
ABSTRACT
Poly(ADP-ribose) polymerase 3 (PARP3) is the third member of the PARP family that catalyze a post-translational modification of proteins to promote, control or adjust numerous cellular events including genome integrity, transcription, differentiation, cell metabolism or cell death. In the late years, PARP3 has been specified for its primary functions in programmed and stress-induced double-strand break repair, chromosomal rearrangements, transcriptional regulation in the zebrafish and mitotic segregation. Still, deciphering the therapeutic value of its inhibition awaits additional investigations. In this review, we discuss the newest advancements on the specific functions of PARP3 in cancer aggressiveness exemplifying the relevance of its selective inhibition for cancer therapy.
Introduction
The successes over the past decades of the use of poly(ADP-ribose) polymerase 1 (PARP1) inhibitors in targeted cancer therapies engendered drugs that for three of them (olaparib, rucaparib, niraparib) have since being FDA approved for clinical trials[Citation1]. These drugs have proven potency in combination with chemotherapeutic regimens and ionizing radiation therapies, or as single agents to selectively kill tumor cells having a dysfunctional homologous recombination pathway [Citation2]. Still, PARP inhibitors inhibit a family of proteins (17 members) with each protein having specific functions [Citation3]. Among them, PARP3 has gained substantial attention in the field revealing unique biological properties in cell response to DNA damage, class-switch recombination, transcriptional regulation during neuronal development in the zebrafish, mitotic progression, and chromosome rearrangements [Citation4–Citation10].
The primary function of PARP3 is to catalyze the covalent attachment of a single ADP-ribose derived from the oxidized form of NAD+ to target proteins in a process called mono-ADP-ribosylation (MARylation) (). The demonstrated protein targets of PARP3 are up till limited to the mitotic components NuMa and Tankyrase 1, the DNA repair proteins Ku80 and Poly(ADP-ribose) polymerase 1 and the histone H2B specifically at Glu2 [Citation4,Citation5,Citation11,Citation12]. More recently, in vitro biochemical experiments have shown that PARP3 can MARylate 5ʹ and 3ʹ-terminal phosphate residues in DNA [Citation13,Citation14].
Figure 1. PARP3 regulated ADP-ribose metabolism. PARP3 catalyzes the addition of a single ADP-ribosyl unit derived from NAD+ to a target substrate in a process defined as mono-ADP-ribosylation (MARylation). The known targets of PARP3 are indicated. The reversal of MARylation can be executed by macro-domain containing hydrolases (macroD1, macroD2), the terminal ADP-ribose protein glycohydrolase 1 (TARG1), the ADP-ribose hydrolases (ARH1, ARH3) or phosphodiester ADP-ribose hydrolases (NUDT16, ENPP1). NUDT16, nucleoside diphosphates linked to moiety-X; ENPP1, ectonucleotide pyrophosphatase/phosphodiesterase 1[Citation43].
![Figure 1. PARP3 regulated ADP-ribose metabolism. PARP3 catalyzes the addition of a single ADP-ribosyl unit derived from NAD+ to a target substrate in a process defined as mono-ADP-ribosylation (MARylation). The known targets of PARP3 are indicated. The reversal of MARylation can be executed by macro-domain containing hydrolases (macroD1, macroD2), the terminal ADP-ribose protein glycohydrolase 1 (TARG1), the ADP-ribose hydrolases (ARH1, ARH3) or phosphodiester ADP-ribose hydrolases (NUDT16, ENPP1). NUDT16, nucleoside diphosphates linked to moiety-X; ENPP1, ectonucleotide pyrophosphatase/phosphodiesterase 1[Citation43].](/cms/asset/04fcb52f-82e4-4216-a08f-0cd2201b3eab/kccy_a_1617454_f0001_oc.jpg)
PARP3 mediates DNA strand break repair
The therapeutic benefit of PARP3 inhibition first emerged in 2011 with the identification of its particular contribution in cell response to double-strand breaks [Citation5,Citation10]. PARP3 acts in cooperation with Ku80 to drive repair pathway choice and promote repair of double-strand breaks using the classical non-homologous end-joining route (C-NHEJ), and it also facilitates the association of APLF to damaged DNA, which in turn accelerates the XRCC4/DNA ligaseIV-mediated ligation during C-NHEJ [Citation4,Citation10,Citation15]. Consequently, the depletion of PARP3 delays the repair efficiency and/or potentiates the cytotoxicity of DNA lesions induced by ionizing radiations, etoposide, and bleomycin (). Of additional therapeutic significance, PARP1 and PARP3 act synergistically in long-term response to ionizing irradiation. Hence, the combined loss of PARP1 and PARP3 significantly sensitize human cells and mice to X-ray irradiation (). These results provide a potential approach for selectively interfering with PARP1 and PARP3 activities by reducing the intracellular concentrations of each active inhibitor and radiotherapy while moderating adverse toxic effects. Moreover, owing to its role in DSB repair, the disruption or chemical inhibition of PARP3 in A549 cells caused high sensitivity to molecules that stabilize G quadruplex (i.e. pyridostatin, PhenDC3) because of extensive accumulation of unresolved DSBs announcing therapeutic promises of PARP3 inhibition combined with G-quadruplex structure binding ligands [Citation6] ().
Table 1. The absence of PARP3 potentiates DNA damage.
A role of PARP3 in single-strand break repair has later being suggested by guided mutagenesis combined with biochemical studies and showing that similarly to PARP2, PARP3 is preferentially activated by 5ʹ phosphorylated nicks often found in DNA ligation intermediates or intermediates of the single-strand break and base excision repair process (SSBR/BER) [Citation16]. Moreover, while the N-terminal domain of PARP3 does not appear strictly required for efficient DNA binding or DNA-dependent activation of these structures, the WGR domain is a central regulator domain. Accumulating in vitro studies suggest that the repair of DNA strand breaks is initiated by a PARP3-dependent mono ADP-ribosylation of the DNA breaks followed by their ligation and repair by the BER system [Citation13,Citation14,Citation17–Citation19]. Furthermore, PARP3 was found to preferentially bind nicked nucleosomes and MARylate histone H2BE in chicken DT40 cells [Citation11]. Its absence reduced the chromosomal SSBR efficiency of γ-rays induced DNA strand breaks (). However, besides an increased sensitivity of PARP3 knockout mouse cells to compounds producing reactive oxygen species (ROS) which induce DNA lesions typically repaired by the SSBR/BER process, the therapeutic value of PARP3 at SSBs in human cells has not been attentively addressed yet [Citation20].
In the late years, the therapeutic advantage of PARP3 in cancer has revealed its specificities (). Below we highlight the recent discoveries that exemplify PARP3 as a prominent beneficial target in a precision medicine approach for the treatment of highly aggressive breast cancers [Citation20,Citation21].
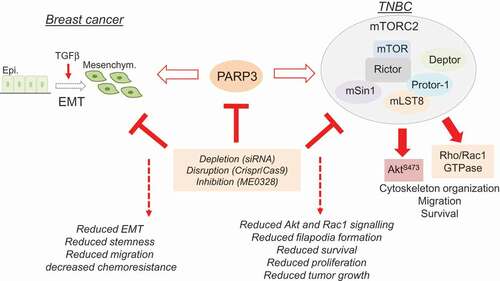
Figure 2. The oncogenic roles of PARP3 in breast cancer. In the context of TGFβ-driven EMT, PARP3 assists EMT properties, stemness, and chemoresistance. In the context of BRCA1-mutated TNBC, PARP3 supports mTORC2-mediated cell proliferation, cell survival, cytoskeleton-associated events, and tumor growth. The inhibition of PARP3 emerges as a leading therapeutic option to treat highly aggressive cancers.
PARP3 promotes TGFβ-induced EMT during breast cancer
High-grade malignancy is largely driven by the epithelial-to-mesenchymal (EMT) program and its connections with the acquisition of stem-like properties. EMT is a transdifferentiation process during which the tumor cell loses epithelial characteristics and converts to a mesenchymal phenotype. By entering this state, tumor cells acquire invasive abilities, drug resistance, and stem cell states. The activation and maintenance of EMT is driven by a panel of external signals generally arising from the tumor environment (stroma), among them the well-described cytokine Transforming Growth Factor (TGFβ) [Citation22].
In the model of breast cancer cells, the expression of PARP3 has been positively associated with the mesenchymal and aggressive basal-like subtypes of these tumors and is notably upregulated during TGFβ-induced EMT [Citation20]. Consequently, the silencing of PARP3 remarkably restrained TGFβ-driven EMT in mammary epithelial and breast cancer cells by preventing the induction of a Snail-E-cadherin axis and the break-down of the ZO-stained tight junctions, by limiting cell motility and by supporting resistance to clastogenic drugs. Conversely, the sustained expression of PARP3 activated the Snail-E-cadherin pathway in response to TGFβ. In comparison, the forced expression of PARP3 in epithelial breast cancer cells was not enough to spontaneously induce EMT, and the depletion of PARP3 in mesenchymal cells did not reverse the process. These findings suggested that PARP3 precisely assists the commitment of TGFβ-driven EMT.
Of additional relevance was the discovery that PARP3 also operates for the maintenance of stem-like cell traits [Citation20]. In the context of breast cancer, PARP3 fosters primary mammosphere formation illustrating stem cell activity, serves stem cell self-renewal, and upholds the frequency of tumor initiating CD44high/CD24low population and the expression of the stemness markers OCT4 and SOX2.
At the mechanistic level, the oncogenic role of PARP3 involves the production of TGFβ-induced Reactive Oxygen Species (ROS) creating a redox imbalance that in turn induces the expression of tissue transglutaminase 2 (TG2), an enzyme with prominent functions in EMT, the acquisition of stemness features and chemoresistance [Citation23–Citation27]. Hence, suppression of TGFβ-induced ROS production using the antioxidant N-acetylcysteine (NAC) was found to significantly weaken the PARP3-dependent transcriptional and protein expression levels of EMT and stemness markers. ROS serve as a prime signal in the commitment to EMT induced by TGFβ and are generated by different sources including mitochondria and the NADPH oxidase (NOX)/dual oxidase (DUOX) enzymes, among them, NOX4 plays a dominant role [Citation28,Citation29]. Noticeably, PARP3 is upregulated in response to ROS produced from exogenous sources and endogenous sources (TGFβ). These findings imply that PARP3 serves as a redox sensor in TGFβ-induced ROS-mediated EMT. The remaining question is to decipher how PARP3 and its catalytic activity supports the expression of TG2. Given the association of PARP3 with chromatin regulators and the selective mono-ADP-ribosylation of specific histones, it seems plausible that the oncogenic role of PARP3 is to establish an appropriate chromatin configuration at specific TGFβ-responsive genes.
The contribution of the EMT program in the resistance to drugs, chemotherapeutic agents and radiation therapies largely limits the success of anticancer therapies for these highly aggressive mesenchymal tumors. For several years, the mechanisms underlying this resistance have been ascribed to the upregulation of genes associated with cell survival, the downregulation of genes associated with cell death or the upregulation of multidrug resistance proteins and transport pumps mediating drug efflux [Citation22]. The contribution of PARP3 supports the relevance of the DNA damage response in the acquisition of this resistance. Correspondingly, several groups have reported that TGFβ maintains genomic stability through its participation in the DNA damage response by facilitating the NHEJ process [Citation30,Citation31].
Whether PARP3 controls EMT owing its role in DNA repair only or because of some other uncharacterized functions such as acting as a co-factor of transcription or a more direct involvement in cell adhesion remains a central question to consider.
Furthermore, besides cancer pathogenesis, EMT contributes to tissue regeneration during wound healing and to specific morphogenetic events during development such as the differentiation of neural crest cells and their migration from the neural tube, thus raising the question of a potential contribution of PARP3 in these physiological events [Citation22]. The first evidence has been suggested by previous studies in the zebrafish, where PARP3 was reported to be required for appropriate neural crest cell differentiation, a process in which NOX4-mediated ROS production plays a leading role [Citation9,Citation32]. It would be interesting to interrogate if the same is true in mice by studying neural crest induction in the PARP3 knockout mice.
PARP3 promotes mTORC2 signaling during breast cancer
The recent work by Beck et al. [Citation21] extended the therapeutic value of PARP3 inhibition. This study reveals that targeting PARP3’s catalytic activity is an encouraging therapeutic route to restrain the oncogenic mTORC2 signaling pathway specifically in BRCA1-mutated Triple Negative Breast Cancers (TNBC) following the principle of synthetic sickness.
mTORC2 together with mTORC1 are two functionally distinct multi-protein complexes of the serine/threonine kinase mammalian target of rapamycin mTOR [Citation33,Citation34]. mTORC1 comprises mTOR, Raptor, mLST8, and PRAS40 and is recognized for its prominent functions in protein and lipid synthesis, mitochondrial activity and autophagy. mTORC2 contains mTOR, Rictor, Sin1, mLST8, Deptor, and Protor and specifically regulates cell survival, chemoresistance, actin cytoskeleton organization and TGFβ-induced EMT, key hallmarks of cancer aggressiveness. Rictor is defined as the core component of the mTORC2 complex [Citation35]. The fine-tuned regulation of its abundance is pivotal for the stability of the mTORC2 complex and adjusted by a GSK3β-dependent, FBXW7-mediated ubiquitination and proteasomal degradation [Citation36]. Because mTORC2 functions as a critical kinase of pAkt-S473, a protein modification often upregulated in various cancers, this complex is gaining attention as a relevant target for cancer therapy. However, unlike mTORC1, mTORC2 is insensitive to acute rapamycin-induced inhibition.
TNBC are clinically negative for the expression of estrogen and progesterone receptors and HER2 protein and thus do not respond to hormonal therapy. Many of the TNBC are of the basal-like subtype, and a high proportion of BRCA1-mutated breast cancers are triple negative.
These tumors are highly aggressive, difficult to treat and represent a major therapeutic challenge [Citation37,Citation38]. Noticeably, PARP3 expression is predominant in breast cancer cell lines of the TNBC phenotype compared to non-TNBC. In these cells, PARP3 provides a means to limit the GSK3β-dependent ubiquitination and degradation of Rictor. Particularly, while the knockdown, knockout or inhibition of its catalytic activity using the selective PARP3 inhibitor ME0328 had no consistent impact in TNBC with normal BRCA1, it was selectively lethal with BRCA1 deficiency in these models. Mechanistically, the knockout of PARP3 or inhibition of its catalytic activity significantly weakened mTORC2-mediated Akt phosphorylation and compromised Rac1 signaling especially in the BRCA1-deficient cells that upregulate both oncogenic events. This study revealed that PARP3 and BRCA1 are synthetic lethal in TNBC. In contrast to PARP1, this property does not associate the repair activities of PARP3 but refers to a singular role of PARP3’s catalytic activity in the stability and activity of the mTORC2 complex.
It is worth stressing that in various cancer cell models, there is strong evidence that mTORC2 contributes to EMT, thus raising the intriguing possibility that the role of PARP3 in TGFβ-induced EMT is also partly mediated by mTORC2 [Citation39,Citation40]. In support of this hypothesis, the absence of PARP3 reduces the TGFβ-dependent activation of mTORC2 independently of the BRCA1 context. This encourages to further explore the precise role of PARP3’s catalytic activity in the ubiquitination and stability of Rictor and to investigate the physiological consequences beyond cancer. Genetic knockouts of Rictor in mice have revealed tissue-specific functions of mTORC2 in T and B lymphocyte development, skeletal and cardiac muscle function, insulin resistance in pancreatic β cells, and brain function [Citation34]. The PARP3 knockout mice offers a powerful genetic model to analyze the contribution of PARP3 in these events.
Conclusion
The connection made between the high expression of PARP3 and tumor aggressiveness in the recent studies places PARP3 as a central driver of tumor malignancy promoting TGFβ-induced EMT and mTORC2 signaling, and prescribes its inhibition as an effective strategy for advanced cancers in a synthetic lethality approach. Expanding on this, the inhibition of PARP3 also revealed promises to sensitize metastatic breast cancers to the vinca alkaloid vinorelbine by raising mitotic defects and inducing cell death [Citation41]. Broadly, these findings set the stage for additional investigations aimed to define the therapeutic relevance of PARP3 inhibition in other cancers. In line with this, upregulated levels of PARP3 have been detected in primary glioblastoma biopsies [Citation42]. Moreover, silencing the expression of PARP3 in glioblastoma cell models showed efficiency in potentializing the cytotoxic impact of radiotherapy suggesting that PARP3 could be a potential target to overcome radioresistance in these primary malignant brain tumors. Overall, from the perspective of clinical implications, these findings foresee the need to screen for more selective PARP3 inhibitors and use PARP3 inhibition to identify synthetic lethal interactions.
Acknowledgments
This work was supported by Association pour la Recherche contre le Cancer, Ligue Nationale Contre le Cancer, CNRS, Université de Strasbourg and Ramon Areces Foundation. This work has been published within the LABEX ANR-10-LABX-0034_Medalis.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- LaFargue CJ, Dal Molin GZ, Sood AK, et al. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019;20:e15–e28.
- Keung MYT, Wu Y, Vadgama JV. PARP inhibitors as a therapeutic agent for homologous recombination deficiency in breast cancers. J Clin Med. 2019;8(4):pii: E435.
- Gupte R, Liu Z, Kraus WL. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 2017;31:101–126.
- Beck C, Boehler C, Guirouilh Barbat J, et al. PARP3 affects the relative contribution of homologous recombination and nonhomologous end-joining pathways. Nucleic Acids Res. 2014;42:5616–5632.
- Boehler C, Gauthier LR, Mortusewicz O, et al. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc Natl Acad Sci USA. 2011;108:2783–2788.
- Day TA, Layer JV, Cleary JP, et al. PARP3 is a promoter of chromosomal rearrangements and limits G4 DNA. Nat Commun. 2017;8:15110.
- Layer JV, Cleary JP, Brown AJ, et al. Parp3 promotes long-range end joining in murine cells. Proc Natl Acad Sci U S A. 2018;115:10076–10081.
- Robert I, Gaudot L, Rogier M, et al. Parp3 negatively regulates immunoglobulin class switch recombination. PLoS Genet. 2015;11:e1005240.
- Rouleau M, Saxena V, Rodrigue A, et al. A key role for poly(ADP-ribose) polymerase 3 in ectodermal specification and neural crest development. PLoS One. 2011;6:e15834.
- Rulten SL, Fisher AE, Robert I, et al. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol Cell. 2011;41:33–45.
- Grundy GJ, Polo LM, Zeng Z, et al. PARP3 is a sensor of nicked nucleosomes and monoribosylates histone H2B(Glu2). Nat Commun. 2016;7:12404.
- Loseva O, Jemth AS, Bryant HE, et al. PARP-3 is a mono-ADP-ribosylase that activates PARP-1 in the absence of DNA. J Biol Chem. 2010;285:8054–8060.
- Zarkovic G, Belousova EA, Talhaoui I, et al. Characterization of DNA ADP-ribosyltransferase activities of PARP2 and PARP3: new insights into DNA ADP-ribosylation. Nucleic Acids Res. 2018;46:2417–2431.
- Munnur D, Ahel I. Reversible mono-ADP-ribosylation of DNA breaks. Febs J. 2017;284:4002–4016.
- Fenton AL, Shirodkar P, Macrae CJ, et al. The PARP3- and ATM-dependent phosphorylation of APLF facilitates DNA double-strand break repair. Nucleic Acids Res. 2013;41:4080–4092.
- Langelier MF, Riccio AA, Pascal JM. PARP-2 and PARP-3 are selectively activated by 5ʹ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 2014;42:7762–7775.
- Belousova EA, Kutuzov MM, Ivankina PA, et al. A new DNA break repair pathway involving PARP3 and base excision repair proteins. Dokl Biochem Biophys. 2018;482:233–237.
- Belousova EA, Lavrik OI. Dna is a new target of PARP3. Sci Rep. 2018;8:4176.
- Dolle C, Ziegler M. ADP-ribosylation of DNA moving into focus. Febs J. 2017;284:3999–4001.
- Karicheva O, Rodriguez-Vargas JM, Wadier N, et al. PARP3 controls TGFbeta and ROS driven epithelial-to-mesenchymal transition and stemness by stimulating a TG2-Snail-E-cadherin axis. Oncotarget. 2016;7:64109–64123.
- Beck C, Rodriguez-Vargas JM, Boehler C, et al. PARP3, a new therapeutic target to alter Rictor/mTORC2 signaling and tumor progression in BRCA1-associated cancers. Cell Death Differ. 2018.
- Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20:69–84.
- Cao L, Shao M, Schilder J, et al. Tissue transglutaminase links TGF-beta, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene. 2012;31:2521–2534.
- Fisher ML, Adhikary G, Xu W, et al. Type II transglutaminase stimulates epidermal cancer stem cell epithelial-mesenchymal transition. Oncotarget. 2015;6:20525–20539.
- Kumar A, Xu J, Brady S, et al. Tissue transglutaminase promotes drug resistance and invasion by inducing mesenchymal transition in mammary epithelial cells. PLoS One. 2010;5:e13390.
- Lin CY, Tsai PH, Kandaswami CC, et al. Role of tissue transglutaminase 2 in the acquisition of a mesenchymal-like phenotype in highly invasive A431 tumor cells. Mol Cancer. 2011;10:87.
- Shao M, Cao L, Shen C, et al. Epithelial-to-mesenchymal transition and ovarian tumor progression induced by tissue transglutaminase. Cancer Res. 2009;69:9192–9201.
- Hiraga R, Kato M, Miyagawa S, et al. Nox4-derived ROS signaling contributes to TGF-beta-induced epithelial-mesenchymal transition in pancreatic cancer cells. Anticancer Res. 2013;33:4431–4438.
- Jiang J, Wang K, Chen Y, et al. Redox regulation in tumor cell epithelial-mesenchymal transition: molecular basis and therapeutic strategy. Signal Transduct Target Ther. 2017;2:17036.
- Kim MR, Lee J, An YS, et al. TGFbeta1 protects cells from gamma-IR by enhancing the activity of the NHEJ repair pathway. Mol Cancer Res. 2015;13:319–329.
- Kirshner J, Jobling MF, Pajares MJ, et al. Inhibition of transforming growth factor-beta1 signaling attenuates ataxia telangiectasia mutated activity in response to genotoxic stress. Cancer Res. 2006;66:10861–10869.
- Lee JE, Cho KE, Lee KE, et al. Nox4-mediated cell signaling regulates differentiation and survival of neural crest stem cells. Mol Cells. 2014;37:907–911.
- Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–3594.
- Oh WJ, Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle. 2011;10:2305–2316.
- Sarbassov DD, Guertin DA, Ali SM, et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101.
- Koo J, Wu X, Mao Z, et al. Rictor undergoes Glycogen Synthase Kinase 3 (GSK3)-dependent, FBXW7-mediated Ubiquitination and Proteasomal degradation. J Biol Chem. 2015;290:14120–14129.
- Turner NC, Reis-Filho JS. Basal-like breast cancer and the BRCA1 phenotype. Oncogene. 2006;25:5846–5853.
- Turner NC, Reis-Filho JS, Russell AM, et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene. 2007;26:2126–2132.
- Chen X, Cheng H, Pan T, et al. mTOR regulate EMT through RhoA and Rac1 pathway in prostate cancer. Mol Carcinog. 2015;54:1086–1095.
- Kwasnicki A, Jeevan D, Braun A, et al. Involvement of mTOR signaling pathways in regulating growth and dissemination of metastatic brain tumors via EMT. Anticancer Res. 2015;35:689–696.
- Sharif-Askari B, Amrein L, Aloyz R, et al. PARP3 inhibitors ME0328 and olaparib potentiate vinorelbine sensitization in breast cancer cell lines. Breast Cancer Res Treat. 2018;172:23–32.
- Quan JJ, Song JN, Qu JQ. PARP3 interacts with FoxM1 to confer glioblastoma cell radioresistance. Tumour Biol. 2015;36:8617–8624.
- O’Sullivan J, Tedim Ferreira M, Gagne JP, et al. Emerging roles of eraser enzymes in the dynamic control of protein ADP-ribosylation. Nat Commun. 2019;10:1182.
- Lindgren AE, Karlberg T, Thorsell AG, et al. A PARP inhibitor with selectivity toward ADP-ribosyltransferase ARTD3/PARP3. ACS Chem Biol. 2013;8:1698–1703.