upon puromycin selection to obtain genomic-integrated stable bulk population. For DNA cutting and repair analysis, Shield-1 (Cheminpharma) was added to a final concentration of 1mM for 3 hours to stabilize dd-I-PpoI followed by another 30-minute incubation with 4mM 4-OHT (Sigma). After treatment, drugs were washed off in PBS to allow the process of DNA repair over time. DNA levels of I-PpoI sites were further quantified by real-time PCR. The primer sequences used for detecting DNA levels were as follows: (SLCO5a1-F-cut: CCCAGTGCTCTGAATGTCAA; SLCO5a1-R-cut: CCATTCATGCGCGTCACTA). DNA levels were normalized to the value of real-time PCR using the GAPDH (GAPDH-F: TCAGCCAGTCCCAGCCCAAG; GAPDH-R: GAGAAAGTAGGGCCCGGCTAC) primer set.
Chromatin immunoprecipitation
The procedure for chromatin immunoprecipitation (ChIP) was performed as described previously [Citation13]. The enriched chromatin was analyzed by real-time PCR analysis. The primer sequences (SLCO5a1F-Left: GCATGAATGG
ATGAACGAGAT; SLCO5a1R-Left: CAAGCTCAACAGGGTCTTCT) for real-time PCR flanked the 5' upstream region which was 200 b.p. distal from the cutting site. The NHEJ factor occupancy and the input at each time point were normalized to GAPDH. All data were normalized to the value of untreated sample as 1.
Author Contributions
JKT, T-HH and BPS guided the project. Z-JS independently verified results. T-HH performed all the remaining experiments. T-HH and JKT wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Supplemental Material
Download PDF (2.6 MB)Acknowledgments
We are grateful to Yuri Moshkin for HA tagged ASF1A plasmids and to Evi Soutoglou for the I-PpoI system. We thank Tej Pandita for the hMOF-FLAG construct and Xuan Li for advice on the I-PpoI system. We thank the shRNA and ORFeome Core Facility at the University of Texas MD Anderson Cancer Center and Memorial Sloan Kettering Cancer Center (MSKCC) RNAi core for providing shRNA clones. This work was supported by grant NCI CA95641 to JKT.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204.
- Mehta A, Haber JE. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol. 2014;6:a016428.
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210.
- Paull TT. Mechanisms of ATM Activation. Annu Rev Biochem. 2015;84:711–738.
- Jette N, Lees-Miller SP. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog Biophys Mol Biol. 2015;117:194–205.
- Zhou Y, Lee JH, Jiang W, et al. Regulation of the DNA damage response by DNA-PKcs inhibitory phosphorylation of ATM. Mol Cell. 2017;65:91–104.
- Chen BP, Uematsu N, Kobayashi J, et al. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J Biol Chem. 2007;282:6582–6587.
- Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184:868–871.
- Williamson EA, Wray JW, Bansal P, et al. Overview for the histone codes for DNA repair. Prog Mol Biol Transl Sci. 2012;110:207–227.
- Chen CC, Carson JJ, Feser J, et al. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008;134:231–243.
- Berkovich E, Monnat RJ Jr., Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9:683–690.
- Goldstein M, Derheimer FA, Tait-Mulder J, et al. Nucleolin mediates nucleosome disruption critical for DNA double-strand break repair. Proc Natl Acad Sci U S A. 2013;110:16874–16879.
- Li X, Tyler JK. Nucleosome disassembly during human non-homologous end joining followed by concerted HIRA- and CAF-1-dependent reassembly. Elife. 2016 Jun 8;5. pii: e15129.
- Kim JA, Haber JE. Chromatin assembly factors Asf1 and CAF-1 have overlapping roles in deactivating the DNA damage checkpoint when DNA repair is complete. Proc Natl Acad Sci U S A. 2009;106:1151–1156.
- Diao LT, Chen CC, Dennehey B, et al. Delineation of the role of chromatin assembly and the Rtt101Mms1 E3 ubiquitin ligase in DNA damage checkpoint recovery in budding yeast. PloS One. 2017;12:e0180556.
- Hammond CM, Stromme CB, Huang H, et al. Histone chaperone networks shaping chromatin function. Nat Rev Mol Cell Biol. 2017;18:141–158.
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506.
- Shia WJ, Osada S, Florens L, et al. Characterization of the yeast trimeric-SAS acetyltransferase complex. J Biol Chem. 2005;280:11987–11994.
- Gupta A, Sharma GG, Young CS, et al. Involvement of human MOF in ATM function. Mol Cell Biol. 2005;25:5292–5305.
- Han J, Zhou H, Li Z, et al. Acetylation of lysine 56 of histone H3 catalyzed by RTT109 and regulated by ASF1 is required for replisome integrity. J Biol Chem. 2007;282:28587–28596.
- Das C, Roy S, Namjoshi S, et al. Binding of the histone chaperone ASF1 to the CBP bromodomain promotes histone acetylation. Proc Natl Acad Sci U S A. 2014;111:E1072–E1081.
- Li L, Wang Y. Cross-talk between the H3K36me3 and H4K16ac histone epigenetic marks in DNA double-strand break repair. J Biol Chem. 2017;292:11951–11959.
- Baldeyron C, Soria G, Roche D, et al. HP1alpha recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J Cell Biol. 2011;193:81–95.
- Stiff T, O’Driscoll M, Rief N, et al. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–2396.
- Yajima H, Lee KJ, Chen BP. ATR-dependent phosphorylation of DNA-dependent protein kinase catalytic subunit in response to UV-induced replication stress. Mol Cell Biol. 2006;26:7520–7528.
- Berkovich E, Monnat RJ Jr., Kastan MB. Assessment of protein dynamics and DNA repair following generation of DNA double-strand breaks at defined genomic sites. Nat Protoc. 2008;3:915–922.
- O’Sullivan RJ, Arnoult N, Lackner DH, et al. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat Struct Mol Biol. 2014;21:167–174.
- Burgess RC, Burman B, Kruhlak MJ, et al. Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 2014;9:1703–1717.
- Sharma GG, So S, Gupta A, et al. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol Cell Biol. 2010;30:3582–3595.
- Meijsing SH, Ehrenhofer-Murray AE. The silencing complex SAS-I links histone acetylation to the assembly of repressed chromatin by CAF-I and Asf1 in Saccharomyces cerevisiae. Genes Dev. 2001;15:3169–3182.
- Osada S, Sutton A, Muster N, et al. The yeast SAS (something about silencing) protein complex contains a MYST-type putative acetyltransferase and functions with chromatin assembly factor ASF1. Genes Dev. 2001;15:3155–3168.
- Han J, Zhou H, Horazdovsky B, et al. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science. 2007;315:653–655.
- Das C, Lucia MS, Hansen KC, et al. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459:113–117.
- Heise F, Chung HR, Weber JM, et al. Genome-wide H4 K16 acetylation by SAS-I is deposited independently of transcription and histone exchange. Nucleic Acids Res. 2012;40:65–74.
- Shogren-Knaak M, Ishii H, Sun JM, et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847.
- Lee KY, Im JS, Shibata E, et al. ASF1a promotes non-homologous end joining repair by facilitating phosphorylation of MDC1 by ATM at double-strand breaks. Mol Cell. 2017;68:61 e5–75 e5.
- Li X, Corsa CA, Pan PW, et al. MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Mol Cell Biol. 2010;30:5335–5347.
- Uematsu N, Weterings E, Yano K, et al. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J Cell Biol. 2007;177:219–229.
- Méndez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602–8612.