
ABSTRACT
L755S, a HER2 kinase domain mutation, is the most common HER2 mutation in breast cancer associated with resistance to anti-HER2 trastuzumab treatment. Here, we showed that HER2-L755S confers hyperactivation of MAPK and PI3K/AKT/mTOR pathways and resistance to both reversible and irreversible HER2 tyrosine kinase inhibitors. We further demonstrated that the HER2 TKIs in combination with MEK inhibitor, AZD6244, or PI3K inhibitor, GDC0941, yield robust killing in HER2-L755S cancer cells, indicating a novel targeted strategy to overcome HER2-L755S resistance to anti-HER2 treatment.
Introduction
Human epidermal growth factor receptor 2 (HER2), a transmembrane receptor with tyrosine kinase activity, belongs to a family of four receptors including EGFR, HER2, HER3 and HER4 [Citation1,Citation2]. Phosphorylation of HER2 kinase domain initiates multiple signaling pathways including PI3K/mTOR to be involved in the regulation of cell growth, survival and differentiation [Citation1,Citation3]. Overexpression of HER2 accounts for about 20% of breast cancer patients and is known to associate with poor prognosis [Citation1]. The HER2-targeted monoclonal antibody, trastuzumab, shows proven efficacy in patients with HER2+ breast cancer [Citation1,Citation2,Citation4]. However, substantial cases of relapsed metastasis have been observed in patients who showed an initial response to the trastuzumab treatment [Citation5–Citation7]. It has been shown that the frequency of HER2 mutations in metastatic breast cancer patients (27.8%) developed after trastuzumab treatment was higher than the 2.24% mutation rate reported in primary tumors, indicating an association of HER2 mutations with trastuzumab resistance [Citation5].
Among the HER2 mutations found in metastatic tumors, HER2-L755S is the most common one that accounts for 60% of HER2 mutations, much higher than the 25% found in the primary tumor [Citation5]. L755S is an activating mutation of HER2, which is also known to be associated with resistance to the reversible HER2 tyrosine kinase inhibitor (TKI) lapatinib. It is believed that L755S mutation stabilizes the active conformation of HER2 while lapatinib could only target the inactive conformation of HER2 [Citation8,Citation9]. Studies have shown that L755S mutation was able to enhance HER2 autophosphorylation as well as the downstream signaling pathway, such as MAPK and JNK/SAPK pathway [Citation9–Citation11]. Another HER2 activating mutation, del.16 (exon 16 deletion in the extracellular domain of HER2), has been linked to the resistance of trastuzumab through the activation of the downstream SRC kinase signaling [Citation12,Citation13].
Neratinib, an irreversible inhibitor of EGFR, HER2 and HER4, has now been approved for the extended adjuvant treatment of HER2+ breast cancer [Citation14,Citation15]. It is shown that neratinib reduces HER2 autophosphorylation by irreversibly targeting the ATP-binding pocket of HER2 [Citation16]. Although neratinib has been suggested to be used in HER2 mutated breast cancer, the response rate is still lower than expected when compared to FDA approved therapies targeting other oncogenic alteration, suggesting that additional mechanism might exist to confer the resistance of HER2-L755S to TKI treatment [Citation17]. Given the increasing consensus that a combination strategy appears to be necessary for complete inhibition of the HER2-mediated growth signal [Citation11], we seek to investigate if HER2-L755S mutation induces additional signaling pathways to confer resistance to HER2 kinase inhibitors, with an aim to develop a new targeted treatment strategy to overcome the HER2-L755S resistance to TKIs.
Results
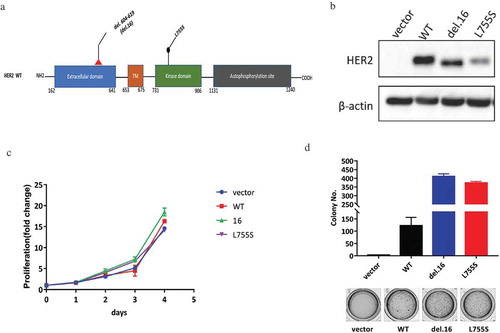
HER2 mutations cause oncogenic transformation of MCF10A
HER2-del.16, an in-frame deletion mutation, was reported as a known oncogenic driver in breast cancer, while the L755S mutation which occurs at the kinase domain was associated with TKI resistance ()). To compare the oncogenic capacity of HER2-del.16 versus L755S, we overexpressed the two mutants together with the wild type HER2 in MCF10A cells, a non-tumorigenic cell line with negative status for ER, PR and HER2 ()) [Citation18]. Although these HER2-modified cell lines showed similar proliferation capacities when cultured in monolayer ()), the two HER2 mutants showed robust growth advantage compared with the wild type HER2 when cultured in soft agar ()). These data showed that HER2-L755S, similar to HER2-del.16, confers a robust oncogenic transformation in MCF10A cells.
Figure 1. HER2 mutations cause oncogenic transformation of MCF10A. (a) Structure visualizations of the HER2 somatic mutations, TM, transmembrane region. (b) Western blotting showing the expression of HER2 in indicated MCF10A cells. β-actin was used as a loading control. (c) Proliferation data for indicated MCF10A cells grown in monolayer for 4 days. (D) Soft agar assay of indicated MCF10A cells for 14 days. Data are expressed as means ± SEM of three technical replicates.
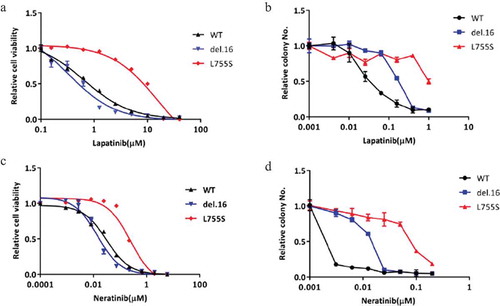
HER2-L755S, compared to HER2-del.16, is highly resistant to both reversible and irreversible TKIs
Next, we used MCF10A-HER2 mutant cell lines as our models to investigate their responses to reversible and irreversible TKIs, lapatinib and neratinib, respectively, in monolayer cell viability assay and soft agar colony formation assay (). Among the HER2-modified cell lines, we found that cells bearing HER2-L755S mutation showed most resistance to lapatinib in both short-term monolayer proliferation assay and longer duration soft agar assay (,b)). Given that neratinib has been suggested for use in patients harboring HER2-L755S mutation, we also tested the sensitivity of our MCF10A HER2 modified cell lines to neratinib. We observed a similar effect that MCF10A bearing HER2-L755S mutation was most resistant to neratinib (,d)). Collectively, these data indicated that HER2-L755S mutation, compared to wild type HER2 or another activating mutation, HER2-del.16, is strongly resistant to both lapatinib and neratinib.
Figure 2. HER2-L755S is highly resistant to both reversible and irreversible TKIs. (a and b) The dose response of indicated MCF10A cells to lapatinib treatment in monolayer for 4 days (a) and in soft-agar for 14 days (b). (c and d). The dose response of indicated MCF10A cells to neratinib treatment in monolayer for 4 days (c) and in soft-agar for 14 days (d). Data are expressed as means ± SEM. of three technical replicates.
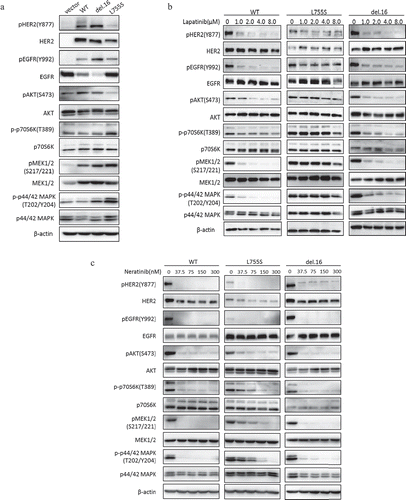
HER2-L755S expressing cells exhibit hyperactivation of MAPK and PI3K/AKT/mTOR pathways
To assess the effect of HER2 mutations on signal transduction pathways, we performed western blot in the MCF10A-transformed cells. Interestingly, we found that MCF10A cells harboring HER2-L755S showed increased levels of phospho-p70S6K, phospho-MEK1/2 and phospho-p42/44-MAPK compared with wild type HER2 and HER2-del.16 ()). While HER2 and EGFR phosphorylation level were highly sensitive to lapatinib in wild type HER2 or HER2-del.16 cells, they were not responsive to lapatinib in L755S-expressing cells ()). Moreover, p70S6K, MEK1/2, and p42/44-MAPK pathways in L755S mutant cells remained high following lapatinib treatment, even though these pathways were easily blocked by lapatinib in other HER2 cells lines ()). The irreversible TKI, neratinib, on the other hand, gave rise to an improved effect of inhibiting HER2 and EGFR phosphorylation in L755S cells as expected. However, the drug remained less effective in inhibiting p70S6K, MEK1/2 and p42/44-MAPK in L755S cells, when compared with other HER2-modified cells ()). These observations indicated that the hyperactivation of PI3K/AKT/mTOR and MAPK pathway which are refractory to both lapatinib and neratinib treatment might be a potential mechanism responsible for HER2 L755S-mediated TKIs resistance.
Figure 3. HER2-L755S expressing cells exhibit hyperactivation of MAPK and PI3K/AKT/mTOR pathways. (a) Western blotting showing the expression of HER2, EGFR, AKT, p70S6K, MEK1/2 and p42/44 MAPK in indicated MCF10A cells. β-actin was used as a loading control. (b and c) Western blotting showing the expression of HER2, EGFR, AKT, p70S6K, MEK1/2 and p42/44 MAPK in indicated MCF10A cells treated by lapatinib (b) and neratinib (c) at indicated concentrations for 4 h. β-actin was used as a loading control.
Lapatinib combination with MEK or PI3K inhibitors induces synergistic growth inhibition in HER2-L755S mutant cells
Given the possible roles of MAPK or PI3K/AKT/mTOR pathway in HER2-L755S expressing cells, we tested the potential benefit of the combination of lapatinib with MEK or PI3K inhibitors in controlling the growth of HER2-modified cell lines. AZD6244 is a second generation MEK inhibitors and is currently at the early stage of clinical development [Citation19,Citation20]. The previous study reported that AZD6244 was an ATP noncompetitive inhibitor of MEK1/2 and could lock MEK1/2 in an inactive conformation despite an increased level of phospho-MEK1/2 [Citation21]. As shown in (,b)), AZD6244 in combination with lapatinib induced a robust and sustained impairment of cell viability in cells bearing HER2-L755S, which was not seen in HER2-WT and HER2-del.16 cells. In accordance with the cellular effect in HER2-L755S cells, the activation of ERK signaling was completely blocked by MEK inhibitor AZD6244 alone and in combination with lapatinib ()).
Figure 4. Lapatinib combination with MEK or PI3K inhibitors induces synergistic growth inhibition in HER2-L755S mutant cells. (a and b) Cell proliferation of indicated MCF10A cells treated with lapatinib (5uM for L755S, 350nM for WT and del.16), AZD6244 (1uM for L755S, 400nM for WT and del.16), or in combination as indicated. Representative data are expressed as log2-fold change normalized to day 0 of three technical replicates. (c) Western blotting showing the expression of HER2, EGFR, MEK1/2 and p42/44 MAPK in MCF10A bearing HER2-L755S treated with MEK inhibitor AZD6244 (1uM) and lapatinib (1uM) for 24 h. (d and e) Cell proliferation of indicated MCF10A cells treated with lapatinib (5uM for L755S, 350nM for WT and del.16), GDC0941 (1uM for L755S, WT and del.16), or in combination as indicated. Representative data are expressed as log2-fold change normalized to day 0 of three technical replicates. (F) Western blotting showing the expression of HER2, EGFR, AKT and p70S6K in MCF10A bearing HER2-L755S treated with PI3K inhibitor GDC0941 (1uM) and/or lapatinib (1uM) for 24 h.
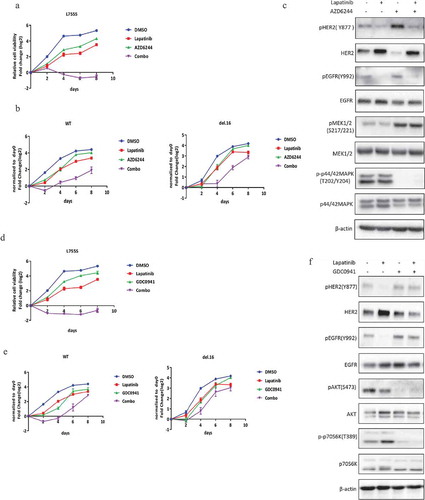
Similarly, PI3K inhibitor, GDC0941, which is currently active in clinical trials on various types of cancers [Citation22], showed a strong effect in HER2-L755S cells when combined with lapatinib in monolayer proliferation assay ()). In contrast, cells harboring HER2-WT and HER2-del.16 only showed an initial response to the combination treatment but recovered after four days of treatment ()). Consistent with this finding, GDC0941 alone or in combination with lapatinib inhibited the activation of PI3K/AKT-mTOR pathway ()). Furthermore, the combined effect of lapatinib with GDC0941 or AZD6244 was also validated in soft agar assay (Figure S2). Together, our data have shown that lapatinib treatment with concurrent inhibition of MAPK or PI3K/AKT/mTOR pathway can lead to robust growth inhibition of HER2-L755S expressing cells.
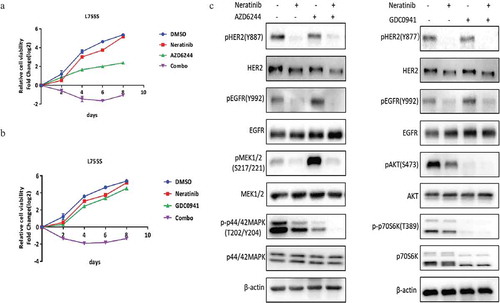
Neratinib combination with MEK or PI3K inhibitors induces synergistic growth inhibition in HER2-L755S mutant cells
Since MAPK and p70S6K remain to be refractory to neratinib in HER2-L755S, we tested the combination inhibition of neratinib in combination with MEK inhibitor or PI3K inhibitor. It turned out that a strong impairment of cell viability in HER2-L755S cells was achieved when neratinib combined with either MEK inhibitor AZD6244 ()) or PI3K inhibitor GDC0941 ()). Western blotting analysis showed a complete inhibition of MAPK or PI3K/AKT-mTOR pathway after combination with related kinase inhibitor ()). Taken together, our data showed that both reversible and irreversible TKIs can be combined with MEK or PI3K inhibitor to induce synergistic growth inhibition in HER2-L755S that is otherwise resistant to TKI alone.
Figure 5. Neratinib combination with MEK or PI3K inhibitors induces synergistic growth inhibition in HER2-L755S mutant cells. (a) Cell proliferation of HER2-L755S treated with neratinib (100nM), AZD6244 (1uM), or in combination as indicated. Representative data are expressed as log2-fold change normalized to day 0 of three technical replicates. (b) Cell proliferation of HER2-L755S treated with neratinib (100nM), GDC0941 (1uM), or in combination as indicated. Representative data are expressed as log2-fold change normalized to day 0 of three technical replicates. (c) Western blotting showing the expression of HER2, EGFR, AKT, p70S6K, MEK1/2 and p42/44 MAPK in MCF10A bearing HER2-L755S treated with MEK inhibitor neratinib (30nM) and AZD6244 (1uM) (left) or PI3K inhibitor GDC0941 (1uM) for 4 h (right).
Discussion
HER2 mutations occur in about 3% of breast cancer patients, among those various mutations, HER2-L755S has been identified as the most frequent HER2 mutation. Another HER2-activating mutation, HER2-del.16, has been identified to promote tumorigenesis in a mouse model by forming constitutively active homodimers which are able to activate oncogenic transduction pathway mediated via SRC family [Citation12,Citation13,Citation23,Citation24]. Of note, HER2-L755S occurs most frequently in trastuzumab treatment-relapsed patients [Citation5] and has been reported to produce lapatinib resistance [Citation9]. It remains unclear if L755S induces any signaling pathways that confer its resistance to these HER2-targeted therapies. Although the irreversible TKIs, neratinib and afatinib have been used with promising efficacy in treating most of the HER2 mutation [Citation25–Citation27], in vitro functional studies demonstrated that HER2-L755S only delivered a partial response to these irreversible TKIs [Citation28]. Clinical studies have also shown that neratinib single agent initially exhibited partial response in a patient harboring HER2-L755S mutation though neratinib-based combination can enhance the response in the progressed patient [Citation29]. Taken together, the response rate of neratinib in treating HER2 mutations remains lower than expected compared with approved targeted therapies for other oncogenic alteration [Citation17]; thus, the effort to identify novel strategies for HER2-L755S to achieve a maximal response is of great importance to HER2 mutated breast cancer.
Herein, we showed that the HER2-transformed cell lines displayed similar proliferation capacities in monolayer culturing condition. However, the two HER2 mutants showed robust growth advantage compared with HER2-WT in soft agar colony formation assay, implying that HER2-del.16 and HER2-L755S are oncogenic HER2 mutation which can cause robust transformation in MCF10A cells. Next, we found, despite comparable oncogenic transformation, HER2-del.16, along with other previously reported HER2 activating mutations such as V777L, V842I, G309A [Citation9,Citation27], is responsive to TKIs while HER2-L755S exhibited strong resistance to both reversible and irreversible TKIs, lapatinib and neratinib. This evidence suggests that HER2-L755S is a unique HER2 mutation in driving resistance to TKIs. Indeed, HER2-L755S expressed a higher level of PI3K/AKT/mTOR and MAPK signaling pathway. Even if neratinib showed a better effect to block HER2 and EGFR phosphorylation compared to lapatinib, the MAPK and p70S6K remained refractory to neratinib, and the current use of neratinib in patients bearing HER2-L755S remains at a high chance of developing relapse. Further combination study demonstrated that indeed the aberrant PI3K/AKT/mTOR and MAPK signaling pathway contributes to the resistance of HER2-L755S to TKIs as co-treatment with the MEK inhibitor, AZD6244, and PI3K inhibitor, GDC0941, clearly delivered the benefit.
In conclusion, these data show that HER2-L755S mutation is an alternative driver event in the resistance of TKIs through the hyperactivation of PI3K/AKT/mTOR and MAPK pathway, and this resistance can be overcome by combination treatment with a related kinase inhibitor. This combination strategy warrants further preclinical or clinical investigation for treatment of patients harboring HER2 L755S mutation.
Materials and methods
Chemicals
Lapatinib, neratinib and AZD6244 were purchased from Selleckchem.com. GDC0941 was purchased from MedChemExpress. Stocks of all drugs were prepared with DMSO.
Cell culture
MCF10A were obtained, authenticated, and cultured according to American Type Culture Collection (ATCC, Manassas, VA) instructions unless otherwise stated. All cell lines used for functional studies were tested and found to be free of mycoplasma contamination. The MCF10A cell line was cultured in DMEM/Ham’s F-12 (supplemented with 5% Horse Serum, 10μg/ml insulin (Sigma), 20 ng/ml EGF (Sigma), 0.5μg/ml hydrocortisone (Sigma) and 5,000 U/ml penicillin-streptomycin (Gibco)). All cell lines were maintained at 37℃in a humidified atmosphere at 5% CO2. The stable cell lines were obtained by infecting the lentivirus carrying PMN-GFP-HER2 WT, PMN-GFP-HER2 del.16 and PMN-GFP-HER2 L755S, selected with 2μg/ml puromycin (Clontech 631305) and sorted by FACS analysis for robust ErbB2 expression. PMN-GFP cell line was generated as reported previously [Citation30].
Cell viability assay
For the cell proliferation assay, optimal cell seeding was first determined empirically for all cell lines by examining the growth of a wide range of seeding densities in a 96-well format to identify conditions that permitted proliferation for eight days.
Cells were seeded at a density of 1000 cells per well of 96-well optical bottom plate (Corning) 24 h before drug treatment. The drug was serially diluted with medium and added into the wells and incubated for 96 h before detection. Three technical replicates were conducted for each sample. CellTiter-Glo Luminescent Cell Viability Assay (Promega) was used to evaluate viable cell numbers. The luminescence signal was measured by a microplate reader. To calculate IC50, nonlinear regression sigmoidal dose–response curves were generated using Graph-Pad PRISM7.
Anchorage-independent colony formation assay.
Experiments were carried out in 24-well plates coated with a base layer of DMEM/F-12 containing 0.75% agar; cells were seeded at a density of 2500 cells per well in DMEM/F-12 containing 0.3% agar, 5%Horse serum for 15 days. Drugs were serially diluted with the medium and added into the wells at day 6. Colonies were stained with iodonitrotetrazolium chloride (INT, Sigma, St Louis, MO) overnight. The number and size of colonies were analyzed using GelCount (Oxford Optronix) according to the manufacturer’s instruction.
Antibodies
The following primary antibodies were used for immunoblotting: anti-phospho-HER2(Y877) (cat. no. 2241), anti-HER2(44E7) (cat. no. 2248), anti-phospho-EGFR(Tyr992) (cat. no. 2235), anti-EGFR (cat. no. 4267), anti-phospho-AKT (Ser473) (cat. no. 9271), anti-AKT (cat. no. 4691), anti-phospho-p70S6K(Tyr389) (cat. no. 9234), anti-p70S6K (cat. no. 9202), anti-phospho-MEK (S217/221) (cat. No. 4370), anti-MEK (61B12) (cat. no. 2352), anti-phospho-p42/44-MAPK (cat. No. 4370), anti-p42/44-MAPK (cat. No. 9101), were purchased from Cell Signaling Technology, and anti-β-actin was purchased from Sigma-Aldrich (cat. no. A5441).
Immunoblot analysis
All immunoblots shown in this paper represent at least two independent experiments. In brief, cells were washed with PBS and lysed in 4% SDS, and further sonicated using an XL2000 Microson Ultrasonic Processor (Misonix). Equal amounts of protein extract were separated on SDS-polyacrylamide gels and transferred to polyvinyl difluoride (PVDF) membranes. Membranes were further blocked with 5% milk or BSA and then probed with the following antibodies.
Statistical analyses
Statistical analysis of this study was performed using GraphPad PRISM7. All values are expressed as mean ± SEM (standard error of the mean) of at least three independent experiments.
Highlights
HER2-L755S, similar to HER2-del.16, confers a robust transformation in MCF10A cells
HER2-L755S is highly resistant to both reversible and irreversible HER2-targeting TKIs compared to wild type HER2 and HER2-del.16
HER2-L755S expressing cells exhibit hyperactivation of MAPK and PI3K/AKT/mTOR pathways
Combination of HER2 targeting TKIs with MEK or PI3K inhibitors induced synergistic growth inhibition in HER2-L755S mutant cells
Supplemental Material
Download MS Power Point (557.3 KB)Acknowledgments
This work is supported by the Graduate Scholarship of Jinan University and OFIRG grant to Q Yu. We thank Min Feng, Gokce Oguz and all the coauthors for their support and helpful suggestions towards our research work.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data can be accessed here.
Related Research Data
References
- Arteaga CL, Sliwkowski MX, Osborne CK, et al. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2011;9(1):16–32.
- Singh JC, Jhaveri K, Esteva FJ. HER2-positive advanced breast cancer: optimizing patient outcomes and opportunities for drug development. Br J Cancer. 2014;111(10):1888–1898.
- Paplomata E, O’Regan R. The PI3K/AKT/mTOR pathway in breast cancer: targets, trials and biomarkers. Ther Adv Med Oncol. 2014;6(4):154–166.
- Esteva FJ, Yu D, Hung M-C, et al. Molecular predictors of response to trastuzumab and lapatinib in breast cancer. Nat Rev Clin Oncol. 2010;7(2):98–107.
- Zuo W-J, Jiang Y-Z, Wang Y-J, et al. Dual characteristics of novel HER2 kinase domain mutations in response to HER2-targeted therapies in human breast cancer. Clin Cancer Res. 2016;22(19):4859–4869.
- Aung KL, Stockley TL, Serra S, et al. Testing ERBB2 p.L755S kinase domain mutation as a druggable target in a patient with advanced colorectal cancer. Cold Spring Harb Mol Case Stud. 2016;2(5):a001016.
- Cousin S, Khalifa E, Crombe A, et al. Targeting ERBB2 mutations in solid tumors: biological and clinical implications. J Hematol Oncol. 2018;11(1):86.
- Wen W, Chen WS, Xiao N, et al. Mutations in the kinase domain of the HER2/ERBB2 gene identified in a wide variety of human cancers. J Mol Diagn. 2015;17(5):487–495.
- Kancha RK, von Bubnoff N, Bartosch N, et al. Differential sensitivity of ERBB2 kinase domain mutations towards lapatinib. PLoS One. 2011;6(10):e26760.
- Zabransky DJ, Yankaskas CL, Cochran RL, et al. HER2 missense mutations have distinct effects on oncogenic signaling and migration. Proc Natl Acad Sci U S A. 2015;112(45):E6205–E14.
- Wang Y-C, Morrison G, Gillihan R, et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2- positive breast cancers-role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 2011;13(6):R121.
- Castagnoli L, Iezzi M, Ghedini GC, et al. Activated d16HER2 homodimers and SRC kinase mediate optimal efficacy for trastuzumab. Cancer Res. 2014;74(21):6248–6259.
- Marchini C, Gabrielli F, Iezzi M, et al. The human splice variant Δ16HER2 induces rapid tumor onset in a reporter transgenic mouse. PLoS One. 2011;6(4):e18727.
- Tiwari SR, Mishra P, Abraham J. Neratinib, a novel HER2-targeted tyrosine kinase inhibitor. Clin Breast Cancer. 2016;16(5):344–348.
- Singh H, Walker AJ, Amiri-Kordestani L, et al. U.S. food and drug administration approval: neratinib for the extended adjuvant treatment of early-stage HER2-positive breast cancer. Clin Cancer Res. 2018;24(15):3486–3491.
- Rabindran SK, Discafani CM, Rosfjord EC, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64:3958–3965.
- Hyman DM, Piha-Paul SA, Won H, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature. 2018;554(7691):189–194.
- Subik K, Lee JF, Baxter L, et al. The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast Cancer (Auckl). 2010;4:35-41.
- Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nat Rev Clin Oncol. 2014;11(7):385–400.
- Caunt CJ, Sale MJ, Smith PD, et al. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer. 2015;15(10):577–592.
- Yeh TC, Marsh V, Bernat BA, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13(5):1576–1583.
- O’Brien C, Wallin JJ, Sampath D, et al. Predictive biomarkers of sensitivity to the phosphatidylinositol 3’ kinase inhibitor GDC-0941 in breast cancer preclinical models. Clin Cancer Res. 2010;16(14):3670–3683.
- Turpin J, Ling C, Crosby EJ, et al. The ErbB2ΔEx16 splice variant is a major oncogenic driver in breast cancer that promotes a pro-metastatic tumor microenvironment. Oncogene. 2016;35(47):6053–6064.
- Mitra D, Brumlik MJ, Okamgba SU, et al. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther. 2009;8(8):2152–2162.
- Xu X, De Angelis C, Burke KA, et al. HER2 reactivation through acquisition of the HER2 L755S mutation as a mechanism of acquired resistance to HER2-targeted therapy in HER2+ breast cancer. Clin Cancer Res. 2017;23(17):5123–5134.
- Kavuri SM, Jain N, Galimi F, et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015;5(8):832–841.
- Bose R, Kavuri SM, Searleman AC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3(2):224–237.
- Nagano M, Kohsaka S, Ueno T, et al. High-throughput functional evaluation of variants of unknown significance in ERBB2. Clin Cancer Res. 2018;24(20):5112–5122.
- Ben-Baruch NE, Bose R, Kavuri SM, et al. HER2 mutated breast cancer responds to treatment with single agent neratinib, a second generation HER2/EGFR tyrosine kinase inhibitor. J Natl Compr Canc Netw. 2015;13(9):1061–1064.
- Wee ZN, Yatim SMJM, Kohlbauer VK, et al. IRAK1 is a therapeutic target that drives breast cancer metastasis and resistance to paclitaxel. Nat Commun. 2015;6:8746.