
ABSTRACT
From early-onset Alzheimer’s disease (EOAD) studies, the amyloid-beta hypothesis emerged as the foremost theory of the pathological causes of AD. However, how amyloid-beta accumulation is triggered and progresses toward senile plaques in spontaneous late-onset Alzheimer’s disease (LOAD) in humans remains unanswered. Various LOAD facilitators have been proposed, and LOAD is currently considered a complex disease with multiple causes. Mice do not normally develop LOAD. Possibly due to the multiple causes, proposed LOAD facilitators have not been able to replicate spontaneous LOAD in mice, representing a disease modeling issue. Recently, we reported spontaneous late-onset development of amyloid-beta accumulation in brains of Shugoshin 1 (Sgo1) haploinsufficient mice, a cohesinopathy-mediated chromosome instability model. The result for the first time expands disease relevance of mitosis studies to a major disease other than cancers. Reverse-engineering of the model would shed light on the process of late-onset amyloid-beta accumulation in the brain and spontaneous LOAD development, and contribute to development of interventions for LOAD. This review will discuss the Sgo1 model, our current “three-hit hypothesis” regarding LOAD development with an emphasis on critical role of prolonged mitosis in amyloid-beta accumulation, and implications for human LOAD intervention and treatment.
Abbreviations: Alzheimer's disease (AD); Late-onset Alzheimer's disease (LOAD); Early-onset Alzheimer's disease (EOAD); Shugoshin-1 (Sgo1); Chromosome Instability (CIN); apolipoprotein (Apoe); Central nervous system (CNS); Amyloid precursor protein (APP); N-methyl-d-aspartate (NMDA); Hazard ratio (HR); Cyclin-dependent kinase (CDK); Chronic Atrial Intestinal Dysrhythmia (CAID); beta-secretase 1 (BACE); phosphor-Histone H3 (p-H3); Research and development (R&D); Non-steroidal anti-inflammatory drugs (NSAIDs); Brain blood barrier (BBB)
Introduction
Spontaneous late-onset Alzheimer’s disease (LOAD) accounts for more than 95% of all human AD. The major biomarkers for AD are categorized as (1) histopathological biomarkers and (2) cognitive/behavioral biomarkers. Histopathological biomarkers include (a) accumulation of amyloid-beta in the central nervous system (CNS), (b) accumulation of TAU and phosphorylated TAU in the CNS, (c) cerebral amyloid angiopathy/congophilic angiopathy (amyloid deposits forming in the walls of the blood vessels of the CNS), and (d) neurodegeneration. Cognitive/behavioral biomarkers include declines in cognitive function and behavioral integrity, which are assessed with interviews and observation [Citation1–Citation4].
The remaining 5% of human AD is early-onset AD (EOAD). The biomarkers of EOAD are identical to those of LOAD, except for earlier onset. EOAD is linked to genetic mutations in amyloid metabolism genes, such as amyloid precursor protein (APP), presenilin, tau, and apolipoprotein E (ApoE) [Citation4]. EOAD researchers made great contributions to establish the contemporary “amyloid beta hypothesis” for AD [Citation5,Citation6]. Although there still is a room for debate in the point how much of AD symptoms solely depends on amyloid metabolism defect [Citation7], amyloid-beta accumulation usually precede TAU accumulation, placing amyloid-beta accumulation as the initial step for AD [Citation1].
As of 2018, approximately 5 million people in the U.S. alone live with AD, and an increase in LOAD incidence is predicted through demographic analysis [Citation8,Citation9]. However, effective intervention and therapeutic measures for LOAD have not been developed. This unfortunate state is not due to lack of efforts. Industrial pharmaceutical coalitions have conducted clinical trials for approximately 400 AD drug candidates; however, more than 98% of the candidate drugs have failed [Citation10–Citation13]. The high failure rate may be because the exact mechanism for spontaneous LOAD development remains unclear. Existing AD drugs in clinics are categorized as 1) cholinesterase inhibitors, such as Donepezil/Aricept, or 2) memantine, which regulates the activity of glutamate, both of which aid neuronal functions. However, these drugs provide only temporarily relief from decline in cognitive functions, and cannot reverse the underlying pathological cause, i.e. amyloid-beta plaques, of LOAD [Citation14]. Concerns about the validity of the drug research and development targets, most of which are components of the amyloid metabolism pathway, persisted. Recent report in July 2018 of phase-II clinical trial with BAN2401 (amyloid-beta protofibril targeting antibody [Citation15,Citation16]) 18-month data, indicating a significant improvement in early phase LOAD patients, provided a hope for drugs targeting amyloid metabolism. Yet, further translational processes including Phase III trials await.
With insufficient knowledge of LOAD development, development of rodent models for LOAD suffered. Rodents do not normally develop LOAD, and few amyloid-beta accumulation is observed in mice at the age of 24 months. The existing AD mouse models are EOAD models that rely on expression(s) of modified EOAD-associated protein(s), including APP, TAU, presenilin, and ApoE [Citation17–Citation21]. EOAD models successfully replicate extracellular amyloid plaques short-term (time for plaque onset: 9–12 months for TG2576, 6 months for 3xTG, 2 months for APP751SL/PS1KI) [Citation22]. However, since the EOAD models are overexpression models, it is questionable whether they recapitulate prerequisites for spontaneous LOAD that may occur with age in humans.
In human LOAD, various aggravating or facilitating factors for AD, including decreased acetylcholine, over-activation of N-methyl-d-aspartate (NMDA) receptors by glutamate, oxidative stress, and inflammation, have been identified [Citation8,Citation9]. Established risk factors/conditions include diabetes (Hazard Ratio [HR] 1.76; 95% confidence interval, 1.50–2.07, p < 0.001), age (HR 1.11), hypertension (HR 1.3), and stroke history (HR1.79) [Citation23]. However, how these factors are translated to amyloid-beta accumulation in the brain at the molecular level must be elucidated. This review will focus on the Sgo1 mouse model, the spontaneous late-onset accumulation of amyloid-beta in the brains of which is newly identified, and depicts the current working hypothesis for the mechanism of amyloid-beta accumulation with an emphasis on the role of prolonged mitosis in the brain. Uncovering the LOAD model provides hopes of rapid translational application toward human LOAD intervention and/or therapy.
Genomic instability, especially chromosome instability, and AD
Chromosome instability (CIN) is a mitotic error-driven type of genomic instability leading to aneuploidy. Although a causal link between CIN, aneuploidy, and AD had not been established, some results suggested such a link between genomic instability and AD. High rates of genomic instability and aneuploidy are present in the human AD brain [Citation24]. Genomic instability biomarkers have been associated with mild cognitive impairment and AD [Citation25]. Aneuploidy may facilitate development of AD-like dementia, since 15% of patients with Down syndrome with chromosome 21 trisomy develop AD-like cognitive dysfunction in their 40s, and the rate increases to 50–70% by age 60 [Citation8,Citation26]. BubR1 is a spindle checkpoint component, and its mutation, knockdown, or haploinsufficiency causes CIN [Citation27–Citation29] and cancer-proneness [Citation30,Citation31]. The hypomorph BubR1h/h mouse was identified as a progeria model [Citation32–Citation34]. siRNA-mediated BubR1 inhibition in mouse brains led to inhibition of neuronal cell division, myelination, and axon growth [Citation35,Citation36], suggesting that BubR1-mediated genomic instability may lead to neurodegenerative disease, including AD. In addition, cohesinopathy is another cause of CIN, showing signs of premature separation of chromosomes. Cohesinopathy in peripheral blood lymphocytes is associated with AD [Citation37–Citation39].
The above findings suggest that genomic instability may play a causal role in AD development. However, in part due to the long maintenance time (24+ months) required for age-associated disease studies, few studies have been conducted on AD-associated brain pathology in genomic instability mouse models.
CIN mouse models have been used to study carcinogenesis
Genomic instability and aneuploidy were theorized to cause cancer [Citation40]. CIN mouse models were generated for carcinogenesis study purposes under a long-standing hypothesis that CIN and aneuploidy would facilitate carcinogenesis [Citation41–Citation45]. Many of the CIN models showed both oncogenic (tumor-prone) and tumor-suppressive characteristics in organ- and targeted gene-specific manners, indicating the influence of CIN and aneuploidy on carcinogenesis in a manner more complex than that suggested by the original hypothesis [Citation46–Citation48]. The current interpretation of the “double-edged sword” effect of genomic instability on carcinogenesis is that a “modest” degree of genomic instability facilitates carcinogenesis through increasing mutational accumulation, while “too high” a degree of genomic instability can lead to cell death or senescence, thus serving as a tumor suppressor [Citation49,Citation50].
Shugoshin 1 haploinsufficient mice (Sgo1-/+) showed cohesinopathy-CIN, cancer-proneness, and amyloid-beta accumulation in the brain in old age
The Sgo1−/+ mouse is one of the CIN models. Sgo1 protein has two functions in the cell cycle: 1) to protect mitotic chromosome cohesion from premature separation [Citation51,Citation52], and 2) to protect centrosome integrity [Citation53–Citation55]. Reduction of Sgo1 by siRNA in cultured cells [Citation51,Citation52,Citation55] by haploinsufficiency (-/+) in transgenic models [Citation56–Citation58] or expression of an aberrant form (e.g. loss, dominant negative) of Sgo1 in human lung, liver, and colon cancer [Citation59–Citation63] leads to cohesinopathy in mitotic chromosomes (premature chromosome separation) and in the centrosome (mitosis with multipolar spindles). The Sgo1 defect-mediated cohesinopathy subsequently provokes the mitotic spindle checkpoint. The mitotically-challenged cells will have prolonged mitosis with high mitotic CDK-cyclin A/B activity, a state that may lead to mitotic catastrophe and cell death. With colonic carcinogen azoxymethane challenge, the Sgo1−/+ mouse also showed enhanced initial development of colonic precancerous lesions and adenocarcinomas compared with wild type [Citation56]. These mice were prone to spontaneous cancers in the liver and lung at middle age [Citation57,Citation58,Citation64]. Using comparative RNAseq, we demonstrated that these cancer-prone organs (i.e. colon, liver, lung) differentially expressed genes with demonstrated cancer-association [Citation64,Citation65]. The results suggest that CIN alone can modify global gene expression and introduce local proneness (a field effect) to carcinogenesis. As aneuploidy alone also can modify global gene expression [Citation66], can give proteotoxic stress to cells [Citation67,Citation68] and can modulate autophagy [Citation69], genomic instability can have a broad impact on cellular physiology. The findings also suggest that such a CIN-specific gene expression signature and modulation of carcinogenic pathways can be targeted for cancer prevention purposes [Citation64,Citation65].
The human homologue is SgoL1. Mutations, dosage alterations, and dominant negative expression of SgoL1 have been reported in various human cancers [Citation59–Citation63]. Congenital mutation in SgoL1 leads to Chronic Atrial Intestinal Dysrhythmia (CAID) syndrome, a rare condition affecting the heart and the digestive system [Citation70]. Human SgoL1-associated diseases outside of the cancer context are under-investigated, in part because of the rarity of congenital diseases. Mouse Sgo1 is highly expressed in the central nervous system and heart during early development [Citation71,Citation72], and the Sgo1 knockout (-/-) in mice [Citation56] and siRNA-mediated SgoL1 knockdown [Citation51,Citation52,Citation59] in human cultured cells are lethal. Thus, it may be difficult to create mutations compatible with development. To our knowledge, no direct LOAD-SgoL1 association has been demonstrated, likely because SgoL1 has not been investigated in the context of LOAD. However, the functional equivalent of SgoL1 mutation (i.e. cohesinopathy, genomic instability, aneuploidy) has been reported frequently in patients with LOAD [Citation26,Citation37–Citation39,Citation73].
We have used Sgo1−/+ and BubR1−/+ models in the context of carcinogenesis study. The models are genomic instability models that commonly show mitotic errors and CIN at the cellular level, and proneness to carcinogenesis in critical organs. In our 2017 review article, we pointed out that links among genomic instability, aging, and cancer were emerging [Citation73], and that we intended to test whether genomic instability facilitates carcinogenesis with age in these models. Thus, we conducted a study on aging and carcinogenesis using these two models and a wild-type control cohort. With the aforementioned rationale stated in the “Genomic instability, CIN, and AD” section, we hypothesized that LOAD development is facilitated by genomic instability in the brain. Taking advantage of the ongoing cancer-and-aging study cohorts, we tested Sgo1−/+, BubR1−/+, and wild-type mice for amyloid-beta accumulation, anticipating that Sgo1−/+ and BubR1−/+ mice would show amyloid-beta accumulation. Unexpectedly, only Sgo1−/+ mice showed amyloid-beta accumulation in the brain in old age [Citation74].
Late-onset accumulation of amyloid-beta in Sgo1 brain
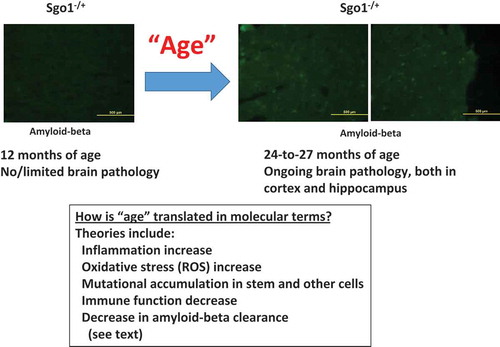
Brains from 24-month-old (equivalent of 65 years and older in humans) Sgo1−/+ mice indicated amyloid-beta accumulation, while brains from 12 month-old (equivalent of middle age in humans) Sgo1 mice did not (). The “late-onset” accumulation of amyloid-beta is a major pathological biomarker for human spontaneous LOAD that precedes noticeable cognitive symptoms [Citation1–Citation3]. The “late-onset” accumulation of amyloid-beta also raises the question of how “age” is translated in terms of cellular/tissue environment, leading to the increase in amyloid-beta accumulation. Existing interrelating theories and interpretations include (i) increased inflammation [Citation75–Citation77], (ii) increased oxidative stress [Citation78–Citation80], (iii) mitochondrial dysfunction [Citation81], (iv) calcium homeostasis defect [Citation82], (v) accumulation of mutations [Citation83], (vi) stem cell fatigue and depletion [Citation84], (vii) decreased proper brain-immune crosstalk [Citation85], (viii) brain blood barrier dysfunction [Citation86], (ix) decreased amyloid-beta clearance [Citation87], (x) increased amyloid-beta deposition [Citation88], and (xi) synaptic senescence [Citation89,Citation90]. In his book The end of Alzheimer’s (2017) [Citation91], Dale Bredesen listed 36 facilitators, which can be roughly categorized as inflammation, lack of nutrition and/or hormone, and toxins. Accordingly, he proposed that there are three subtypes in human AD (i.e. inflammatory, non-inflammatory, and atypical), and that the type of each patient can be determined by profiling a set of biomarkers dominant in each type (e.g. APOE type, homocysteine, hs-CRP, IL-6, TNF-α, HbA1C, insulin, progesterone, estradiol, cortisol, vitamin D, zinc, heavy metal, mycotoxin) [Citation92]. Factors with prominent effects in the Sgo1−/+ model are being investigated, and will be determined with further time-course study focusing on these biomarkers and proposed facilitators.
Figure 1. Effects of age on AD-like pathology with amyloid-beta accumulation. In Sgo1−/+ model, amyloid-beta accumulation in the brain was not observed at the age of 12 months, but was observed at the age of 24 months and older. The accumulation occurred both in the hippocampus and in the cortex, with significant accumulation indicated in the cortex. Various factors that represent “age” have been proposed (see text).
Genomic instability alone was insufficient; apparent importance of prolonged mitosis
The accumulation of amyloid-beta in Sgo1−/+ mice was an exciting finding, while the lack of accumulation in BubR1−/+ mice was puzzling. Although both models are CIN models that indicate cancer-proneness [e.g. 30, 56], a major difference between the two models is the function of the mitotic spindle checkpoint. The Sgo1−/+ cohesinopathy model has an intact spindle checkpoint. Upon mitotic defect, Sgo1−/+ cells show prolonged mitosis. In contrast, the BubR1−/+ model is spindle-checkpoint-defective, and little delay is observed during flawed mitosis. The difference in mitotic spindle checkpoint function may have been the key. Thus, we hypothesized that prolonged mitosis is a critical step for the accumulation of amyloid-beta. Consistently, the mitotic marker phosphor-Histone H3 (pH3) indicated increases in Sgo1−/+, but not in BubR1−/+, in the brain (cerebrum) extract and in immunofluorescence analyses [Citation74].
The literature reports parallel observations suggesting mitotic involvement in human AD brains. For example, human neurofibrillary tangles co-localized with MPM2 antigens, another mitotic marker [Citation93]. Further, abnormal Tau phosphorylation of the Alzheimer-type also occurred during mitosis in human neuroblastoma SY5Y cells overexpressing Tau [Citation94]. APPThr668 phosphorylation in mitosis was correlated with increased processing of APP to generate amyloid-beta and the C-terminal fragment of APP [Citation95]. Finally, although p-H3 localization is usually limited to chromatin in many other organs, human AD brains showed a cytoplasmic, diffused pattern of p-H3 [Citation96], which was recapitulated in the Sgo1−/+ mouse brain [Citation74].
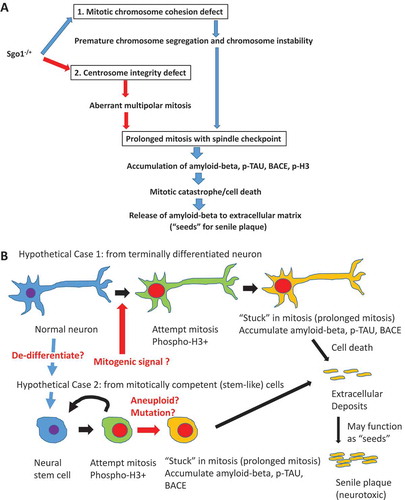
depicts our current working hypothesis. Sgo1 −/+ defects lead to spindle checkpoint-mediated prolonged mitosis, or a prolonged mitosis-like state with activation of mitotic kinase(s), which is followed by amyloid-beta, BACE, and pH3 accumulation in the cell. Once cell death occurs, the accumulated amyloid-beta is released to extracellular matrix, where the amyloid-beta can serve as “seeds” for further amyloid-beta aggregation and formation of deposits ()). Currently, the origin of “mitotic cells” and whether these cells originate from differentiated neurons or cycling stem-like cells, is unclear and is under investigation ()). The next section explores possible causes of mitotic re-entry.
Figure 2 Critical role of prolonged mitosis in amyloid-beta accumulation, suggested by Sgo1−/+ model. (a) Two consequences of Sgo1−/+ with common destinationSgo1−/+ induces two defects in the cell cycle; 1) Mitotic chromosome cohesion defect followed by premature chromosome segregation and chromosome instability, and 2) Centrosome integrity defect followed by aberrant multipolar mitosis. Both defects provoke the mitotic spindle checkpoint that leads to prolonged mitosis. Amyloid-beta (and p-TAU and BACE) accumulation occurred in mitotic marker p-H3-positive, live cells. We propose that the amyloid-beta accumulating cells in prolonged mitosis state eventually die of mitotic catastrophe, releasing amyloid-beta to extracellular matrix. As amyloid-beta possesses prion-like self-aggregating property, the released amyloid-beta may serve as “seeds” for further aggregation and eventual development of neurotoxic senile plaques. (b) “Mitotic origin of deposits” model. Whether amyloid-beta accumulating cells are originated from (case1) terminally differentiated neuron or (case 2) mitotically competent (stem-like) cells has not been determined. In “Hypothetical Case 1: from terminally differentiated neuron”, terminally differentiated neuronal cells de-differentiate to stem-like cells, or receive mitogenic signal and re-enter mitotic cycle. In “Hypothetical Case 2: from mitotically competent (stem-like) cells”, mitotically-competent stem (-like) cells “got stuck in mitosis” possibly due to aneuploidy or other mutation/stimulus that provokes spindle checkpoint leading to mitotic prolongation. In either case, cells in the state of prolonged mitosis are the source of amyloid-beta accumulation. When these cells die (of mitotic catastrophe), the accumulated amyloid-beta is released to extracellular matrix, followed by senile plaque development.
To have prolonged mitosis, mitotic re-entry should occur …but how?
Mitotic re-entry has been proposed as a critical event in human AD development [Citation97]. Based on human studies, a few hypotheses emphasizing the role of mitotic re-entry were proposed. One such hypothesis is the “simple linear model”, which states that human AD pathology develops from mitotic cycle-reentering neurons that later die [Citation98]. Another hypothesis is the “two-hit model” that purports that LOAD development occurs with (i) oxidative stress and (ii) mitotic re-entry [Citation99–Citation101]. These two models were based on observations in human LOAD brain tissues indicating increased mitotic cells.
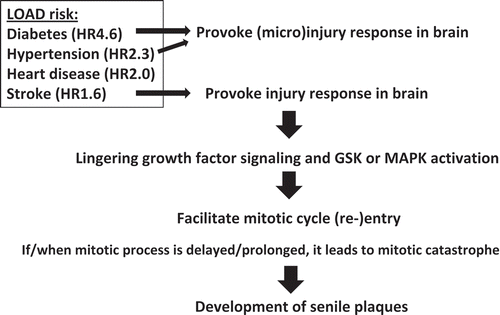
Consistent with increased number of cycling cells, mitotic cdk1 activators Cdc25A and Cdc25B show higher activity in degenerating neurons [Citation102,Citation103]. The Cdk1 inhibitor Wee1 showed lower activity in these neurons [Citation104]. Although the trigger for mitotic re-entry remains unclear, a few candidates exist. Soluble Aβ itself could trigger neuronal cell cycle re-entry [Citation105]. Tissue-injury-inducing insult, such as stroke, and micro-injury-inducing conditions, such as diabetes or hypertension [Citation106–Citation108], represent risk factors for AD [Citation8,Citation23]. Injuries can provoke growth signaling that is normally associated with wound healing and inflammation (). Indeed, tissue inflammation with TNF-alpha, NOS2, IL1-beta upregulations, and activation of mitogenic signaling via JNK/NFkB have been reported in human AD and in mouse models of AD [Citation109–Citation111].
Figure 3. “Injury-wound healing response” model for mitotic re-entry. How mitotic re-entry occur is a key question. Known LOAD risk factors (diabetes, hypertension, stroke) all provoke small or large injuries in the brain. Genomic instability including Sgo1−/+ can also cause cell death, a form of micro-injury. Such injuries activate growth signaling associated with wound-healing (NFkB/JNK, GSK or MAPK). The wound/injury-activated growth signaling may play a significant role in facilitating mitotic re-entry in the brain.
At the cellular level, genomic instability can provoke cell death, which can be considered a sterile micro injury for the organ. Sgo1−/+ mice showed more colonic cell death with carcinogen treatment [Citation58], and dominant-negative Sgo1 peptide triggered cell death in HeLa cells [Citation112]. We speculate that mitotic re-entry may have been achieved in Sgo1−/+ mice via cell death/micro injury that activated a post-translational growth signaling.
Mitotic cycle entry occurs as a part of normal adult neurogenesis, which is an established part of normal homeostasis. In the adult rodent brain, the two main regions of active neurogenesis are in the subgranular zone (SGZ) of the dentate gyrus in the hippocampus and the subventricular zone (SVZ) of the lateral ventricles, where neural stem cells are enriched [Citation113]. However, overall mitotic cells in the normal brain are rare (approximately 1% of cells). In the mitosis-scarce environment, re-entry to the cell cycle in adult neurons is an early hallmark of neurodegeneration [Citation114]. In the LOAD brain, there is an increase in mitotic re-entry and (prolonged) mitosis. However, it remains to be distinguished whether the cycling cells originate from terminally-differentiated mature neurons that went through de-differentiation to re-enter the mitotic cycle, or whether the cycling cells arise from a reserve of stem(-like) cells ().
The three-hit hypothesis: age, mitotic re-entry, and prolonged mitosis
Based on our results, we posit three factors that play key roles in amyloid-beta accumulation in the Sgo1−/+ model ().
Age. Age is a necessary ingredient for spontaneous LOAD. In addition, age may be translated to various biomarkers. The Sgo1−/+ model would be a useful tool with which to identify age-associated factors for amyloid-beta accumulation. For example, simple questions, such as whether anti-oxidants or anti-inflammatories prevent amyloid-beta accumulation, can be easily tested in the model.
Mitotic re-entry. [see previous section]
Prolonged mitosis. Prolonged mitosis is a signature phenotype in Sgo1−/+ mice, and is a major difference from the BubR1−/+ model. However, prolonged mitosis has been thus far unexplored. Although SgoL1 mutation itself remains under-characterized in the human LOAD brain, many other causes, e.g. spindle checkpoint-activating events, such as the aneuploidy that is prevalent in aged brains [Citation115] or dysfunction in mitosis-regulatory genes that also induce cohesinopathy (genes of cohesins or centrosome components), can produce prolonged mitosis in the human brain that results in the functional equivalent of Sgo1 mutation [Citation74,Citation116–Citation119]
Figure 4. The “three-hit” hypothesis. Based on results from Sgo1−/+ mouse model and existing theories from human LOAD studies, we propose the “three-hit” hypothesis for LOAD. When the “Three-hit” (age, mitotic re-entry, and mitotic prolongation) occurs simultaneously, brain cells accumulate amyloid-beta during prolonged mitosis. Prolonged mitosis may be followed by cell death via mitotic catastrophe, leading to release of amyloid-beta to extracellular matrix (see legend). Various factors lead to each “hit”. “Age” can be translated to a variety of insults (see text). Mitotic re-entry may have been caused by injuries and activation of growth signaling associated with wound/injury healing. Mitotic prolongation is caused by spindle checkpoint activation. Spindle checkpoint activation can occur by conditions commonly found in LOAD brains, such as aneuploidy or cohesinopathy.

Implications of LOAD mouse model on AD drug research and development (R&D)
Previously, mammalian models for AD drug R&D were limited to apes, which also develop LOAD-like pathology, and EOAD mouse models. However, there are many restrictions in using ape models [Citation120]. Facility requirements, high cost, selective expertise, and the long time frame for experiments make the ape model rather impractical for drug R&D. EOAD mouse models have been used widely for AD drug R&D purposes, yet the modeling is usually based on forcible expression of amyloid metabolism component(s). Although current AD drug R&D is conducted with the premise that drugs effective for EOAD would be effective for LOAD, the majority of the AD drug candidates that showed ameliorative effects in the EOAD model mice failed in human clinical trials. Thus, concerns regarding whether the AD aspects recapitulated in EOAD models optimally represent human LOAD and its druggable targets remain. The recent identification of the Sgo1−/+ mouse model as a LOAD mouse model candidate would represent a breakthrough in LOAD modeling, which may further generate a new set of drug target candidates.
Mouse models are highly valuable as test models. For example, mouse models are indispensable for carcinogenesis and cancer prevention studies. The efficacies of various environmental (e.g. arsenic, pesticide), lifestyle (e.g. smoking, diet), and dietary components (e.g. anti-oxidants, NSAIDs) have been tested using these models. [e.g. Citation121–Citation123] With the same experimental setting, potential interventions or facilitators for LOAD can be directly tested. With a genetically uniform mouse model, the biological effects of environmental, lifestyle, dietary components, or intervention and therapeutic medicine on LOAD development can be examined in a quantifiable and conclusive manner.
Implications for human LOAD intervention and treatment
Recent and current AD drug development efforts are based on the amyloid hypothesis. Thus, core components of the amyloid metabolism pathway, such as APP-amyloid-beta conversion enzyme beta-secretase 1 (BACE), have been primary targets. Thus far, this line of investigation has not been successful. Only future studies will tell us whether targeting amyloid metabolism components will yield effective LOAD drugs.
Instead of core amyloid metabolism components, prolonged mitosis in the Sgo1−/+ model suggests a different set of potential drug targets. One prediction from our “mitotic origin model of amyloid beta accumulation” () is that reducing mitotic entry and/or resolving prolonged mitosis will reduce amyloid-beta generation and accumulation. Here we propose prolonged mitosis and/or mitotic re-entry as newly implicated targets.
Re-purposing potential of mitosis-targeting anti-cancer drugs for LOAD treatment
From the three-hit hypothesis, we predict that drugs that target mitotic entry and/or prolonged mitosis would represent a new class of LOAD drug, and would receive more attention for repurposing to LOAD therapy or intervention purposes. Many cancer cells are actively going through the cell cycle, and compounds identified as effective in anti-cancer efficacy turned out to be targeting a phase of cell cycle (e.g. mitosis-targeting anti-microtubule drugs). As such, two major transitions in the cell cycle, mitosis and G1/S cell cycle progression, have been targets for cancer drug development [Citation124–Citation126]. With the new hypothesis of mitosis involving LOAD, repurposing existing FDA-approved mitosis- or G1/S-targeting cancer drugs may be applicable for rapid translation.
For example, the CDK inhibitor Flavopiridol/Alvocidib has been used for in vitro studies and showed blockage in G1/S transition and facilitation of mitotic exit [Citation127]. Thus, this agent can simultaneously inhibit mitotic re-entry and prolonged mitosis. Flavopiridol is an FDA-approved drug for the treatment of acute myeloid leukemia [Citation128,Citation129]. Neuroprotective action of Flavopiridol was previously reported [Citation130]. In an amyloid-beta injection mouse model of AD, i.p. injection of Flavopiridol improved memory, supporting the notion that inhibiting mitotic re-entry and/or prolonged mitosis can ameliorate AD [Citation131]. In the mouse model, Flavopiridol passed through the Brain Blood Barrier (BBB) and showed efficacy, although Flavopiridol is a substrate of the BBB [e.g. Citation132]. Tests of Flavopiridol in Sgo1−/+ mice have been proposed. If Flavopiridol ameliorates existing amyloid-beta accumulation in the Sgo1−/+ model, or prevents the accumulation, this result would open possibilities for rapid clinical translation through drug repurposing, which can significantly cut costs and time for drug research and development [Citation133,Citation134].
The use of cell-cycle-targeting cancer drugs as AD drugs is an emerging, yet promising approach. For example, AstraZeneca and NIH/NCATS-led repurposing of Fyn-kinase inhibitor Saracatinib for AD treatment in a mouse-based study that has provided promising results of reversal of memory function [Citation133,Citation135], and phase IIa clinical trial is ongoing as of 2017/2018 (clinicaltrials.gov). Targeting Fyn kinase is rationalized as targeting the inhibition of the downstream Amyloid-beta signaling, particularly at the synapse, in which Fyn kinase is involved [Citation136]. However, Fyn kinase inhibitors may show their efficacy through cell cycle manipulation, at least in part.
Summary
The Sgo1−/+ model mouse has demonstrated spontaneous, late-onset amyloid-beta accumulation in the brain. The LOAD-associated pathology of amyloid-beta accumulation in the mouse model will serve as a useful biomarker for drug testing and development. With confirmation of cognitive and/or behavioral dysfunctions, the model will represent the first, genetically-defined mouse model for spontaneous LOAD.
Sgo1 functions and the cohesinopathy-mediated chromosome instability phenotype led us to emphasize the critical role of mitotic cycle re-entry and prolonged mitosis in the accumulation of amyloid-beta in the brain. Once these critical roles are further demonstrated in AD model systems, drugs originally developed to target mitosis, most of which were intended to target cancers, may be repurposed for rapid translational use for LOAD intervention and/or therapy.
Acknowledgments
We thank Ms. Kathy Kyler for editorial aid and Mr. Nathan Goad for administrative aid.
Disclosure statement
In accordance with Taylor & Francis policy and my ethical obligation as a researcher, I am reporting that I have a financial and/or business interests in a company that may be affected by the research reported in the enclosed paper. I have disclosed those interests fully to Taylor & Francis, and I have in place an approved plan for managing any potential conflicts arising from that involvement.
Additional information
Funding
References
- Jack CR Jr., Bennett DA, Blennow K, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–562.
- Frisoni GB, Boccardi M, Barkhof F, et al. Strategic roadmap for an early diagnosis of Alzheimer’s disease based on biomarkers. Lancet Neurol. 2017;16:661–676.
- Jansen WJ, Ossenkoppele R, Tijms BM, et al. Association of cerebral amyloid-β aggregation with cognitive functioning in persons without dementia. JAMA Psychiatry. 2018;75:84–95.
- Jan AT, Azam M, Rahman S, et al. Perspective insights into disease progression, diagnostics, and therapeutic approaches in Alzheimer’s disease: a judicious update. Front Aging Neurosci. 2017;9:356.
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356.
- Hardy J. The discovery of Alzheimer-causing mutations in the APP gene and the formulation of the “amyloid cascade hypothesis”. FEBS J. 2017;284:1040–1044.
- Kametani F, Hasegawa M. Reconsideration of amyloid hypothesis and Tau hypothesis in Alzheimer’s disease. Front Neurosci. 2018;12:25.
- Alzheimer’s organization [cited 2018 July 20]. Available from: https://www.alz.org/research/science/alzheimers_research.asp
- Scheltens P, Blennow K, Breteler MM, et al. Alzheimer’s disease. Lancet. 2016;388:505–517.
- Cummings J, Aisen PS, DuBois B, et al. Drug development in Alzheimer’s disease: the path to 2025. Alzheimers Res Ther. 2016;8:39.
- Qian X, Hamad B, Dias-Lalcaca G. The Alzheimer disease market. Nat Rev Drug Discov. 2015;14:675–676.
- Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6:37.
- Carroll J Another Alzheimer’s drug flops in pivotal clinical trial Endpoints News. cited 2017 Feb 15. Available from: http://www.sciencemag.org/news/2017/02/another-alzheimers-drug-flops-pivotal-clinical-trial
- Szeto JY, Lewis SJ. Current treatment options for Alzheimer’s disease and Parkinson’s disease dementia. Curr Neuropharmacol. 2016;14(4):326–338.
- Logovinsky V, Satlin A, Lai R, et al. Safety and tolerability of BAN2401–a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res Ther. 2016;8:14.
- Söllvander S, Nikitidou E, Gallasch L, et al. The Aβ protofibril selective antibody mAb158 prevents accumulation of Aβ in astrocytes and rescues neurons from Aβ-induced cell death. J Neuroinflammation. 2018;15:98.
- Hall AM, Roberson ED. Mouse models of Alzheimer’s disease. Brain Res Bull. 2012;88:3–12.
- Sasaguri H, Nilsson P, Hashimoto S, et al. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017;36:2473–2487.
- Kitazawa M, Medeiros R, Laferla FM. Transgenic mouse models of Alzheimer disease: developing a better model as a tool for therapeutic interventions. Curr Pharm Des. 2012;18:1131–1147.
- Ameen-Ali KE, Wharton SB, Simpson JE, et al. Review: neuropathology and behavioural features of transgenic murine models of Alzheimer’s disease. Neuropathol Appl Neurobiol. 2017;43:553–570.
- Egan KJ, Vesterinen HM, Beglopoulos V, et al. From a mouse: systematic analysis reveals limitations of experiments testing interventions in Alzheimer’s disease mouse models. Evid Based Preclin Med. 2016;3:e00015.
- Wirths O. Altered neurogenesis in mouse models of Alzheimer disease. Neurogenesis (Austin). 2017;4:e1327002.
- Huang CC, Chung CM, Leu HB, et al. Diabetes mellitus and the risk of Alzheimer’s disease: a nationwide population-based study. PLoS One. 2014;9:e87095.
- Hou Y, Song H, Croteau DL, et al. Genome instability in Alzheimer disease. Mech Ageing Dev. 2017;161:83–94.
- Lee SL, Thomas P, Fenech M. Genome instability biomarkers and blood micronutrient risk profiles associated with mild cognitive impairment and Alzheimer’s disease. Mutat Res. 2015;776:54–83.
- Hartley D, Blumenthal T, Carrillo M, et al. Down syndrome and Alzheimer’s disease: common pathways, common goals. Alzheimers Dement. 2015;11(6):700–709.
- Chan GK, Jablonski SA, Sudakin V, et al. Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J Cell Biol. 1999;146:941–954.
- Tang Z, Bharadwaj R, Li B, et al. Mad2-independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev Cell. 2001;1:227–237.
- Kapanidou M, Lee S, Bolanos-Garcia VM. BubR1 kinase: protection against aneuploidy and premature aging. Trends Mol Med. 2015;21:364–372.
- Dai W, Wang Q, Liu T, et al. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res. 2004;64:440–445.
- Rao CV, Yang YM, Swamy MV, et al. Colonic tumorigenesis in BubR1±ApcMin/+ compound mutant mice is linked to premature separation of sister chromatids and enhanced genomic instability. Proc Natl Acad Sci U S A. 2005;102(12):4365–4370.
- Baker DJ, Jeganathan KB, Cameron JD, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36(7):744–749.
- Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236.
- Baker DJ, Dawlaty MM, Wijshake T, et al. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat Cell Biol. 2013;15:96–102.
- Choi CI, Yoo KH, Hussaini SM, et al. The progeroid gene BubR1 regulates axon myelination and motor function. Aging (Albany NY). 2016;8:2667–2688.
- Yang Z, Jun H, Choi CI, et al. Age-related decline in BubR1 impairs adult hippocampal neurogenesis. Aging Cell. 2017;16:598–601.
- Zivković L, Spremo-Potparević B, Djelić N, et al. Analysis of premature centromere division (PCD) of the chromosome 18 in peripheral blood lymphocytes in Alzheimer disease patients. Mech Ageing Dev. 2006;127:892–896.
- Zivković L, Spremo-Potparević B, Plecas-Solarović B, et al. Premature centromere division of metaphase chromosomes in peripheral blood lymphocytes of Alzheimer’s disease patients: relation to gender and age. J Gerontol A Biol Sci Med Sci. 2010;65:1269–1274.
- Bajic V, Spremo-Potparevic B, Zivkovic L, et al. Cohesion and the aneuploid phenotype in Alzheimer’s disease: a tale of genome instability. Neurosci Biobehav Rev. 2015;55:365–374.
- Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci. 2008;121(Suppl 1):1–84.
- Schvartzman JM, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer. 2010;10(2):102–115.
- Rao CV, Yamada HY, Yao Y, et al. Enhanced genomic instabilities caused by deregulated microtubule dynamics and chromosome segregation: a perspective from genetic studies in mice. Carcinogenesis. 2009;30:1469–1474.
- Foijer F, Draviam VM, Sorger PK, et al. Studying chromosome instability in the mouse. Biochim Biophys Acta. 2008;1786:73–82.
- Ricke RM, van Ree JH, van Deursen JM. Whole chromosome instability and cancer: a complex relationship. Trends Genet. 2008;24:457–466.
- Rao CV, Yamada HY. Genomic instability and colon carcinogenesis: from the perspective of genes. Front Oncol. 2013;3:130.
- Weaver BA, Silk AD, Montagna C, et al. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36.
- Weaver BA, Cleveland DW. The role of aneuploidy in promoting and suppressing tumors. J Cell Biol. 2009;185:935–937.
- Zasadil LM, Britigan EM, Ryan SD, et al. High rates of chromosome missegregation suppress tumor progression but do not inhibit tumor initiation. Mol Biol Cell. 2016;27:1981–1989.
- Silk AD, Zasadil LM, Holland AJ, et al. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci USA. 2013;110:E4134–41.
- Funk LC, Zasadil LM, Weaver BA. Living in CIN: mitotic infidelity and its consequences for tumor promotion and suppression. Dev Cell. 2016;39:638–652.
- Salic A, Waters JC, Mitchison TJ. Vertebrate shugoshin links sister centromere cohesion and kinetochore microtubule stability in mitosis. Cell. 2004;118:567–578.
- McGuinness BE, Hirota T, Kudo NR, et al. Shugoshin prevents dissociation of cohesin from centromeres during mitosis in vertebrate cells. PLoS Biol. 2005;3:e86.
- Schöckel L, Möckel M, Mayer B, et al. Cleavage of cohesin rings coordinates the separation of centrioles and chromatids. Nat Cell Biol. 2011;13:966–972.
- Wang X, Yang Y, Duan Q, et al. sSgo1, a major splice variant of Sgo1, functions in centriole cohesion where it is regulated by Plk1. Dev Cell. 2008;14:331–341.
- Tsang WY, Dynlacht BD. sSgo1, a guardian of centriole cohesion. Dev Cell. 2008;14:320–322.
- Yamada HY, Yao Y, Wang X, et al. Haploinsufficiency of SGO1 results in deregulated centrosome dynamics, enhanced chromosomal instability and colon tumorigenesis. Cell Cycle. 2012;11:479–488.
- Yamada HY, Zhang Y, Reddy A, et al. Tumor-promoting/progressing role of additional chromosome instability in hepatic carcinogenesis in Sgo1 (Shugoshin 1) haploinsufficient mice. Carcinogenesis. 2015;36:429–440.
- Rao CV, Sanghera S, Zhang Y, et al. Antagonizing pathways leading to differential dynamics in colon carcinogenesis in Shugoshin1 (Sgo1)-haploinsufficient chromosome instability model. Mol Carcinog. 2016;55:600–610.
- Iwaizumi M, Shinmura K, Mori H, et al. Human Sgo1 downregulation leads to chromosomal instability in colorectal cancer. Gut. 2009;58:249–260.
- Kim MS, An CH, Yoo NJ, et al. Frameshift mutations of chromosome cohesion-related genes SGOL1 and PDS5B in gastric and colorectal cancers with high microsatellite instability. Hum Pathol. 2013;44:2234–2240.
- Kahyo T, Iwaizumi M, Shinmura K, et al. A novel tumor-derived SGOL1 variant causes abnormal mitosis and unstable chromatid cohesion. Oncogene. 2011;30:4453–4463.
- Matsuura S, Kahyo T, Shinmura K, et al. SGOL1 variant B induces abnormal mitosis and resistance to taxane in non-small cell lung cancers. Sci Rep. 2013;3:3012.
- Wang LH, Yen CJ, Li TN, et al. Sgo1 is a potential therapeutic target for hepatocellular carcinoma. Oncotarget. 2015;6:2023–2033.
- Yamada HY, Kumar G, Zhang Y, et al. Systemic chromosome instability in Shugoshin-1 mice resulted in compromised glutathione pathway, activation of Wnt signaling and defects in immune system in the lung. Oncogenesis. 2016;5:e256.
- Rao CV, Sanghera S, Zhang Y, et al. Systemic chromosome instability resulted in colonic transcriptomic changes in metabolic, proliferation, and stem cell regulators in Sgo1-/+ mice. Cancer Research. 2016;76:630–642.
- Sheltzer JM, Torres EM, Dunham MJ, et al. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci U S A. 2012;109:12644–12649.
- Torres EM, Sokolsky T, Tucker CM, et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–924.
- Williams BR, Prabhu VR, Hunter KE, et al. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–709.
- Santaguida S, Vasile E, White E, et al. Aneuploidy-induced cellular stresses limit autophagic degradation. Genes Dev. 2015;29:2010–2021.
- Chetaille P, Preuss C, Burkhard S, et al. Mutations in SGOL1 cause a novel cohesinopathy affecting heart and gut rhythm. Nat Genet. 2014;46(11):1245–1249.
- Song AT, Galli A, Leclerc S, et al. Characterization of Sgo1 expression in developing and adult mouse. Gene Expr Patterns. 2017;25-26:36–45.
- Song AT, Galli A, Leclerc S, et al. Dataset of Sgo1 expression in cardiac, gastrointestinal, hepatic and neuronal tissue in mouse. Data Brief. 2017;13:731–737.
- Rao CV, Asch AS, Yamada HY. Emerging links among Chromosome Instability (CIN), cancer, and aging. Molecular Carcinogenesis. 2017;56:791–803.
- Rao CV, Farooqui M, Zhang Y, et al. Spontaneous development of Alzheimer’s disease-associated brain pathology in a Shugoshin 1 mouse cohesinopathy model. Aging Cell. 2018;17(4):e12797. PMID: 29943428.
- Valero J, Bernardino L, Cardoso FL, et al. Impact of neuroinflammation on hippocampal neurogenesis: relevance to aging and Alzheimer’s disease. J Alzheimers Dis. 2017;60:S161–S168.
- Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405.
- Sawikr Y, Yarla NS, Peluso I, et al. Neuroinflammation in Alzheimer’s disease: the preventive and therapeutic potential of polyphenolic nutraceuticals. Adv Protein Chem Struct Biol. 2017;108:33–57.
- Praticò D. Oxidative stress hypothesis in Alzheimer’s disease: a reappraisal. Trends Pharmacol Sci. 2008;29:609–615.
- Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid Med Cell Longev. 2013;2013:316523.
- García-Blanco A, Baquero M, Vento M, et al. Potential oxidative stress biomarkers of mild cognitive impairment due to Alzheimer disease. J Neurol Sci. 2017;373:295–302.
- Connolly NMC, Theurey P, Adam-Vizi V, et al. Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ. 2018;25:542–572.
- Wang JM, Sun C. Calcium and neurogenesis in Alzheimer’s disease. Front Neurosci. 2010;4:194.
- Leija-Salazar M, Piette C, Proukakis C. Review: somatic mutations in neurodegeneration. Neuropathol Appl Neurobiol. 2018;44(3):267–285.
- Duncan T, Valenzuela M. Alzheimer’s disease, dementia, and stem cell therapy. Stem Cell Res Ther. 2017;8:111.
- Schwartz M, Deczkowska A. Neurological disease as a failure of brain-immune crosstalk: the multiple faces of neuroinflammation. Trends Immunol. 2016;37:668–679.
- Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133–150.
- Nalivaeva NN, Turner AJ. Role of ageing and oxidative stress in regulation of amyloid-degrading enzymes and development of neurodegeneration. Curr Aging Sci. 2017;10:32–40.
- Adlard PA, Tran BA, Finkelstein DI, et al. A review of β-amyloid neuroimaging in Alzheimer’s disease. Front Neurosci. 2014;8:327.
- Avila J, Llorens-Martín M, Pallas-Bazarra N, et al. Cognitive decline in neuronal aging and Alzheimer’s disease: role of NMDA receptors and associated proteins. Front Neurosci. 2017;11:626.
- Foster TC, Kyritsopoulos C, Kumar A. Central role for NMDA receptors in redox mediated impairment of synaptic function during aging and Alzheimer’s disease. Behav Brain Res. 2017;322:223–232.
- Bredesen D. The end of Alzheimer’s: the first program to prevent and reverse cognitive decline. New York (NY): Avery; 2017. ISBN-10: 0735216207.
- Bredesen DE. Metabolic profiling distinguishes three subtypes of Alzheimer’s disease. Aging (Albany NY). 2015;7:595–600.
- Kondratick CM, Vandré DD. Alzheimer’s disease neurofibrillary tangles contain mitosis-specific phosphoepitopes. J Neurochem. 1996;67:2405–2416.
- Delobel P, Flament S, Hamdane M, et al. Abnormal Tau phosphorylation of the Alzheimer-type also occurs during mitosis. J Neurochem. 2002;83:412–420.
- Judge M, Hornbeck L, Potter H, et al. Mitosis-specific phosphorylation of amyloid precursor protein at threonine 668 leads to its altered processing and association with centrosomes. Mol Neurodegener. 2011;6:80.
- Ogawa O, Zhu X, Lee HG, et al. Ectopic localization of phosphorylated histone H3 in Alzheimer’s disease: a mitotic catastrophe? Acta Neuropathol. 2003;105:524–528.
- Folch J, Junyent F, Verdaguer E, et al. Role of cell cycle re-entry in neurons: a common apoptotic mechanism of neuronal cell death. Neurotox Res. 2012;22:195–207.
- Yang Y, Herrup K. Cell division in the CNS: protective response or lethal event in post-mitotic neurons? Biochim Biophys Acta. 2007;1772:457–466.
- Zhu X, Lee HG, Perry G, et al. Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta. 2007;1772:494–502.
- Zhu X, Raina AK, Perry G, et al. Alzheimer’s disease: the two-hit hypothesis. Lancet Neurol. 2004;3:219–226.
- Webber KM, Raina AK, Marlatt MW, et al. The cell cycle in Alzheimer disease: a unique target for neuropharmacology. Mech Ageing Dev. 2005;126:1019–1025.
- Vincent I, Bu B, Hudson K, et al. Constitutive Cdc25B tyrosine phosphatase activity in adult brain neurons with M phase-type alterations in Alzheimer disease. Neuroscience. 2001;105:639–650.
- Ding XL, Husseman J, Tomashevski A, et al. The cell cycle Cdc25A tyrosine phosphatase is activated in degenerating postmitotic neurons in Alzheimer disease. Am J Pathol. 2000;157:1983–1990.
- Tomashevski A, Husseman J, Jin LW, et al. Constitutive Wee1 activity in adult brain neurons with M phase-type alterations in Alzheimer neurodegeneration. J Alzheimers Dis. 2001;3:195–207.
- Seward ME, Swanson E, Norambuena A, et al. Amyloid-β signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J Cell Sci. 2013;126:1278–1286.
- Csiszar A, Tarantini S, Fülöp GA, et al. Hypertension impairs neurovascular coupling and promotes microvascular injury: role in exacerbation of Alzheimer’s disease. Geroscience. 2017. PMID: 28853030.
- Tucsek Z, Noa Valcarcel-Ares M, Tarantini S, et al. Hypertension-induced synapse loss and impairment in synaptic plasticity in the mouse hippocampus mimics the aging phenotype: implications for the pathogenesis of vascular cognitive impairment. Geroscience. 2017. PMID: 28664509.
- Orehek AJ. The micron stroke hypothesis of Alzheimer’s disease and dementia. Med Hypotheses. 2012;78:562–570.
- Shi S, Liang D, Chen Y, et al. Gx-50 reduces β-amyloid-induced TNF-α, IL-1β, NO, and PGE2 expression and inhibits NF-κB signaling in a mouse model of Alzheimer’s disease. Eur J Immunol. 2016;46(3):665–676.
- Li X, Long J, He T, et al. Integrated genomic approaches identify major pathways and upstream regulators in late onset Alzheimer’s disease. Sci Rep. 2015;5:12393.
- Arendt T, Holzer M, Stöbe A, et al. Activated mitogenic signaling induces a process of dedifferentiation in Alzheimer’s disease that eventually results in cell death. Ann N Y Acad Sci. 2000;920:249–255.
- Yang Y, Wang X, Dai W. Human Sgo1 is an excellent target for induction of apoptosis of transformed cells. Cell Cycle. 2006;5:896–901.
- Bordiuk OL, Smith K, Morin PJ, et al. Cell proliferation and neurogenesis in adult mouse brain. PLoS One. 2014;9:e111453.
- Wang W, Bu B, Xie M, et al. Neural cell cycle dysregulation and central nervous system diseases. Prog Neurobiol. 2009;89:1–17.
- Rosenkrantz JL, Carbone L. Investigating somatic aneuploidy in the brain: why we need a new model. Chromosoma. 2017;126:337–350.
- Kline AD, Krantz ID, Deardorff MA, et al. Cornelia de Lange syndrome and molecular implications of the cohesin complex: abstracts from the 7th biennial scientific and educational symposium 2016. Am J Med Genet A. 2017;173:1172–1185.
- Kumar R, Corbett MA, Van Bon BW, et al. Increased STAG2 dosage defines a novel cohesinopathy with intellectual disability and behavioral problems. Hum Mol Genet. 2015;24:7171–7181.
- Nigg EA, Čajánek L, Arquint C. The centrosome duplication cycle in health and disease. FEBS Lett. 2014;588:2366–2372.
- Bajić VP, Spremo-Potparević B, Zivković L, et al. Is the time dimension of the cell cycle re-entry in AD regulated by centromere cohesion dynamics? Biosci Hypotheses. 2008;1:156–161.
- Van Dam D, De Deyn PP. Non human primate models for Alzheimer’s disease-related research and drug discovery. Expert Opin Drug Discov. 2017;12(2):187–200.
- Chikara S, Nagaprashantha LD, Singhal J, et al. Oxidative stress and dietary phytochemicals: role in cancer chemoprevention and treatment. Cancer Lett. 2018;413:122–134.
- Wilson DW, Nash P, Buttar HS, et al. The role of food antioxidants, benefits of functional foods, and influence of feeding habits on the health of the older person: an overview. Antioxidants (Basel). 2017;6:E81.
- Rao CV, Pal S, Mohammed A, et al. Biological effects and epidemiological consequences of arsenic exposure, and reagents that can ameliorate arsenic damage in vivo. Oncotarget. 2017;8:57605–57621.
- O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13:417–430.
- Salmela AL, Kallio MJ. Mitosis as an anti-cancer drug target. Chromosoma. 2013;122:431–449.
- Rao CV, Kurkjian CD, Yamada HY. Mitosis-targeting natural products for cancer prevention and therapy. Curr Drug Targets. 2012;13:1820–1830.
- Motwani M, Li X, Schwartz GK. Flavopiridol, a cyclin-dependent kinase inhibitor, prevents spindle inhibitor-induced endoreduplication in human cancer cells. Clin Cancer Res. 2000;6:924–932.
- Srikumar T, Padmanabhan J. Potential use of flavopiridol in treatment of chronic diseases. Adv Exp Med Biol. 2016;929:209–228.
- Zeidner JF, Karp JE. Clinical activity of alvocidib (flavopiridol) in acute myeloid leukemia. Leuk Res. 2015;39(12):1312–1318.
- Jorda EG, Verdaguer E, Canudas AM, et al. Neuroprotective action of flavopiridol, a cyclin-dependent kinase inhibitor, in colchicine-induced apoptosis. Neuropharmacology. 2003;45:672–683.
- Leggio GM, Catania MV, Puzzo D, et al. The antineoplastic drug flavopiridol reverses memory impairment induced by Amyloid-ß1-42 oligomers in mice. Pharmacol Res. 2016;106:10–20.
- Kodaira H, Kusuhara H, Ushiki J, et al. Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther. 2010;333:788–796.
- Wadman M. NIH gambles on recycled drugs. Nature. 2013;499:263–264.
- National NIH Center for Advancing Translational Science [cited 2018 July 20]. Available from: https://ncats.nih.gov/preclinical/repurpose
- Kaufman AC, Salazar SV, Haas LT, et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol. 2015;77:953–971.
- Folch J, Petrov D, Ettcheto M, et al. Current research therapeutic strategies for Alzheimer’s disease treatment. Neural Plast. 2016;2016:8501693.