
ABSTRACT
DAPK1 and DAPK2 are calmodulin (CaM)-regulated protein kinases that share a high degree of homology in their catalytic and CaM regulatory domains. Both kinases function as tumor suppressors, and both have been implicated in autophagy regulation. Over the years, common regulatory mechanisms for the two kinases as well as kinase-specific ones have been identified. In a recent work, we revealed that DAPK2 is phosphorylated on Ser289 by the metabolic sensor AMPK, and that this phosphorylation enhances DAPK2 catalytic activity. Notably, Ser289 is conserved between DAPK1 and DAPK2, and was previously found to be phosphorylated in DAPK1 by RSK. Intriguingly, Ser289 phosphorylation was conversely reported to inhibit the pro-apoptotic activity of DAPK1 in cells. However, as the direct effect of this phosphorylation on DAPK1 catalytic activity was not tested, indirect effects were not excluded. Here, we compared Ser289 phosphorylation of the two kinases in the same cells and found that the intracellular signaling pathways that lead to Ser289 phosphorylation are mutually-exclusive and different for each kinase. In addition, we found that Ser289 phosphorylation in fact enhances DAPK1 catalytic activity, similar to the effect on DAPK2. Thus, Ser289 phosphorylation activates both DAPK1 and DAPK2, but in response to different intracellular signaling pathways.
DAPK1 (Death-associated protein kinase 1) and DAPK2 (Death-associated protein kinase 2; also named DRP-1) are both Ca2+/CaM-regulated Ser/Thr kinases that belong to the DAPK family of proteins (which also includes DAPK3/ZIPK, DRAK1, and DRAK2) [Citation1–Citation4]. The founding and most well-studied member of the family is DAPK1, a large (160kDa) multi-domain protein implicated in a wide range of biological activities [Citation5]. Importantly, DAPK1 functions as a tumor suppressor gene whose expression is lost in many cancers due to promoter hypermethylation [Citation6–Citation16]. DAPK2 is a much smaller protein (42kDa), which also mediates various cellular processes [Citation17], and was identified as a tumor suppressor [Citation18]. As their name implies, DAPK1 and DAPK2 were shown to mediate cell death, a function likely contributing to their tumor suppressive function. Several works have linked DAPK1 [Citation19–Citation23] and DAPK2 [Citation18,Citation24] to caspase-dependent apoptosis. In addition, DAPK1 and DAPK2 were found to promote autophagy induction [Citation25,Citation26].
Autophagy is a highly regulated intracellular degradation process in which a double-membrane vesicle, termed an autophagosome, is formed in the cytosol, engulfing cytosolic material. Autophagy induction is mediated by the Ulk1 kinase complex, which is negatively and positively regulated by the cellular energy sensors mTOR (mammalian target of rapamycin) [Citation27,Citation28] and AMPK (AMP-activated protein kinase) [Citation29,Citation30], respectively. In its active state, Ulk1 triggers autophagosome biogenesis through phosphorylation and activation of a class III PI(3)K (phosphoinositide 3-kinase) multi-protein complex containing Beclin-1, Atg14, Vps15, and the lipid kinase Vps34 (Beclin-1/Vps34 complex) [Citation31,Citation32]. This complex is responsible for the production of PI(3)P (phosphatidylinositol 3-phosphate) at the autophagosome assembly site, which in turn serves to recruit other autophagic proteins. The autophagy-promoting activity of Beclin-1 is suppressed, among other mechanisms, by direct binding of anti-apoptotic members of the Bcl-2 family to the BH3 domain of Beclin-1 [Citation33]. The fully formed autophagosome later fuses with the lysosome, where its content is degraded and then recycled (reviewed in [Citation34,Citation35]). Under steady-state conditions, basal levels of autophagy are maintained within the cell to eliminate damaged organelles, protein aggregates, and long-lived proteins. Under certain stress conditions, such as nutrient deprivation, autophagy is highly enhanced in order to provide the cell with essential building blocks for its survival [Citation36]. However, accumulating evidence over recent years has led to the characterization of “autophagy-dependent cell death” under certain circumstances [Citation37], thus possibly linking DAPK1 and DAPK2’s function in autophagy to their role in promoting cell death.
DAPK1 and DAPK2 were initially linked to autophagy in a study in which overexpression of DAPK1 or DAPK2 induced the appearance of autophagosomes [Citation38]. This link was later strengthened by works showing that depletion of DAPK1 [Citation39,Citation40] and DAPK2 [Citation41,Citation42] reduced autophagy induced by different triggers. Two mechanisms have been proposed so far for DAPK1-mediated activation of autophagy, both involving activation of the Beclin-1/Vps34 complex responsible for PI(3)P production at the autophagosome assembly site. The first is a kinase cascade, in which DAPK1 phosphorylates PKD, which then directly phosphorylates and activates Vps34 to induce autophagy [Citation40]. The second mechanism involves DAPK1-mediated phosphorylation of Beclin-1 on Thr119 located in its BH3 domain, consequent dissociation of the inhibitory interaction with Bcl-XL/Bcl-2, and activation of the Beclin-1/Vps34 complex [Citation43,Citation44].
Several different mechanisms have been proposed for DAPK2-mediated activation of autophagy. DAPK2 phosphorylation of the mTOR binding partner raptor was suggested to inhibit mTORC1 (mTOR complex 1) activity and thus promote autophagy induction [Citation41]. DAPK2 was also found to phosphorylate the autophagy cargo receptor protein p62/SQSTM, which recognizes specific intracellular targets and recruits them to the nascent autophagosome for degradation. This phosphorylation was suggested to promote selective autophagy [Citation45]. Most recently, we have found that similarly to DAPK1, DAPK2 can phosphorylate Beclin-1 on Thr119 and promote its dissociation from the inhibitory interaction with Bcl-XL, causing activation of the Beclin-1/Vps34 complex [Citation46]. Thus, DAPK2 is linked in multiple ways to various stages of the autophagy pathway, highlighting autophagy as one of its main cellular functions.
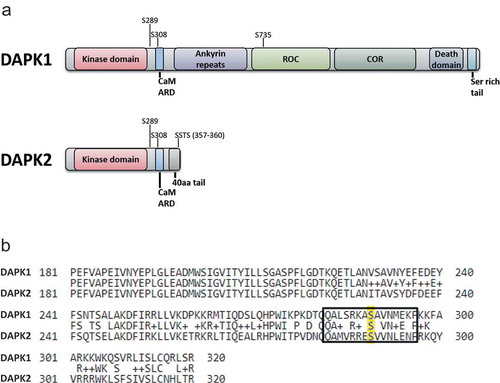
DAPK1 and DAPK2 show a high degree of homology in their N-terminal catalytic domains (~80% identity) and the adjacent CaM auto-regulatory domains [Citation4], yet outside of these domains, they differ substantially. DAPK2, at 42 kDa, possesses only a short C-terminal tail of 40 amino acids that follows the kinase and the CaM auto-regulatory domain. DAPK1, at 160 kDa, possesses many additional extra-catalytic domains, including eight ankyrin repeats, a cytoskeletal-binding region overlapping with a ROC-COR (Ras of complex proteins-C-terminal of ROC) domain (which regulates catalytic activity through GTP binding [Citation47]), a death domain and a serine-rich C-terminal tail ()). These major differences in extra-catalytic domain composition are thought to provide specific roles for DAPK1 and DAPK2 in cells, despite their highly identical kinase domains and hence similar substrate preferences. Indeed, previous work has found that DAPK1 and DAPK2 reside in different intracellular localizations [Citation4] and have different profiles of interacting proteins [Citation5,Citation48]. DAPK1 and DAPK2 also share similar regulatory mechanisms in their common domains. Mainly, both are positively regulated by binding of calcium-activated CaM to their CaM auto-regulatory domains and are negatively regulated by an inhibitory auto-phosphorylation on Ser308 within this domain [Citation49,Citation50]. Yet, outside of their shared domains, they are regulated by different mechanisms, including several-specific trans-phosphorylation events ()). Primarily, DAPK1 is positively regulated by ERK, which phosphorylates Ser735 in DAPK1’s ROC domain, thereby activating its cellular functions [Citation51]. DAPK2, on the other hand, is negatively regulated by phosphorylation of one or more of the last four amino acids of the protein (SSTS), which constitute a 14-3-3 binding site [Citation48]. Consequent binding of 14-3-3 to the C-terminal tail reduces DAPK2 catalytic activity. The kinase(s) responsible for this inhibitory phosphorylation are yet unknown, though AKT was proposed as a possible kinase [Citation52].
Figure 1. Comparison between DAPK1 and DAPK2 sequence and domain structure. (a) A schematic representation of the different domains of DAPK1 and DAPK2. Major phosphorylation sites are marked. CaM ARD – CaM autoregulatory domain. (b) Alignment of DAPK1 and DAPK2. The amino acid sequences of human DAPK1 (UniProt IP P53355) and human DAPK2 (UniProt ID Q9UIK4) were aligned using the Protein BLAST algorithm (amino acids 181–320 shown). Ser289 is highlighted in yellow and the surrounding sequence marked by a rectangle.
In a recent study, we identified the metabolic sensor AMPK as an upstream kinase of DAPK2 in cells and in starved mice [Citation46]. AMPK phosphorylates DAPK2 on Ser289, which resides at a critical site in the protein structure, between the catalytic and the CaM-binding domains. We showed that this phosphorylation activates DAPK2 by functionally mimicking CaM binding and mitigating the inhibitory autophosphorylation on Ser308, providing a novel, alternative mechanism for DAPK2 activation during metabolic stress. Importantly, we found that Ser289 phosphorylation enhances DAPK2’s ability to phosphorylate its autophagic substrate Beclin-1. At the cellular level, we found that depletion of DAPK2 reduced AMPK-dependent autophagy, thus highlighting the importance of the AMPK-DAPK2 axis in promoting autophagy in response to metabolic stress [Citation46].
Ser289 is also conserved in DAPK1, and was previously reported to be phosphorylated by RSK (ribosomal s6 kinase) following activation of the MAPK cascade [Citation53]. Interestingly, we found that despite the fact that the sequence surrounding DAPK1 Ser289 is highly similar to that of DAPK2, and also constitutes a possible AMPK consensus motif ()), DAPK1 is not phosphorylated on this site upon AMPK activation in cells [Citation46]. Thus, Ser289 phosphorylation by AMPK is specific to DAPK2. Here, we tested the reciprocal setting, i.e., whether DAPK2 Ser289 can also be phosphorylated by activation of the MAPK cascade, or whether this signaling pathway is exclusive to DAPK1. Additionally, since it has been reported that Ser289 phosphorylation by RSK conversely inhibits the pro-apoptotic function of DAPK1 in 293E cells [Citation53], we also measured here for the first time the direct effect of Ser289 phosphorylation on the catalytic activity of DAPK1, to determine whether phosphorylation of this site might have opposite effects on each kinase.
Materials and methods
Cell culture
HEK293T and HCT116 cells were grown in Dulbecco’s Modified Eagle’s medium (Biological Industries, Beit Haemek, Israel), supplemented with 10% fetal bovine serum (FBS, GibcoBRL), 4mM glutamine (GibcoBRL) and combined antibiotics (100µg/ml penicillin and 0.1mg/ml streptomycin).
DNA constructs
HA-tagged DAPK2 or DAPK1 WT and different mutants were expressed from pcDNA3 expression vectors. Point mutations were generated by PCR-mediated site-directed mutagenesis.
Transfection procedures
HEK293T cells were transfected by standard calcium phosphate technique. HCT116 cells were transfected using JetPEI (PolyPlus), according to the manufacturer’s protocol.
Protein analysis
HCT116 cells were lysed with PLB buffer (10mM NaPO4 pH 7.5, 5mM EDTA, 100mM NaCl, 1% Triton X-100, 1% Na deoxycholate, 0.1% SDS). 293T cells were lysed with TNE buffer (1% NP-40, 20 mM Tris–HCl pH 7.5, 150mM NaCl, 1mM EDTA). Lysis buffer was supplemented with 1μM PMSF and protease and phosphatase inhibitor cocktails (Sigma). Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes, which were incubated with antibodies against HA, vinculin, ERK1/2, active diphosphorylated ERK1/2 (Thr183/Tyr185) (Sigma), phospho-MLC (Ser19), AMPK, phospho-AMPK (Thr172), phospho-Akt substrate (RXRXXS*/T*) (Cell Signaling). Polyclonal antiphospho-Ser289 DAPK2 antibody was raised in rabbits immunized with the phosphorylated peptide CMVRRE(pS)VVNLEN (Bethyl Laboratories). Detection was done with either HRP-conjugated goat anti-mouse or anti-rabbit secondary antibodies (Jackson ImmunoResearch), followed by enhanced chemiluminescence (EZ-ECL, Biological Industries Israel Beit-Haemek Ltd.). Protein densitometric analysis was performed using ImageJ (NIH Imaging Software) on scanned blots.
Immunoprecipitation
Protein extracts were incubated with anti-HA beads (Sigma–Aldrich). After multiple washes in lysis buffer, the immunoprecipitated protein was eluted from the beads with an excess of HA peptides (Sigma–Aldrich).
Kinase assay
30 ng HA-DAPK1 immunoprecipitated from 293T cells were incubated with 2 µg MLC in DAPK1 kinase assay buffer (50 mM Hepes pH 7.5, 20 mM MgCl2, 50 µM ATP, protease and phosphatase inhibitor mix) for 5 min at 30ºC. Reactions were stopped with sample buffer and resolved by SDS-PAGE.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software. Values of P< 0.05 were considered significant.
Results
DAPK1 and DAPK2 are phosphorylated on Ser289 in response to different triggers
In order to simultaneously compare the status of Ser289 phosphorylation of the two kinases in response to the different triggers, HCT116 cells were transfected with either DAPK1 or DAPK2 and treated with either PMA (Phorbol 12-myristate 13-acetate) to activate the MAPK cascade, or resveratrol to activate AMPK. Untreated cells were used as control. DAPK1 and DAPK2 were immunoprecipitated and phosphorylation of Ser289 was assessed with phospho-specific antibodies. Notably, DAPK1 was phosphorylated on Ser289 solely in response to PMA treatment, and not in response to resveratrol treatment, while the opposite occurred for DAPK2, which was phosphorylated on Ser289 in response to resveratrol, but not in response to PMA ()). This remarkable difference between the two kinases is especially noteworthy, as not only is Ser289 conserved between the two kinases, but its surrounding sequence is also highly similar ()), and the overall structure of this region is identical between the two kinases. This suggests high specificity of the upstream kinases responsible for Ser289 phosphorylation, and possible regulation of this phosphorylation by specific protein–protein interactions or subcellular localization.
Figure 2. Ser289 phosphorylation activates DAPK1 and DAPK2 in response to different triggers. (a) HCT116 cells were transfected with either HA-DAPK1 or HA-DAPK2 and treated with either 200µM resveratrol (RSV) for 2 h or serum-starved overnight and then treated with 100nM PMA for 30 min. Untreated cells were used as control. Anti-HA immunoprecipitates were resolved by SDS-PAGE and Ser289 phosphorylation of the immunoprecipitated proteins was measured by specific antibodies. DAPK1 Ser289 phosphorylation was monitored using an antibody that recognizes the sequence RXRXXpSer/Thr, which was previously shown to specifically detect phosphorylation of DAPK1 on Ser289 [Citation53]. DAPK2 Ser289 phosphorylation was monitored using an antibody specifically raised against pSer289 DAPK2 [Citation46]. Two representative immunoprecipitation results, from two separate biological replicates (IP #1 and IP #2, left panel), are shown. Total cell extract (TCE, right panel) was used to monitor activation of the relevant pathways by the triggers, and to monitor the expression of HA-DAPK1/DAPK2. The TCE results shown belong to experiment #2 and are representative of both experiments. (b) HA-DAPK1 WT, S289A or S289D was incubated in a kinase reaction mixture with MLC, and phosphorylation assessed by western blotting of the reaction mixtures. pMLC band intensity was quantified using NIH ImageJ software. Bar graph represents pMLC intensity as mean ± SD of three independent repeats. Statistical analyses were performed using one-way ANOVA with post hoc Dunnett’s multiple comparison test. **P< 0.01.
![Figure 2. Ser289 phosphorylation activates DAPK1 and DAPK2 in response to different triggers. (a) HCT116 cells were transfected with either HA-DAPK1 or HA-DAPK2 and treated with either 200µM resveratrol (RSV) for 2 h or serum-starved overnight and then treated with 100nM PMA for 30 min. Untreated cells were used as control. Anti-HA immunoprecipitates were resolved by SDS-PAGE and Ser289 phosphorylation of the immunoprecipitated proteins was measured by specific antibodies. DAPK1 Ser289 phosphorylation was monitored using an antibody that recognizes the sequence RXRXXpSer/Thr, which was previously shown to specifically detect phosphorylation of DAPK1 on Ser289 [Citation53]. DAPK2 Ser289 phosphorylation was monitored using an antibody specifically raised against pSer289 DAPK2 [Citation46]. Two representative immunoprecipitation results, from two separate biological replicates (IP #1 and IP #2, left panel), are shown. Total cell extract (TCE, right panel) was used to monitor activation of the relevant pathways by the triggers, and to monitor the expression of HA-DAPK1/DAPK2. The TCE results shown belong to experiment #2 and are representative of both experiments. (b) HA-DAPK1 WT, S289A or S289D was incubated in a kinase reaction mixture with MLC, and phosphorylation assessed by western blotting of the reaction mixtures. pMLC band intensity was quantified using NIH ImageJ software. Bar graph represents pMLC intensity as mean ± SD of three independent repeats. Statistical analyses were performed using one-way ANOVA with post hoc Dunnett’s multiple comparison test. **P< 0.01.](/cms/asset/412b4ba5-85de-467b-bb76-665330718423/kccy_a_1617616_f0002_b.gif)
Ser289 phosphorylation enhances DAPK1 catalytic activity in vitro
We have previously shown that Ser289 phosphorylation enhances DAPK2 catalytic activity (Fig. 3 in [Citation46]). Conversely, it was reported that Ser289 phosphorylation of DAPK1 reduces its pro-apoptotic activity in 293E cells [Citation53], which suggests that the effect of Ser289 phosphorylation on the catalytic activity of the two kinases might be opposite. Thus, it became of interest to measure the direct effect of Ser289 phosphorylation on DAPK1 catalytic activity. To this end, we performed an in vitro kinase assay of DAPK1 WT and Ser289 mutants using MLC (myosin II regulatory light chain) as substrate. Remarkably, the DAPK1 S289D phospho-mimicking mutant showed significantly higher catalytic activity compared to WT DAPK1 ()). These results imply that Ser289 phosphorylation in fact enhances the catalytic activity of DAPK1.
Discussion
Both DAPK1 and DAPK2 were shown to positively regulate autophagy. Specifically, both were shown to phosphorylate Beclin-1 on Thr119 located in its BH3 domain, causing dissociation of its inhibitor Bcl-XL [Citation44,Citation46]. The fact that both kinases can perform the same function to promote autophagy raises the question of whether they have kinase-specific or redundant roles in the cell.
The link between AMPK and DAPK2 is the first connection between metabolic stress and activation of any member of the DAPK family. The fact that this pathway is unique to DAPK2 implies that it may have a specific role in promoting autophagy in response to metabolic stress. DAPK1, however, was previously shown to promote autophagy in response to ER stress [Citation39] and oxidative stress [Citation40]. As we have now shown that activation of the MAPK cascade enhances DAPK1 catalytic activity not only by Ser735 phosphorylation [Citation51] but also by Ser289 phosphorylation, it would be interesting to study whether the MAPK-DAPK1 cascade also promotes autophagy. Overall, it seems that while both DAPK1 and DAPK2 are able to promote autophagy by phosphorylation of shared substrates, they may do so in response to different stimuli.
The in vitro kinase assay results suggest that the effect of Ser289 phosphorylation on DAPK1 and DAPK2 catalytic activity is similar, consistent with the fact that DAPK1 and DAPK2 are essentially identical in terms of structure in this region of the protein. We have previously suggested that the activating effect of Ser289 phosphorylation on DAPK2 catalytic activity is a result of a conformational change causing a shift of the CaM auto-regulatory domain away from the catalytic cleft, similar to the effect of CaM binding [Citation46]. It is plausible to assume that a similar effect would occur in DAPK1 structure as a result of phosphorylation on the same site, inducing the same activating effect on its catalytic activity.
The finding that the DAPK1 Ser289 phospho-mimicking mutant has enhanced catalytic activity is intriguing in light of the previous report of the reduced pro-apoptotic activity of this mutant in 293E cells. However, that report was based solely on the phenotypic effect of overexpression of DAPK1 WT and Ser289 mutants in 293E cells, while no phosphorylation of direct DAPK1 substrates was measured. Thus, the reduced apoptotic phenotype measured might have resulted from changes in protein–protein interactions or subcellular localization, rather than from reduced catalytic activity. An especially interesting possibility is that the reduced apoptotic phenotype might result from enhanced phosphorylation of pro-autophagic substrates, and consequent antagonistic interactions between survival autophagy and apoptosis. This possibility should be further studied.
Acknowledgments
This work was supported by a grant from European Research Council under the European Union’s Seventh Framework Program (FP7/2007-2013/ERC grant agreement 322709). A.K. is the Incumbent of the Helena Rubinstein Chair of Cancer Research.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Deiss LP, Feinstein E, Berissi H, et al. Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev. 1995 Jan 1;9(1):15–30. PubMed PMID: 7828849; eng.
- Inbal B, Shani G, Cohen O, et al. Death-associated protein kinase-related protein 1, a novel serine/threonine kinase involved in apoptosis. Mol Cell Biol. 2000 Feb;20(3):1044–1054. PubMed PMID: 10629061; eng.
- Kawai T, Nomura F, Hoshino K, et al. Death-associated protein kinase 2 is a new calcium/calmodulin-dependent protein kinase that signals apoptosis through its catalytic activity. Oncogene. 1999 Jun 10;18(23):3471–3480. PubMed PMID: 10376525; eng.
- Shiloh R, Bialik S, Kimchi A. The DAPK family: a structure-function analysis. Apoptosis. 2014 Feb;19(2):286–297. PubMed PMID: 24220854; eng.
- Bialik S, Kimchi A. The DAP-kinase interactome. Apoptosis. 2014 Feb;19(2):316–328. PubMed PMID: 24220855.
- Gozuacik D, Kimchi A. DAPk protein family and cancer. Autophagy. 2006 Apr-Jun;2(2):74–79. PubMed PMID: 17139808; eng.
- Esteller M, Corn PG, Baylin SB, et al. A gene hypermethylation profile of human cancer. Cancer Res. 2001 Apr 15;61(8):3225–3229. PubMed PMID: 11309270.
- Collins Y, Dicioccio R, Keitz B, et al. Methylation of death-associated protein kinase in ovarian carcinomas. Int J Gynecol Cancer. 2006 Jan-Feb;16(Suppl 1):195–199. PubMed PMID: 16515590.
- Mittag F, Kuester D, Vieth M, et al. DAPK promotor methylation is an early event in colorectal carcinogenesis. Cancer Lett. 2006 Aug 18;240(1):69–75. PubMed PMID: 16246486.
- Qian J, Wang YL, Lin J, et al. Aberrant methylation of the death-associated protein kinase 1 (DAPK1) CpG island in chronic myeloid leukemia. Eur J Haematol. 2009 Feb;82(2):119–123. PubMed PMID: 19018866.
- Kilinc D, Ozdemir O, Ozdemir S, et al. Alterations in promoter methylation status of tumor suppressor HIC1, SFRP2, and DAPK1 genes in prostate carcinomas. DNA Cell Biol. 2012 May;31(5):826–832. PubMed PMID: 22136354.
- Kristensen LS, Asmar F, Dimopoulos K, et al. Hypermethylation of DAPK1 is an independent prognostic factor predicting survival in diffuse large B-cell lymphoma. Oncotarget. 2014 Oct 30;5(20):9798–9810. PubMed PMID: 25229255; PubMed Central PMCID: PMCPMC4259438.
- Zhang J, Yu XL, Zheng GF, et al. DAPK promoter methylation status correlates with tumor metastasis and poor prognosis in patients with non-small cell lung cancer. Cancer Biomark. 2015;15(5):609–617. PubMed PMID: 26406950.
- Dai L, Ma C, Zhang Z, et al. DAPK promoter methylation and bladder cancer risk: a systematic review and meta-analysis. PLoS One. 2016;11(12):e0167228. PubMed PMID: 27907054; PubMed Central PMCID: PMCPMC5132202.
- Yuan W, Chen J, Shu Y, et al. Correlation of DAPK1 methylation and the risk of gastrointestinal cancer: A systematic review and meta-analysis. PLoS One. 2017;12(9):e0184959. PubMed PMID: 28934284; PubMed Central PMCID: PMCPMC5608298.
- Cai F, Xiao X, Niu X, et al. Association between promoter methylation of DAPK gene and HNSCC: A meta-analysis. PLoS One. 2017;12(3):e0173194. PubMed PMID: 28249042; PubMed Central PMCID: PMCPMC5332095.
- Geering B. Death-associated protein kinase 2: regulator of apoptosis, autophagy and inflammation. Int J Biochem Cell Biol. 2015 Aug;65:151–154. PubMed PMID: 26055515; eng.
- Tur MK, Neef I, Jost E, et al. Targeted restoration of down-regulated DAPK2 tumor suppressor activity induces apoptosis in Hodgkin lymphoma cells. J Immunother. 2009 Jun;32(5):431–441. PubMed PMID: 19609235; eng.
- Raveh T, Droguett G, Horwitz MS, et al. DAP kinase activates a p19ARF/p53-mediated apoptotic checkpoint to suppress oncogenic transformation. Nat Cell Biol. 2001 Jan;3(1):1–7. PubMed PMID: 11146619; eng.
- Jang CW, Chen CH, Chen CC, et al. TGF-beta induces apoptosis through Smad-mediated expression of DAP-kinase. Nat Cell Biol. 2002 Jan;4(1):51–58. PubMed PMID: 11740493.
- Wang WJ, Kuo JC, Yao CC, et al. DAP-kinase induces apoptosis by suppressing integrin activity and disrupting matrix survival signals. J Cell Biol. 2002 Oct 14;159(1):169–179. PubMed PMID: 12370243; eng.
- Llambi F, Lourenco FC, Gozuacik D, et al. The dependence receptor UNC5H2 mediates apoptosis through DAP-kinase. Embo J. 2005 Mar 23;24(6):1192–1201. PubMed PMID: 15729359; eng.
- Guenebeaud C, Goldschneider D, Castets M, et al. The dependence receptor UNC5H2/B triggers apoptosis via PP2A-mediated dephosphorylation of DAP kinase. Mol Cell. 2010 Dec 22;40(6):863–876. PubMed PMID: 21172653; eng.
- Li H, Ray G, Yoo BH, et al. Down-regulation of death-associated protein kinase-2 is required for beta-catenin-induced anoikis resistance of malignant epithelial cells. J Biol Chem. 2009 Jan 23;284(4):2012–2022. PubMed PMID: 18957423; eng.
- Bialik S, Kimchi A. Lethal weapons: DAP-kinase, autophagy and cell death: DAP-kinase regulates autophagy. Curr Opin Cell Biol. 2010 Apr;22(2):199–205. PubMed PMID: 20005690.
- Levin-Salomon V, Bialik S, Kimchi A. DAP-kinase and autophagy. Apoptosis. 2014 Feb;19(2):346–356. PubMed PMID: 24264886.
- Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009 Apr;20(7):1992–2003. PubMed PMID: 19225151; PubMed Central PMCID: PMCPMC2663920.
- Ganley IG, Lam Du H, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009 May 1;284(18):12297–12305. PubMed PMID: 19258318; PubMed Central PMCID: PMCPMC2673298.
- Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011 Jan 28;331(6016):456–461. PubMed PMID: 21205641; PubMed Central PMCID: PMC3030664.
- Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011 Feb;13(2):132–141. PubMed PMID: 21258367; eng.
- Itakura E, Kishi C, Inoue K, et al. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008 Dec;19(12):5360–5372. PubMed PMID: 18843052; eng.
- Sun Q, Fan W, Chen K, et al. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2008 Dec 9;105(49):19211–19216. PubMed PMID: 19050071; eng.
- He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol. 2010 Apr;22(2):140–149. PubMed PMID: 20097051; eng.
- Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014 Jan;24(1):24–41. PubMed PMID: 24366339; eng.
- Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014 Jan;24(1):9–23. PubMed PMID: 24366340; PubMed Central PMCID: PMC3879711.
- Mizushima N. A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol. 2018 May;20(5):521–527. PubMed PMID: 29686264.
- Bialik S, Dasari SK, Kimchi A. Autophagy-dependent cell death - where, how and why a cell eats itself to death. J Cell Sci. 2018 Sep 20;131(18). PubMed PMID: 30237248. DOI:10.1242/jcs.215152
- Inbal B, Bialik S, Sabanay I, et al. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol. 2002 Apr 29;157(3):455–468. PubMed PMID: 11980920; eng.
- Gozuacik D, Bialik S, Raveh T, et al. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008 Dec;15(12):1875–1886. PubMed PMID: 18806755; eng.
- Eisenberg-Lerner A, Kimchi A. PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell Death Differ. 2012 May;19(5):788–797. PubMed PMID: 22095288; PubMed Central PMCID: PMCPMC3321617.
- Ber Y, Shiloh R, Gilad Y, et al. DAPK2 is a novel regulator of mTORC1 activity and autophagy. Cell Death Differ. 2015 Mar;22(3):465–475. PubMed PMID: 25361081; eng.
- Soussi H, Reggio S, Alili R, et al. DAPK2 downregulation associates with attenuated adipocyte autophagic clearance in human obesity. Diabetes. 2015 Oct;64(10):3452–3463. PubMed PMID: 26038578; eng.
- Zalckvar E, Berissi H, Eisenstein M, et al. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy. 2009 Jul;5(5):720–722. PubMed PMID: 19395874; eng.
- Zalckvar E, Berissi H, Mizrachy L, et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009 Mar;10(3):285–292. PubMed PMID: 19180116; PubMed Central PMCID: PMCPMC2658558.
- Chi HC, Chen SL, Tsai CY, et al. Thyroid hormone suppresses hepatocarcinogenesis via DAPK2 and SQSTM1-dependent selective autophagy. Autophagy. 2016 Sep 21;1–15. PubMed PMID: 27653365. DOI:10.1080/15548627.2016.1230583
- Shiloh R, Gilad Y, Ber Y, et al. Non-canonical activation of DAPK2 by AMPK constitutes a new pathway linking metabolic stress to autophagy. Nat Commun. 2018 May 1;9(1):1759. PubMed PMID: 29717115; PubMed Central PMCID: PMCPMC5931534.
- Carlessi R, Levin-Salomon V, Ciprut S, et al. GTP binding to the ROC domain of DAP-kinase regulates its function through intramolecular signalling. EMBO Rep. 2011 Sep;12(9):917–923. PubMed PMID: 21738225; eng.
- Gilad Y, Shiloh R, Ber Y, et al. Discovering protein-protein interactions within the programmed cell death network using a protein-fragment complementation screen. Cell Rep. 2014 Aug 7;8(3):909–921. PubMed PMID: 25066129; eng.
- Shohat G, Spivak-Kroizman T, Cohen O, et al. The pro-apoptotic function of death-associated protein kinase is controlled by a unique inhibitory autophosphorylation-based mechanism. J Biol Chem. 2001 Dec 14;276(50):47460–47467. PubMed PMID: 11579085; eng.
- Shani G, Henis-Korenblit S, Jona G, et al. Autophosphorylation restrains the apoptotic activity of DRP-1 kinase by controlling dimerization and calmodulin binding. Embo J. 2001 Mar 1;20(5):1099–1113. PubMed PMID: 11230133; eng.
- Chen CH, Wang WJ, Kuo JC, et al. Bidirectional signals transduced by DAPK-ERK interaction promote the apoptotic effect of DAPK. Embo J. 2005 Jan 26;24(2):294–304. PubMed PMID: 15616583; eng.
- Yuasa K, Ota R, Matsuda S, et al. Suppression of death-associated protein kinase 2 by interaction with 14-3-3 proteins. Biochem Biophys Res Commun. 2015 Aug 14;464(1):70–75. PubMed PMID: 26047703; eng.
- Anjum R, Roux PP, Ballif BA, et al. The tumor suppressor DAP kinase is a target of RSK-mediated survival signaling. Curr Biol. 2005 Oct 11;15(19):1762–1767. PubMed PMID: 16213824; eng.