
ABSTRACT
Lung cancer remains one of the world’s deadliest cancers, with effective targeted treatment options available for only a small subset of patients. The rapid expansion of cancer genomics in recent years has provided insight into the genetic landscape of all major lung cancer subtypes and led to new discoveries on the heterogeneous biology underlying lung tumorigenesis. Interestingly, these studies have revealed a high frequency of alterations in the Kelch-like ECG-associated protein 1 (KEAP1)–Nuclear factor erythoid–2–related factor 2 (NRF2) stress response pathway, for which no targeted treatments are currently available. In this review, we describe the molecular mechanisms underlying NRF2 pathway activation in lung cancer cells, with a focus on in vivo functional studies in genetically engineered mouse models. Importantly, potential avenues and implications for therapeutic targeting of KEAP1-NRF2 pathway vulnerabilities for lung cancer patients will be highlighted.
Lung cancer
Lung cancers constitute the highest proportion of cancer-related deaths worldwide, accounting for over 1.5 million deaths annually, an estimated one in five of all cancer-related mortality [Citation1]. Moreover, the predicted five-year survival rate for lung cancer is a mere 17.8%, a figure that is significantly lower than many other types of cancer, such as prostate (99.6%), breast (90.5%) and colon (65.4%) cancer. Lung cancer is classified into two main pathological subgroups: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), comprising 80% and 20% of lung cancers, respectively. NSCLC is a heterogeneous group of diseases encompassed of adenocarcinoma (ADC), squamous cell carcinoma (SqCC) and large cell carcinoma. Recently, comprehensive genomic datasets have been generated for all major lung cancer subtypes [Citation2–Citation4], with attention now focused on validating targetable driver mutations. Among these, mutations in the KEAP1-NRF2 stress response pathway have been identified in 23% of lung ADC [Citation4] and 34% of SqCC [Citation3], for which no targeted treatments are currently available. In this review, we describe recent advances in delineating KEAP1-NRF2 pathway biology in lung cancer. Specifically, we will discuss mechanistic insights learnt from in vivo genetically engineered mouse models (GEMMs) and address how these vulnerabilities can be harnessed for therapeutic intervention in this subset of lung cancer patients.
The KEAP1-NRF2 pathway
One of the most critical stress-response pathways in mammals is mediated by the transcription factor Nuclear factor erythoid-2-related factor 2 (NFE2L2/NRF2), which is negatively regulated by Kelch-like ECG-associated protein 1 (KEAP1). Under homeostatic conditions, NRF2 is degraded via the proteasome through binding to KEAP1, an adaptor protein of the CUL3 E3 ubiquitin ligase (). Oxidative stress induces the oxidation of KEAP1 at key cysteine residues which causes a conformational change in KEAP1 releasing NRF2, resulting in translocation and nuclear accumulation of NRF2. In the nucleus, NRF2 forms a heterodimer with its partner sMAF (v-Maf avian musculoaponeurotic fibrosarcoma oncogene homolog) and binds to antioxidant responsive element (ARE) sequences to regulate the transcription of target genes (). NRF2 binding activates expression of over 200 genes, many of which are involved in cellular antioxidant, detoxification and metabolic pathways [Citation5]. A major NRF2 transcriptional target is NAD(P)H:quinone dehydrogenase 1 (NQO1), which serves as a robust marker of NRF2 activation in vitro and in vivo [Citation6,Citation7]. It is however important to note that, activation of NRF2 can also occur through KEAP1-independent mechanisms (). These mechanisms include competitive binding of NRF2 by proteins such as p53-induced p21 [Citation8] or nutrient sensor p62 [Citation9,Citation10]; inhibition of GSK3β-mediated NRF2 degradation via the activity of the PI3K pathway [Citation11]; nuclear sequestration of NRF2 through phosphorylation at Serine 558 by AMP-activated protein kinase (AMPK) in response to cellular energy stress [Citation12]; among others. For a more detailed description of the additional level of complexity of the regulation of the KEAP1-NRF2 pathway, we refer readers to relevent reviews [Citation13–Citation16].
Figure 1. The KEAP1-NRF2 pathway is altered in human lung cancers.
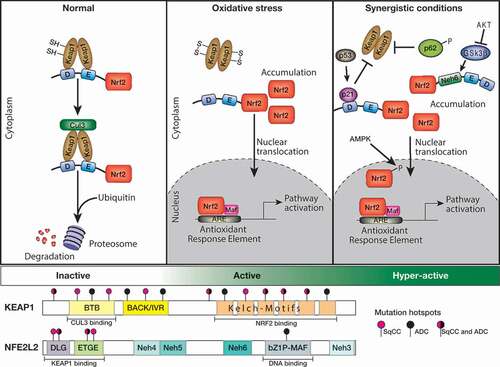
Deregulation of the KEAP1-NRF2 pathway in lung cancer
Comprehensive genomic sequencing studies have reported deregulation of the KEAP1-NRF2 pathway in human cancer, through both genetic and epigenetic mechanisms (reviewed in [Citation17,Citation18]). Since the initial identification of loss of function mutations in the KEAP1 gene in two lung ADC cell lines [Citation19], multiple studies have revealed the presence of somatic alterations in NSCLC cell lines and lung tumor tissue from patients [Citation3,Citation4,Citation20,Citation21]. KEAP1 inactivating mutations have been detected throughout the protein, with an increased frequency within the Kelch domains (), involved in mediating protein-protein interactions with NRF2 [Citation22] and result in nuclear accumulation of NRF2 [Citation19]. In human lung ADC, KEAP1 is the third most commonly mutated gene, behind the tumor suppressor TP53 and KRAS oncogene, where it is found mutated in 19% of patients [Citation4,Citation23]. Interestingly, emerging evidence also suggests that mutations in the KEAP1 gene are less prevalent in non-smokers compared to former or current smokers [Citation24,Citation25]. Recent genomic profiling studies of pulmonary large-cell neuroendocrine carcinomas (LCNECs) revealed the presence of KEAP1 somatic alterations (22%) [Citation26]. Interestingly, KEAP1 alterations are mutually exclusive with inactivating mutations in RB1, and together with somatic mutations of STK11 define a distinct subgroup of LCNECs that exhibit an ASCL1low/DLL3low/NOTCHhigh neuroendocrine profile, akin to SCLC [Citation26,Citation27].
In lung cancer, gain-of-function mutations in NRF2 appear to be largely restricted to lung SqCC and are mutually exclusive with mutations in KEAP1 and CUL3, which occur less frequently in this cancer subtype [Citation3,Citation28]. Interestingly, high mutation frequencies of NRF2 are also detected in squamous cell cancers of the esophagus, larynx and skin [Citation29], suggesting that augmented NRF2 regulates transcriptional programs that are associated with squamous cell differentiation. Indeed, newborn Keap1-null mice die from severe hyperkeratosis of the esophagus and forestomach, rescued by loss of Nrf2 [Citation30]. Notably, all mutations in the NRF2 gene are localized within the DLG (43%) and ETGE (57%) motifs, which are critical sites for the binding of NRF2 to KEAP1 [Citation31] (). Thus, mutations in NRF2 disrupt the high affinity binding to KEAP1, resulting in the stabilization and nuclear accumulation of NRF2 [Citation31].
In lung ADC, recent studies have revealed a significant correlation between increased NQO1 expression and NRF2 pathway activation, suggesting that NQO1 is a clinically relevant biomarker for KEAP1– and/or NRF2-mutant tumors [Citation6,Citation7]. Furthermore, an NRF2 gene signature, composed of 108 high-confidence NRF2 target genes could stratify lung ADC patients that exhibited high-grade tumors and poor survival [Citation7]. Together, these studies reveal an unappreciated role of the KEAP1-NRF2 pathway in lung cancer, suggesting that this pathway is a critical player in protecting lung epithelium from cellular transformation.
Unveiling the function of the KEAP1-NRF2 pathway in lung cancer using genetically engineered mouse models (GEMMs)
GEMMs of lung cancer have emerged as critical bridging strategies between understanding pathogenic mechanism and clinical translation. Importantly, they reveal insights on the events and processes underlying tumor initiation and progression, studies which are not possible utilizing transplantation or chemically-induced model systems. The “gold-standard” lung cancer models are based on Cre-LoxP recombination technology that enable the formation of autochthonous tumors from a limited number of somatic cells. Critically, tumors form in their natural location in the presence of their normal tissue microenvironment. This approach has not only allowed investigators to interrogate the functional consequences of genetic alterations found in human lung cancer but has also provided key insights into the cellular origin of the disease [Citation32]. A loss-of-function (LOF) Keap1 conditional allele (Keap1f/f) was generated by Blake et al, to study constitutive activation of the Keap1-Nrf2 pathway [Citation33]. Using this GEMM, prolonged activation of the Nrf2 pathway was shown to result in no adverse events in lung function or pathology (aged up to 12 months) [Citation6] (). However, when combined with mutations that enhance proliferative signals, Keap1 inactivation can drive the malignant transformation of lung epithelium, as discussed below.
Table 1. Mouse models engineered to harbor KEAP1-NRF2 pathway alterations.
Adenocarcinoma
KRAS, TP53 and KEAP1 are the three most common genetic mutations in lung ADC, and mutations in TP53 and KEAP1 occur in 26% and 18% of KRAS-mutant lung ADC, respectively [Citation34]. The functional consequences of Keap1 inactivation on KrasG12D-induced lung cancer formation has been explored using the KrasG12D conditional knock-in mouse model, whereby a glycine to aspartic acid substitution at codon 12 was engineered in the endogenous Kras locus. Expression of oncogenic KrasG12D is controlled by a LoxP-Stop-LoxP (LSL) cassette, which consists of transcriptional and translational stop elements flanked by LoxP sites [Citation35] (KrasLSL-G12D/+; hereafter KrasG12D). Intra-nasal delivery of an adenovirus or lentivirus expressing Cre-recombinase under the control of the ubiquitous cytomegalovirus (CMV) promoter (Ad5-CMV-Cre) results in the removal of the stop elements and formation of lung ADC in KrasG12D mice [Citation36]. Critically, additional loss of the p53 tumor suppressor gene accelerates tumorigenesis. Tumors that develop in KrasG12D;p53f/f mice (hereafter KP) more closely mimic their human counterpart and exhibit an aggressive phenotype, which promotes the local dissemination of tumor cells [Citation37,Citation38]. While combined loss of Lkb1, detected in 17% of KRAS-mutant lung ADC [Citation4,Citation34], accelerates KrasG12D tumorigenesis and promotes ADC to SqCC transformation [Citation39,Citation40].
A bespoke in vivo screening approach based on CRISPR-Cas9 gene editing combined with high-throughput barcode sequencing (Tuba-seq) [Citation41] was employed to interrogate the functional consequences of validated and candidate tumor suppressor genes found commonly altered in human lung ADC [Citation4,Citation42]. KrasG12D, KP and KrasG12D;Lkb1f/f mice engineered to carry a Cre-inducible tdTomato reporter allele, and a LoxP-Stop-LoxP-Cas9 allele were infected with pool libraries and lung tumor size was evaluated 15 weeks following Lenti-sgRNA/Cre infection. While this approach was successful in identifying new genetic interactions, Keap1 inactivation did not provide a significant growth advantage to KrasG12D tumors, nor to KrasG12D tumors that had concomitant loss of either p53 or Lkb1 [Citation41,Citation42] (). Interestingly, sgRNAs targeting p53 were also not enriched in KT;Cas9 mice under these experimental conditions, which contradicts findings using conventional Cre/loxP-based mouse models [Citation37]. The absence of an effect of sgRNAs targeting Keap1 in the background of Lkb1-deficiency contradict recent bioinformatic analyses that revealed a strong co-occurrence of KEAP1 inactivation and loss of function mutations in STK11 (Lkb1) [Citation28,Citation34].
In a more targeted approach, Romero et al., intra-tracheally infected KP mice with pSECC lentiviral vector expressing a single sgRNA against Keap1 [Citation7,Citation43]. Notably, compared to KP mice infected with a control sgRNA (tdTomato), sgKeap1-infected mice exhibited increased tumor burden suggesting that Keap1 loss accelerates KrasG12D-driven lung cancer (). Keap1 LOF tumors displayed augmented KEAP1-NRF2 pathway activation, characterized by increased NRF2 nuclear localization and elevated expression of Nqo1 [Citation7]. Moreover, the levels of reactive oxygen species (ROS) were significantly lower in Keap1 LOF tumors. Tumorigenesis was also augmented in the presence of sgRNAs targeting Nrf2, which induced gain-of-function mutations. Together, these findings implicate a crucial role for the oxidative stress response in KrasG12D-driven lung cancer. Curiously, LOF mutations in KEAP1 occur in 18% of KRAS-mutant lung ADC and are largely mutually exclusive with mutations in TP53 [Citation34]. The findings of Romero et al., would therefore argue against the idea that TP53 and KEAP1 are synthetic lethal. Further research is therefore required to dissect whether the mechanisms of p53 and Keap1 tumor suppression overlap. An in-depth comparative analysis of tumorigenesis in KrasG12D;Keap1f/f and KP mice would assist in addressing these outstanding questions.
A functional interaction between the KEAP1-NRF2 and phosphatidylinositide 3-kinase (PI3K) pathways has been reported to promote the malignant evolution of tumors [Citation25,Citation44]. Given that genetic inactivation of both KEAP1 and PTEN have been identified in lung ADC and lung SqCC, a GEMM based on conditional loss of Keap1 and Pten was generated [Citation6]. Consistent with previous reports, significant synergy in Nrf2 activity, measured by augmented expression of Nqo1, was identified in Keap1f/f/Ptenf/f (hereafter K1P) tumor cells, compared to the modest increase in Nqo1 following the loss of Keap1 alone [Citation6]. These findings are in line with the regulation of Nrf2 by GSK3β [Citation11,Citation45] (). Critically, combined loss of Keap1 and Pten led to bronchiolocentric ADC formation [Citation6] (). On a molecular level, the tumors that developed in this model resemble the salient features of human ADC stratified on the basis of high NQO1 mRNA expression [Citation4], a robust biomarker of KEAP1-NRF2 pathway activity [Citation6,Citation7].
Squamous cell carcinoma
The transcriptome of lung basal cells most closely correlates with the transcriptional profile of human lung SqCC [Citation46]. Indeed, landmark studies have revealed that different lung cancer subtypes arise from distinct lung epithelial cell lineages [Citation47,Citation48], suggesting that basal cells are the cells-of-origin of lung SqCC [Citation49]. Given that recent studies demonstrated that Keap1 loss promoted the self-renewal of basal stem cells in vitro [Citation50], we hypothesized that combined loss of Keap1 and Pten specifically in basal progenitor cells would drive lung SqCC formation in vivo. To directly test this, we infected K1P mice treated with polidocanol one day prior to infection with the Ad5-K14-Cre virus infection that directs Cre-recombinase expression to basal cells [Citation51,Citation52] (). We, and others have shown that pre-treatment with polidocanol causes damage of the luminal epithelium, enhancing the infection of the exposed basal cell layer [Citation53,Citation54]. Critically, when combined loss of Keap1 and Pten was restricted to basal progenitor cells, lung SqCC was observed, 12 months following Ad5-K14-Cre infection (). These tumors, like their human counterpart, stained positive for keratin 5 (K5), expressed high levels of Nqo1 and Sox2, but stained negative for TTF-1/Nkx2.1, a marker of lung ADC (). Interestingly, tumors exhibiting histological and morphological features of lung ADC were also observed in the lungs of K1P mice (), likely resulting from “switched” Club cells repopulated following polidocanol-induced injury (). Taken together, these findings suggest that combined loss of Keap1 and Pten has the ability to drive SqCC formation when inactivated in basal progenitor cells, highlighting the importance of the cell-of-origin in dictating the tumor phenotype.
Figure 2. Directed loss of Keap1 and Pten in lung basal progenitor cells promotes lung SqCC formation.
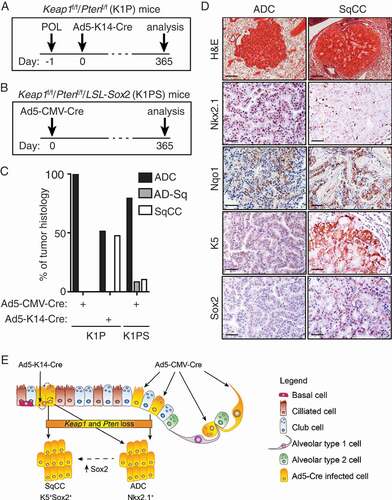
Genomic profiling studies have enabled the stratification of lung SqCC into 4 molecular subgroups: classical (36%), basal (25%), secretory (24%) and primitive (15%) [Citation3,Citation55]. The classical subtype is characterized by alterations in the KEAP1-NRF2 pathway, as well as inactivation and/or loss of the tumor suppressor gene PTEN and amplification of chromosome 3q, containing the SOX2, TP63 and PIK3CA genes [Citation3]. SOX2 has been hypothesized to act as a lineage-specific oncogene in human lung SqCC, where is found to be amplified and/or overexpressed in 56% of cancers [Citation56]. Indeed, more recently, we and others have shown that overexpression of SOX2 is a potent driver of lung SqCC formation when combined with either loss of Pten and Cdkn2ab [Citation51] or loss of Lkb1 alone [Citation57]. SOX2 overexpression, however, can induce SqCC formation from Club (CC10-expressing), alveolar type 2 (AT2; SPC-expressing), in addition to basal progenitor cells, suggesting that enforced SOX2 expression, rather than the initiating cell, dictates the resulting tumor phenotype [Citation51]. We therefore hypothesized that overexpression of Sox2 in combination with loss of both Keap1 and Pten in basal cells, would promote lung SqCC with high penetrance. To address this, we crossed LSL-Sox2 mice [Citation51] with Keap1f/f;Ptenf/f mice [Citation6] (LSL-Sox2;Keap1f/f;Ptenf/f, hereafter K1PS mice) (). Consistent with observations made in K1P mice following Ad5-CMV-Cre infection, pronounced bronchiolar hyperplasia was observed throughout the large and small airways of K1PS mice (data not shown). In addition however, tumors exhibiting characteristics of both ADC and SqCC were observed in K1PS mice, suggesting that enhanced Sox2 expression promotes ADC-SqCC transdifferentiation [Citation58] (), with the most progessed tumors detected in the lungs of K1PS mice exhibiting a pure SqCC tumor phenotype. These data suggest that alterations in the KEAP1-NRF2 pathway can promote SqCC development, but this is dependent upon on the cellular origins and/or the genotype of co-occurring mutations ().
NRF2 regulation of tumor metabolism
Tumor cells utilize a high rate of glycolysis to generate macromolecules essential for the synthesis of new DNA, RNA, cellular membranes and protein [Citation59,Citation60]. Indeed, lung cancers express high levels of enzymes involved in glycolysis, with a significant correlation between the glucose transporter GLUT1 and aggressive ADC [Citation61]. In addition to regulating genes that confer the inducible resistance to xenobiotic and oxidative stress, NRF2 activity alters the expression of key metabolic enzymes [Citation15,Citation16]. Key studies of altered lung cancer metabolism have utilized the A549 cell line, a human lung ADC with KEAP1 LOF via point mutation (KEAP1G333C) and loss of heterozygosity (19p13.2) [Citation21]. Using A549 cells, in vitro studies identified a key role for NRF2-activated transcription in enhancing serine biosynthesis and pentose phosphate pathway (PPP) activity to generate key metabolic intermediates for tumor cell proliferation [Citation44,Citation62,Citation63]. Recently, in vivo studies have detected the altered metabolism of Keap1-mutant lung tumors as a distinct metabolite signature peripherally in the plasma of tumor-bearing GEMMs [Citation6]. Specifically, serum levels of lactate and sugar phosphates (Glc6P, Rib5P, and Ru5P), as well as a number of nucleotides and nucleosides, were significantly decreased in tumor-bearing mice, consistent with an increase in PPP enzyme transaldolase 1 (Taldo1) expression.
Taldo1 is a key enzyme in the non-oxidative arm of the PPP, generating ribose 5-phosphate (Rib5P) and NADPH, essential for cell proliferation. In rapidly proliferating cancer cells, over 85% of ribonucleotides are derived from the non-oxidative PPP [Citation64]. Enhanced PPP activity is achieved through the constitutive activation of NRF2, and results in enhanced proliferation rates and ribose synthesis in A549 cells [Citation44,Citation63]. Tumor studies using a YFP reporter of Keap1 loss, and thus Nrf2 nuclear localization, have identified key regulatory patterns of enzyme regulation in NSCLC [Citation6]. Though expression of PPP enzymes G6PD, Tkt, Pgd and Taldo1 were increased in the tumor as a whole compared to untransformed lung epithelial cells, only the expression of Taldo1 was specific to YFP positive tumor cells [Citation6]. Indeed, the expression of G6PD, Tkt and Pgd are regulated generally by an oxidative stress axis involving HDAC4 with miR-1 and miR-206 [Citation63]. Moreover, while G6PD, TKT and PGD promoters contain an ARE that can be bound by NRF2 [Citation58], the location is distant from the transcription start site suggesting that additional elements are required for robust gene activation. TALDO1 alone has been confirmed in two independent reports to be directly regulated by NRF2 [Citation44,Citation63], highlighting the direct relationship between NRF2 and the non-oxidative arm of the PPP.
In addition to the PPP, NRF2 regulates the expression of enzymes involved in the serine/glycine biosynthetic pathway [Citation62]. Serine and glycine are important metabolic intermediates in the synthesis of proteins, nucleic acids and lipids that support tumor processes [Citation15,Citation65]. Through the NRF2-dependent regulation of Activating Transcription Factor 4 (ATF4), the enzymes PHGDH, PSAT1 and SHMT2 are transcribed in KEAP1–mutant NSCLC cells that render these cancer cells self-sufficient in the biosynthesis of serine and glycine [Citation62]. Importantly, the production of serine and glycine was capable of supporting glutathione (GSH) biosynthesis, purine biosynthesis, thymidine production and NADPH levels, supporting NSCLC cell growth [Citation62]. Furthermore, KEAP1 LOF renders mouse and human cells dependent on the non-essential amino acid glutamine, crucial for the generation of the antioxidant GSH [Citation66].
The NRF2 pathway is intimately linked to metabolic processes that are essential for cancer cell proliferation and survival. The enhanced utility of these pathways creates unique characteristics of KEAP1– and/or NRF2-mutant NSCLC, that can be harnessed as both a biomarker [Citation6] and exploited for targeted therapy, discussed below.
Targeting the KEAP1-NRF2 pathway in the treatment of cancer
Cancers with high NRF2 levels are associated with poor prognosis [Citation67], due to their chemotherapy and radiotherapy resistance and proliferative nature [Citation50,Citation68–Citation70]. This may, in part, be due to the NRF2 mediated up-regulation of Multidrug Resistance-associated Protein 1 (MRP1/ABCC1) [Citation71]. MRP1 is an energy-dependent efflux pump that exports bulky hydrophobic molecules from the cell, such as Paclitaxel, Methotrexate, Doxorubicin and Flutamide chemotherapeutic classes [Citation72]. MRP1 is expressed frequently in lung cancer and is a predictor of poor response to chemotherapy [Citation73,Citation74]. NRF2-mediated upregulation of MRP1 may be a direct mediator of resistance to standard lung cancer treatment and thus targeted treatment to mitigate the activity of NRF2 are urgently required.
Direct inhibition of NRF2
The activity of NRF2 promotes resistance to platinum-based chemotherapy, the current first-line treatment of NSCLC, and is thus itself a molecular target for therapy prompting the identification of NRF2 inhibitors [Citation75]. In cancer cell lines, Brusatol [Citation76], Trigonelline [Citation77], Ascorbic acid [Citation78], Chrysin (5,7-digydroxyflavone) [Citation79], Apigenin (4ʹ,5,7-trihydroxyflavone) [Citation80] and Luteolin (3ʹ,4ʹ,5,7-tetrahydroxyflavone) [Citation81] were found to be effective inhibitors of NRF2 pathway activity. Specifically, the activity of brusatol and luteolin were investigated in A549 lung ADC cells. Brusatol acts to reduce the abundance of NRF2 through enhanced ubiquitination and degradation, reducing NRF2-protective response in chemotherapy treated cell lines. Significantly, brusatol sensitized A549 xenograft models to the activity of cisplatin in a KEAP1-NRF2 dependent mechanism [Citation76]. Similarly, the flavonoid luteolin accelerates NRF2 mRNA turnover and sensitizes A549 cells to oxaliplatin, bleomycin and doxorubicin in cell line viability assays [Citation81]. Additionally, targeted chemical screening approaches have identified NRF2 inhibitors, such as ML385 [Citation82] and AEM1 [Citation83], that similarly sensitize KEAP1-mutant NSCLC cell lines to combination therapy. Though these compounds provide strong evidence of activity in cell lines, no clinical trials in lung cancer have yet been performed to validate the beneficial effect of NRF2 inhibition on sensitizing ADC to chemotherapy. These studies strongly suggest there is an advantage to decreasing NRF2 stabilization in promoting sensitivity of lung ADC cells to platinum-based chemotherapeutic agents.
Inhibition of NRF2 downstream metabolic pathways
Lung cancer with high NRF2 activity, determined via the expression level of NQO1 [Citation6,Citation7], have enhanced PPP and serine biosynthetic pathway enzyme expression, consistent with preclinical studies [Citation6,Citation7,Citation62]. Abrogation of these pathways in culture have indicated that metabolic targets may be viable treatment options in lung cancer with highly active NRF2. Indeed, blocking the oxidative and non-oxidative arms of the PPP using combined knockdown of G6PD and TKT repressed growth of A549 in vitro and in xenograft models in vivo [Citation44]. Similarly, knockdown of serine and glycine biosynthesis enzyme PHGDH reduced colony formation and xenograft growth in A549 KEAP1-mutant cell lines [Citation62]. Utilizing a translational approach, a small molecule inhibitor of PHGDH has been developed (CBR-5884) and was found to be efficacious in cancer cells with high serine biosynthetic activity [Citation84]. Compellingly, drug studies in cell lines and orthotopic transplant models of KP and KrasG12D/p53/Keap1 spontaneous lung tumors found that Keap1-mutant lung tumors are sensitive to the glutaminase inhibitor CB-839 [Citation7,Citation66]. In addition to rational drug design, an unbiased chemical screen recently identified a GLUT8-dependent pathway that selectively sensitized KEAP1-mutant cells based on their consumption of glucose to support serine biosynthetic pathway activity [Citation85]. These data, while still preliminary, provide evidence that targeting downstream GSH and PPP metabolic pathways are viable in NRF2-active lung cancers, and highlight the dependence of these cancers on NRF2-driven metabolic pathways.
Immunotherapy in KEAP1-mutant lung cancers
Immunotherapy provides additional promise in KEAP1-mutant lung tumors. Unleashing a patient’s autologous T cells to target tumors has received attention in lung cancer due to a high mutation load and level of immune infiltration [Citation86,Citation87]. In a recent clinical trial (KEYNOTE-189), non-squamous NSCLC (excluding EGFR or ALK mutant ADC) found that the addition of Pembrolizumab to chemotherapy enhanced both the progression-free and overall survival of lung cancer patients [Citation88]. GEMMs are crucial for preclinical studies of immunotherapy, due to the spontaneous generation of a primary tumor in an intact immune microenvironment. The efficacy of checkpoint inhibition using monoclonal antibodies that target programmed cell death protein-1 (PD-1) has been tested in a number of lung cancer GEMMs, and while the efficacy was low in KP GEMMs [Citation89,Citation90], sensitivity to immunotherapy was achieved in the NRF2-active K1P GEMM [Citation6]. This suggests that activation of the NRF2 pathway may provide additional immunomodulation that sensitizes these ADC to immunotherapy. Significantly, RNA sequencing data provide insight into the immune landscape of ADC [Citation4]. Analysis of lung ADC with high NQO1 expression identified significantly reduced immune infiltration signatures [Citation6]. Similarly, mutation in KEAP1 correlates with reduced leukocyte infiltration in lung ADC [Citation91]. Together, these findings suggest that aberrant NRF2 pathway activity alters the immune microenvironment of lung ADC, which improves the efficacy of immunotherapy. Indeed, the K1P GEMM exhibited increased PD-L1 tumor cell expression [Citation6], and enhanced serine biosynthesis results in phosphotidyl-serine (PS) exposure on epithelial cells [Citation15], together suggesting that NRF2-active tumor cells can mediate immune evasion in the tumor microenvironment. Further studies are required to verify whether NRF2 pathway activation correlates with immunotherapy efficacy in clinical trials, however preclinical evidence suggests that there is significant merit in selecting patients with high NQO1 expression for treatment with immunotherapy.
On multiple levels of tumor progression and resistance to treatment, NRF2 transcriptional activity provides advantages to the cancer cell. Proliferative advantages arise from resistance to oxidative stress, enhanced NAPDH production and metabolic pathway activity. In addition to the advantages of these cellular systems under toxic chemotherapy insult, enhanced efflux pump expression exports from the cell the classes of chemotherapeutic drugs that are most commonly used to treat lung cancer. Thus, alternative treatments to target this aggressive subset of lung cancer are urgently required. While metabolic targets and immunotherapy provide limited tumor control, additional studies are required to identify synergistic combinations to control KEAP1-NRF2 altered tumors. This review highlights recent findings in the role of KEAP1-NRF2 pathway in lung cancer and provides hope for additional advances in the field for the treatment of this aggressive disease.
Acknowledgments
We are grateful to A. Kersbergen for technical assistance and C. Alvarado, L. Scott and H. Johnson for animal husbandry; E. Tsui in the WEHI Histology Facility for expert support; A. Berns (Netherlands Cancer Institute) for LSL-Sox2 conditional mice; and The University of Iowa Gene Transfer Core Facility for purified Ad5-Cre viruses. All experiments presented in this manuscript were conducted according to regulatory standards approved by the Walter and Eliza Hall Institute Animal Ethics Committee. We are thankful to A. Cox (Peter MacCallum Cancer Centre) and Giustina Ferone (Netherlands Cancer Institute) for critical reading of the manuscript. This work was supported by an Australian Health and Medical Research Council (NHMRC) grant awarded to K.D.S (APP1138275). S.A.B is supported by a Victorian Cancer Agency (VCA) Early Career Seed Grant (ECSG16001); K.D.S is supported by the Peter and Julie Alston Centenary Fellowship. This work is made possible through Victorian Government Operational Infrastructure Support and the Australian government.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Fitzmaurice C, Allen C, Rm B, et al.; Global Burden of Disease Cancer C. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: A systematic analysis for the global burden of disease study. JAMA Oncol. 2017;3:524–548.
- George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53.
- Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525.
- Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550.
- Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39:199–218.
- Best SA, De Souza DP, Kersbergen A, et al. Synergy between the KEAP1/NRF2 and PI3K Pathways Drives Non-Small-Cell Lung Cancer with an Altered Immune Microenvironment. Cell Metab. 2018;27:935–43 e4.
- Romero R, Sayin VI, Davidson SM, et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med. 2017;23:1362–1368.
- Chen W, Sun Z, Wang X-J, et al. Direct interaction between Nrf2 and p21 Cip1/WAF1 upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009;34:663–673.
- Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213.
- Lau A, Wang X-J, Zhao F, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol. 2010;30:3275–3285.
- Salazar M, Rojo AI, Velasco D, et al. Glycogen synthase kinase-3β inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem. 2006;281:14841–14851.
- Joo MS, Kim WD, Lee KY, et al. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol Cell Biol. 2016;36:1931–1942.
- Alexander C-M. The involvement of NRF2 in lung cancer. Oxid Med Cell Longev. 2013;2013.
- Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–2191.
- Lee SB, Sellers BN, DeNicola GM. The Regulation of NRF2 by Nutrient-Responsive Signaling and Its Role in Anabolic Cancer Metabolism. Antioxid Redox Signal. 2017 Oct 16.
- Menegon S, Columbano A, Giordano S. The dual roles of NRF2 in cancer. Trends Mol Med. 2016;22:578–593.
- Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–188.
- Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564.
- Padmanabhan B, Tong KI, Ohta T, et al. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006;21:689–700.
- Ohta T, Iijima K, Miyamoto M, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008;68:1303–1309.
- Singh A, Misra V, Thimmulappa RK, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420.
- Canning P, Sorrell FJ, Bullock AN. Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med. 2015;88:101–107.
- Campbell JD, Alexandrov A, Kim J, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016;48:607–616.
- Frank R, Scheffler M, Merkelbach-Bruse S, et al. Clinical and Pathological Characteristics of KEAP1-and NFE2L2-Mutated Non–small Cell Lung Carcinoma (NSCLC). Clin Cancer Res. 2018: 24:3087-3096.
- Luo W, Tian P, Wang Y, et al. Characteristics of genomic alterations of lung adenocarcinoma in young never‐smokers. International Journal of Cancer. 2018 Apr 18.
- George J, Walter V, Peifer M, et al. Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nat Commun. 2018;9:1048.
- Mollaoglu G, Guthrie MR, Böhm S, et al. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell. 2017;31:270–285.
- Sanchez-Vega F, Mina M, Armenia J, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018;173(321–37):e10.
- Kim YR, Oh JE, Kim MS, et al. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J Pathol. 2010;220:446–451.
- Wakabayashi N, Itoh K, Wakabayashi J, et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet. 2003;35:238–245.
- Shibata T, Ohta T, Tong KI, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci. 2008; 105:13568–13573.
- Kwon M-C, Berns A. Mouse models for lung cancer. Mol Oncol. 2013;7:165–177.
- Blake DJ, Singh A, Kombairaju P, et al. Deletion of Keap1 in the lung attenuates acute cigarette smoke-induced oxidative stress and inflammation. Am J Respir Cell Mol Biol. 2010;42:524–536.
- Skoulidis F, Byers LA, Diao L, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5:860–877.
- Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248.
- DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009;4:1064–1072.
- Jackson EL, Olive KP, Tuveson DA, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–10288.
- Winslow MM, Dayton TL, Verhaak RG, et al. Suppression of lung adenocarcinoma progression by Nkx2–1. Nature. 2011;473:101.
- Ji H, Ramsey MR, Hayes DN, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807.
- Li F, Han X, Li F, et al. LKB1 inactivation elicits a redox imbalance to modulate non-small cell lung cancer plasticity and therapeutic response. Cancer Cell. 2015;27:698–711.
- Rogers ZN, McFarland CD, Winters IP, et al. A quantitative and multiplexed approach to uncover the fitness landscape of tumor suppression in vivo. Nat Methods. 2017;14:737.
- Rogers ZN, McFarland CD, Winters IP, et al. Mapping the in vivo fitness landscape of lung adenocarcinoma tumor suppression in mice. Nat Genet. 2018;50:483-486.
- Sánchez-Rivera FJ, Papagiannakopoulos T, Romero R, et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature. 2014;516:428.
- Mitsuishi Y, Taguchi K, Kawatani Y, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79.
- Taguchi K, Hirano I, Itoh T, et al. Nrf2 enhances cholangiocyte expansion in Pten-deficient livers. Mol Cell Biol. 2014;34:900–913.
- Weeden CE, Chen Y, Ma SB, et al. Lung Basal Stem Cells Rapidly Repair DNA Damage Using the Error-Prone Nonhomologous End-Joining Pathway. PLoS Biol. 2017;15:e2000731.
- Sutherland KD, Proost N, Brouns I, et al. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell. 2011;19:754–764.
- Sutherland KD, Song JY, Kwon MC, et al. Multiple cells-of-origin of mutant K-Ras-induced mouse lung adenocarcinoma. Proc Natl Acad Sci U S A. 2014;111:4952–4957.
- Sutherland KD, Berns A. Cell of origin of lung cancer. Mol Oncol. 2010;4:397–403.
- Jeong Y, Hoang NT, Lovejoy A, et al. Role of KEAP1/NRF2 and TP53 mutations in lung squamous cell carcinoma development and radiation resistance. Cancer Discov. 2017;7:86–101.
- Ferone G, Song JY, Sutherland KD, et al. SOX2 Is the Determining Oncogenic Switch in Promoting Lung Squamous Cell Carcinoma from Different Cells of Origin. Cancer Cell. 2016;30:519–532.
- Best SA, Kersbergen A, Asselin-Labat ML, et al. Combining Cell Type-Restricted Adenoviral Targeting with Immunostaining and Flow Cytometry to Identify Cells-of-Origin of Lung Cancer. Methods Mol Biol. 2018;1725:15–29.
- Borthwick DW, Shahbazian M, Todd Krantz Q, et al. Evidence for stem-cell niches in the tracheal epithelium. Am J Respir Cell Mol Biol. 2001;24:662–670.
- Parsons DW, Grubb BR, Johnson LG, et al. Enhanced in vivo airway gene transfer via transient modification of host barrier properties with a surface-active agent. Hum Gene Ther. 1998;9:2661–2672.
- Wilkerson MD, Yin X, Hoadley KA, et al. Lung squamous cell carcinoma mRNA expression subtypes are reproducible, clinically important, and correspond to normal cell types. Clinical Cancer Research: an Official Journal of the American Association for Cancer Research. 2010;16:4864–4875.
- Bass AJ, Watanabe H, Mermel CH, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41:1238–1242.
- Mukhopadhyay A, Berrett KC, Kc U, et al. Sox2 cooperates with Lkb1 loss in a mouse model of squamous cell lung cancer. Cell Rep. 2014;8:40–49.
- Han X, Li F, Fang Z, et al. Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat Commun. 2014;5:3261.
- Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519.
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033.
- Giatromanolaki A, Sivridis E, Arelaki S, et al. Expression of enzymes related to glucose metabolism in non-small cell lung cancer and prognosis. Exp Lung Res. 2017;43:167–174.
- DeNicola GM, Chen PH, Mullarky E, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47:1475–1481.
- Singh A, Happel C, Manna SK, et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Invest. 2013;123:2921–2934.
- Boros LG, Puigjaner J, Cascante M, et al. Oxythiamine and dehydroepiandrosterone inhibit the nonoxidative synthesis of ribose and tumor cell proliferation. Cancer Res. 1997;57:4242–4248.
- Amelio I, Cutruzzolá F, Antonov A, et al. Serine and glycine metabolism in cancer. Trends Biochem Sci. 2014;39:191–198.
- Sayin VI, LeBoeuf SE, Singh SX, et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife. 2017 Oct 2;6. e28083.
- Solis LM, Behrens C, Dong W, et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clinical Cancer Research: an Official Journal of the American Association for Cancer Research. 2010;16:3743–3753.
- Bai X, Chen Y, Hou X, et al. Emerging role of NRF2 in chemoresistance by regulating drug-metabolizing enzymes and efflux transporters. Drug Metab Rev. 2016;48:541–567.
- McDonald J, Kim K, Norris A, et al. Ionizing radiation activates the Nrf2 antioxidant response. Cancer Res. 2010;70:8886–8895.
- Krall EB, Wang B, Munoz DM, et al. KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. eLife. 2017 Feb 1;6. e18970.
- Ji L, Li H, Gao P, et al. pathway regulates multidrug-resistance-associated protein 1 in small cell lung cancer. PloS One. 2013;8:e63404.
- Munoz M, Henderson M, Haber M, et al. Role of the MRP1/ABCC1 multidrug transporter protein in cancer. IUBMB Life. 2007;59:752–757.
- Berger W, Setinek U, Hollaus P, et al. Multidrug resistance markers p-gp, MRP1 and lung resistance protein in non-small cell lung cancer: prognostic implications. J Cancer Res Clin Oncol. 2005;131:355–363.
- Hsia T-C, Lin -C-C, Wang -J-J, et al. Relationship between chemotherapy response of small cell lung cancer and P-glycoprotein or multidrug resistance-related protein expression. Lung. 2002;180:173–179.
- No J, Kim Y, Song Y. Targeting nrf2 signaling to combat chemoresistance. J Cancer Prev. 2014;19:111–117.
- Ren D, Villeneuve NF, Jiang T, et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci. 2011;108:1433–1438.
- Arlt A, Sebens S, Krebs S, et al. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene. 2013;32:4825.
- Tarumoto T, Nagai T, Ohmine K, et al. Ascorbic acid restores sensitivity to imatinib via suppression of Nrf2-dependent gene expression in the imatinib-resistant cell line. Exp Hematol. 2004;32:375–381.
- Gao A-M, Ke Z-P, Shi F, et al. Chrysin enhances sensitivity of BEL-7402/ADM cells to doxorubicin by suppressing PI3K/Akt/Nrf2 and ERK/Nrf2 pathway. Chem Biol Interact. 2013;206:100–108.
- Gao A-M, Ke Z-P, Wang J-N, et al. Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway. Carcinogenesis. 2013;34:1806–1814.
- Tang X, Wang H, Fan L, et al. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radic Biol Med. 2011;50:1599–1609.
- Singh A, Venkannagari S, Oh KH, et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem Biol. 2016;11:3214–3225.
- Bollong MJ, Yun H, Sherwood L, et al. A small molecule inhibits deregulated NRF2 transcriptional activity in cancer. ACS Chem Biol. 2015;10:2193–2198.
- Mullarky E, Lucki NC, Beheshti Zavareh R, et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc Natl Acad Sci U S A. 2016;113:1778–1783.
- McMillan EA, Ryu M-J, Diep CH, et al. Chemistry-first approach for nomination of personalized treatment in lung cancer. Cell. 2018;173(864–78):e29.
- Chalmers Z, Connelly C, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017; 9(1):34.
- Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128.
- Gandhi L, Rodríguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus Chemotherapy in Metastatic Non–small-Cell Lung Cancer. New England J Med. 2018;378(22):2078-2092.
- Akbay EA, Koyama S, Carretero J, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355–1363.
- Pfirschke C, Engblom C, Rickelt S, et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity. 2016;44:343–354.
- Thorsson V, Gibbs DL, Brown SD, et al. The immune landscape of cancer. Immunity. 2018;48(812–30):e14.