
ABSTRACT
Objective: To determine the underlying mechanism of miR-34b/c in regulating doxorubicin (Dox)-induced myocardial cell injury.
Methods: The viability of mouse myocardial cells HL-1 was detected by MTT assay. The apoptosis of HL-1 cells was detected by TUNEL assay. mRNA expressions of ITCH, TNF-α and IL-6 were measured by qRT-PCR. Protein levels of ITCH, NF-κB, TNF-α and IL-6 were measured by western blot. Dual luciferase assay was performed to detect the regulation of miR-34b/c on ITCH. Mouse model of cardiomyopathy was induced by intraperitoneal injection of Dox.
Results: Dox reduced HL-1 cell viability and activated NF-κB pathway in HL-1 cells. miR-34b/c expressions were gradually up-regulated and ITCH expression was gradually down-regulated in Dox-treated HL-1 cells. miR-34b/c expression had negative correlation with the mRNA expression of ITCH. Besides, ITCH was a target of miR-34b/c. miR-34b/c mimic reduced cell viability, suppressed ITCH expression, increased TNF-α and IL-6 level, and promoted NF-κB expression in nucleus and cytoplasm of HL-1 cells. Whereas silencing miR-34 protected HL-1 cells through regulating ITCH. Finally, we demonstrated miR-34 antagomir-protected myocardial cells in mouse model of cardiomyopathy.
Conclusion: miR-34b/c decreased HL-1 cell viability and promoted the secretion of proinflammatory cytokines in Dox-induced myocardial cells through ITCH/NF-κB pathway.
Introduction
Congestive heart failure is a clinical syndrome caused by inflammation, myocardial injury, myocardial stretch and matrix remodeling, and is the final stage in the development of most cardiovascular diseases, which becomes the leading cause of death and disability [Citation1]. Doxorubicin (Dox) is an effective broad-spectrum anti-cancer drug, which is used to treat leukemia and various solid tumors [Citation2,Citation3]. However, Dox can cause cumulative, dose-dependent and irreversible myocardial injury, which will progress to heart failure [Citation4]. Researchers have shown that the main performances of heart failure are myocardial cell inflammation and apoptosis, which may be the important mechanisms of cardiac remodeling and dysfunction [Citation5,Citation6]. Myocardial cell apoptosis has been considered to play a critical role in the progression of Dox-induced heart failure [Citation7,Citation8]. Excessive inflammatory response can lead to myocardial cell death and dysfunction which finally cause heart failure [Citation9]. Increasing cytokines and cytokine receptors indicate increased mortality and undesirable outcomes in patients with heart failure [Citation10]. However, the mechanisms mediating myocardial cell inflammation and apoptosis are not fully understood.
Nuclear factor (NF)-κB is a key transcription factor that controls inflammation in heart failure [Citation11]. Excessive activation of NF-κB pathway induces tumor necrosis factor α (TNF-α), interleukin-1 (IL-1) and interleukin-6 (IL-6), which leads to myocardial cell death [Citation11]. Researchers found that FGF21 down-regulated the secretion of inflammatory factors (TNF-α and IL-6) through inhibiting NF-κB pathway in Dox-induced cardiotoxicity models in H9c2 cells [Citation12]. ITCH is a homologous to the E6-AP carboxyl terminus (HECT)-type ubiquitin E3 ligase that plays important roles in cell apoptosis via ubiquitin proteasomal protein degradation [Citation13,Citation14]. ITCH can reduce the apoptosis of myocardial cells through interacting with thioredoxin-interacting protein (TXNIP) to protect myocardium [Citation15]. Moreover, it has been reported that ITCH could inhibit Dox-induced NF-κB activation [Citation16]. Therefore, ITCH might inhibit inflammatory response and myocardial cell apoptosis through negatively regulating NF-κB.
microRNAs (miRNAs) are a kind of non-coding RNAs (<22 nt) that can be involved in the regulation of myocardial cell apoptosis, microvessel angiogenesis, and metabolism of failing myocardium [Citation17]. miR-34 family is highly conserved miRNAs containing three members, namely miR-34a, miR-34b and miR-34c [Citation18]. It has been reported that inhibition of miR-34a could protect myocardial cells from hypoxic stress [Citation19]. miR-34a silencing could reduce Dox-induced damage to cardiac progenitor cells [Citation20]. miR-34a and miR-34b were found to be up-regulated in Dox-treated myocardial cells [Citation21]. Thus, miR-34 family may play a protective role in Dox-induced myocardial injury, but the role of miR-34b and miR-34c in Dox-induced myocardial injury is still not known. According to the online bioinformatics software (TargetScan and miRNADA), there were binding sites between miR-34b/c and ITCH 3ʹUTR. Therefore, we speculated that miR-34b/c regulates Dox-induced myocardial cell injury through ITCH/NF-κB pathway.
Materials and methods
Cell culture and transfection
Mouse myocardial cell line HL-1 was purchased from the American Type Culture Collection (ATCC, VA, USA), and cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, CA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, CA, USA) and 1% penicillin-streptomycin (Gibco, CA, USA) at 37℃ in a 5% CO2 incubator. For the observation of Doxorubicin (Dox; Sigma, MO, USA) on HL-1 cells, HL-1 cells were treated with 0, 0.01, 0.1, 1 or 10 μM Dox for 20 h. To observe the effect of NF-kB inhibitor on the changes of TNF-α and IL-6 levels, 5 μM Bay-11-7082 (Sigma) was added to HL-1 cells.
For silencing ITCH, small interference RNA targeting ITCH (si-ITCH) was synthesized by GENECHEM (Shanghai, China). HL-1 cells were seeded at 2.5 × 105 cells/well in a six-well plate and cultured until 50% confluence, and transfected with 60 nmol si-ITCH, miR-34b mimic, miR-34c mimic, miR-34b inhibitor, miR-34c inhibitor or empty vectors using Lipofectamine transfection reagent (Invitrogen, CA, USA) according to the manufacturer’s instructions.
Cell viability
HL-1 cell viability was detected by 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. HL-1 cells were seeded in a 96-well plate, then 10 μl MTT solution (5 mg/ml; Beyotime Biotechnology, Nantong, China) was added to each well and incubated at 37°C for 4 h. The formazan crystals were dissolved in dimethyl sulfoxide (DMSO; 100 μl/well) and added to each well. The absorbance was read at 570 nm by a microplate reader (Bio-Rad, CA, USA).
Quantitative real-time PCR
Total RNA was extracted from HL-1 cells using TRIzol Reagent (Invitrogen, CA, USA), and reversely transcribed into cDNA using High-Capacity RNA-to-cDNA Kit (Applied Biosystems, CA, USA). PCR was performed by the SuperScript III Platinum SYBR Green One-Step qRT-PCR Kit (Invitrogen, CA, USA). The mRNA expression of the target genes was quantified by qRT-PCR in a QuantStudio 3 Real-Time PCR System (Applied Biosystems, CA, USA). The relative expression of mRNAs and microRNA was normalized with GAPDH or U6. The sequences of primers were as follows: ITCH, F: 5ʹ-AGGATGGTAGCTGAGTGGAGGTTG-3ʹ, R: 5ʹ-TCTGGTGTAGTGGCGGTAGATGG-3ʹ; TNF-α, F: 5ʹ-GCGACGTGGAACTGGCAGAAG-3ʹ, R: 5ʹ-GCCACAAGCAGGAATGAGAAGAGG-3ʹ; IL-6, F: 5ʹ-AGGAGTGGCTAAGGACCAAGACC-3ʹ, R: 5ʹ-CTGACCACAGTGAGGAATGTCCAC-3ʹ; miR-34b, F: 5ʹ-CGAGGCAGTGTAATTAGCTGATTGT-3ʹ, R: 5ʹ-TGGTGTCGTGGAGTCG-3ʹ; miR-34c, F: 5ʹ-GCTGCTGTAGGCAGTGTAGTTAG-3ʹ, R: 5ʹ-CTCAACTGGTGTCGTGGAGTC-3ʹ; GAPDH, F: 5ʹ-GCATCTTCTTGTGCAGTGCC-3ʹ, R: 5ʹ-GAGAAGGCAGCCCTGGTAAC-3ʹ; U6, F: 5ʹ-CTCGCTTCGGCAGCACA-3ʹ, R: 5ʹ-AACGCTTCACGAATTTGCGT-3ʹ.
TUNEL assay
HL-1 cells were collected to perform TdT-mediated dUTP nick-end labeling (TUNEL) staining assay. Cells were fixed in 4% paraformaldehyde, and stained with nucleo-tide Mix and rTdT enzyme (DeadEnd Colorimetric TUNEL system, Promega) to label the fragmented DNA of apoptotic cells for 60 min at 37°C. The percent of apoptotic cells (TUNEL positive) was calculated by flow cytometry and expressed as the ratio of TUNEL-positive cells/all cells.
Western blot
HL-1 cells were digested with pancreatin (Sigma, Mo, USA), and nucleus and cytoplasm extracts were prepared using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, CA, USA). The concentration of protein was measured by the Pierce BCA Protein Assay kit (Thermo Scientific, CA, USA). Total protein was separated on 12% sodium dodecyl sulfate -polyacrylamide gel electrophoresis (SDS-PAGE), and electro-transferred to polyvinylidene difluoride membranes (Invitrogen, CA, USA). The membranes were probed with primary antibodies against ITCH (1:1000; Abcam, Mass, USA), NF-κB (1:1000; Abcam, Mass, USA), p-NF-κB (1:1000; Abcam, Mass, USA), TNF-α (1:500; Abcam, Mass, USA), IL-6 (1:1000; Abcam, Mass, USA), c-caspase 3 (1:1000; Abcam, Mass, USA), and GAPDH (1:2000; Abcam, Mass, USA) and PCNA (1:5000; Abcam, Mass, USA) were served as a loading control. Then, the blots were incubated with the HRP-conjugated secondary antibody (1:5000; Abcam, Mass, USA) for 2 h. The bands were visualized with the Novex ECL Chemiluminescent Substrate Reagent Kit (Invitrogen, CA, USA), and images were obtained by Gel Imaging Systems (Bio-Rad, CA, USA).
ELISA
HL-1 cells treated with 0.05 μM Dox were centrifuged and washed with PBS for twice, then resuspended in DMEM supplemented with 10% FBS. Cells (2.5 × 105 cells/well) were seeded in a 24-well plate for 24 h. Then, TNF alpha Mouse ELISA Kit (Invitrogen, CA, USA) and IL-6 Mouse ELISA Kit were used to measure the expression of TNF-α and IL-6 in the supernatant of HL-1 cells. The absorbance of each well was measured at 450 nm on a Microplate Reader (Bio-Rad, CA, USA).
Dual luciferase assay
The sequence of ITCH or the mutated sequence was predicted to bind with miR-34b and miR-34c. These sequences were inserted into pmirGLO vector (Promega, MI, USA) to construct recombined luciferase report vectors with ITCH, which were noted as pmirGLO-ITCH or pmirGLO-mut-ITCH. For Luciferase reporter assay, the reporter plasmid (pmirGLO-ITCH or pmirGLO-mut-ITCH, 360 ng) and microRNAs (miR-34b/c mimic or negative control, 720 ng) were transfected into HL-1 cells for 48 h using Lipofectamine 2000 (Invitrogen, CA, USA). Cells were collected, and the luciferase activity was measured by the Dual Glo Luciferase Assay System (Promega, MI, USA).
In vivo experiments
Male BALB/c mice (7–8 weeks old, weighing 25–28 g) were purchased from the laboratory animal center of Zhengzhou University, and maintained under controlled environmental conditions with a 12/12 h light/dark cycle. The animal experiments were approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University. Mice were randomly divided into four groups: control group (n = 5), received normal feeding; Dox group (n = 6), received an intraperitoneal injection of 5 mg/kg Dox every week for 8 weeks to induce mouse model of cardiomyopathy, and heart tissue was obtained at the 10th week; Dox+miR-34b antagomir group (n = 6), received an intraperitoneal injection of 5 mg/kg Dox every week for 8 weeks, miR-34b antagomir (5 nmol/g) was injected 4 days before Dox injection and booster injection were conducted every 2 weeks for 8 weeks, and heart tissue was obtained at the 10th week; Dox+miR-34c antagomir group (n = 6), received an intraperitoneal injection of 5 mg/kg Dox every week for 8 weeks, miR-34c antagomir (5 nmol/g) was injected 4 days before Dox injection and booster injection were conducted every 2 weeks for 8 weeks, and heart tissue was obtained at the 10th week.
Histopathological examination
Heart tissues were fixed in 10% neutral-buffered formalin for 48 h, dehydrated in ethanol, embedded in paraffin, and cut into slices (5 μm). Thick sections (5 μm) were prepared and stained with hematoxylin and eosin (H&E) and Masson, and observed under a light microscope (Olympus, Tokyo, Japan). Col I (1:50; Abcam, Mass, USA), col III (1:100; Abcam, Mass, USA), TNF-α (1:50; Abcam, Mass, USA) and IL-6 (1:100; Abcam, Mass, USA) were stained according to the manufacturer’s instructions.
Statistical analysis
All data were presented as mean±SD, and analyzed using SPSS 18.0 (Chicago, USA). Statistical significance among groups was determined by one-way ANOVA or Student’s t-test. P values of <0.05 were considered significant.
Results
miR-34b/c and ITCH are required for Dox-induced myocardial cell injury
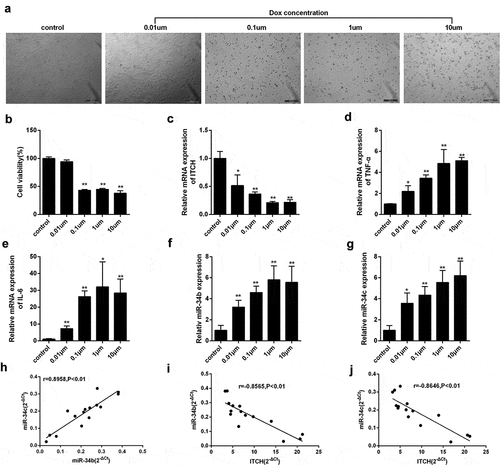
To determine the effect of Dox at different concentrations on myocardial cell injury, we selected 0, 0.01, 0.1, 1 and 10 μM Dox for the treatment of HL-1 cells. We found that HL-1 cell viability was gradually reduced after the treatment of Dox at the indicated concentrations (,)). Besides, mRNA expression of ITCH was gradually down-regulated after the treatment of Dox at the indicated concentrations ()). mRNA expressions of proinflammatory cytokine TNF-α and IL-6 were gradually up-regulated after the treatment of Dox at the indicated concentrations, indicating myocardial cells were injured (,)). In addition, miR-34b and miR-34c expressions were gradually up-regulated after the treatment of Dox (,)). Moreover, we found miR-34b expression had a positive correlation with the expression of miR-34c ()), and miR-34b/c expression had a negative correlation with the mRNA expression of ITCH (,)).
Figure 1. miR-34b/c and ITCH are required for Dox-induced myocardial injury. (a-b). HL-1 cells treated by Dox at the indicated concentrations (0, 0.01, 0.1, 1 and 10 μM) were observed under an inverted microscope, and HL-1 cell viability was gradually reduced after the treatment of Dox at the indicated concentrations. (c). mRNA expression of ITCH was gradually down-regulated after the treatment of Dox at the indicated concentrations. (d-e). mRNA expressions of TNF-α and IL-6 were gradually up-regulated after the treatment of Dox at the indicated concentrations. (f-g). miR-34b and miR-34c expressions were gradually up-regulated after the treatment of Dox at the indicated concentrations. (h). miR-34b expression had positive correlation with the expression of miR-34c. (i). miR-34b expression had negative correlation with the mRNA expression of ITCH. (j). miR-34c expression had negative correlation with the mRNA expression of ITCH. *P < 0.05 vs control; **P < 0.01 vs control
Doxorubicin activates Nf-κB pathway in HL-1 cells
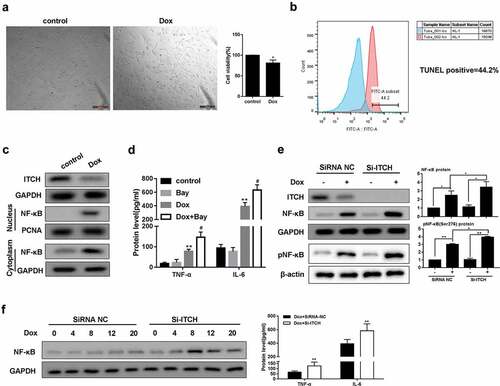
It has been reported that FGF21 could remarkably inhibited TNF-α and IL-6 levels through suppressing NF-κB p65 in Dox-treated myocardial cells, indicating that NF-κB played a key role in cardiac inflammation [Citation12]. After the treatment of Dox, HL-1 cell viability was significantly reduced ()), and TUNEL-positive rate was found to be 44.2% ()). In addition, protein level of ITCH was down-regulated in HL-1 cells treated by Dox, whereas protein level of NF-κB was up-regulated in nucleus and cytoplasm of HL-1 cells treated by Dox ()). TNF-α and IL-6 levels were higher in the supernatant of HL-1 cells treated by Dox, and NF-κB inhibitor Bay 11–7082 further increased TNF-α and IL-6 levels ()). Importantly, silencing ITCH in HL-1 cells further promoted Dox-induced total NF-κB and pNF-κB protein levels ()). Under the treatment of Dox for 0, 4, 8, 12 and 20 h in HL-1 cells, silencing ITCH promoted protein levels of total NF-κB, TNF-α and IL-6 ()). These findings indicated that ITCH could inhibit NF-κB pathway in Dox-treated HL-1 cells.
Figure 2. Doxorubicin activates NF-κB pathway in HL-1 cells. HL-1 cells were treated by Dox (0.05 μM). (a). MTT assay showed that HL-1 cell viability was reduced after the treatment of Dox. (b). TUNEL staining showed that TUNEL positive rate was 44.2%. (c). Protein level of ITCH was reduced in HL-1 cells treated by Dox, whereas protein level of NF-κB was increased in nucleus or cytoplasm of HL-1 cells treated by Dox. (d). Under the treatment of Dox, BAY 11–7082 (5 μM) was added to HL-1 cells and cultured for 20 h. TNF-α and IL-6 levels increased in the supernatant of HL-1 cells. (e). Silencing ITCH in HL-1 cells promoted Dox-induced total NF-κB and pNF-κB protein levels. (f). HL-1 cells were treated by Dox for 0, 4, 8, 12 and 20 h. Silencing ITCH in HL-1 cells promoted protein levels of NF-κB, TNF-α and IL-6. **P < 0.01 vs control
The regulation of miR-34b/c on ITCH
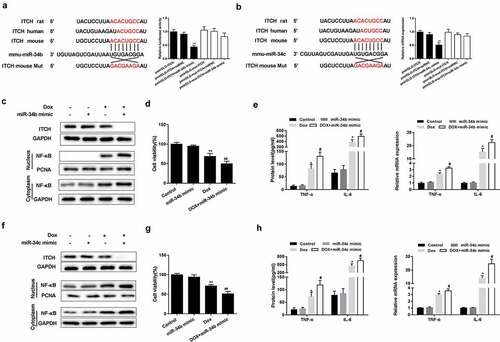
Bioinformatics software miRNADA and TargetScan predicted the binding sites between miR-34b/c and ITCH 3ʹUTR (,)). According to dual-luciferase reporter assay, miR-34b/c mimic significantly decreased the luciferase activity of pmirGLO-ITCH, whereas miR-34b/c mimic did not significantly change the luciferase activity of pmirGLO-mut-ITCH (,)). Under the treatment of Dox, miR-34b/c mimic suppressed protein level of ITCH, and promoted protein level of NF-κB in nucleus and cytoplasm of HL-1 cells (,)). miR-34b/c mimic further decreased Dox-treated HL-1 cell viability (,)). miR-34b/c mimic increased protein and mRNA expression of TNF-α and IL-6 in Dox-induced HL-1 cells (,)).
Figure 3. The regulation of miR-34b/c on ITCH. (a-b). There were binding sites between miR-34b/c and ITCH. Dual-luciferase reporter assay showed that miR-34b/c mimic significantly decreased the luciferase activity of pmirGLO-ITCH, whereas miR-34b/c mimic did not significantly change the luciferase activity of pmirGLO-mut-ITCH. **P < 0.01 vs pmirGLO-ITCH+miRNC. (c-h). Under the treatment of Dox, miR-34b/c mimic reduced protein level of ITCH, promoted nucleus and cytoplasm protein level of NF-κB, increased protein and mRNA expression of TNF-α and IL-6, and decreased HL-1 cell viability. **P < 0.01 vs Dox; ##P < 0.01 vs miR-34b/c mimic
Silencing miR-34 protected HL-1 cells through regulating ITCH
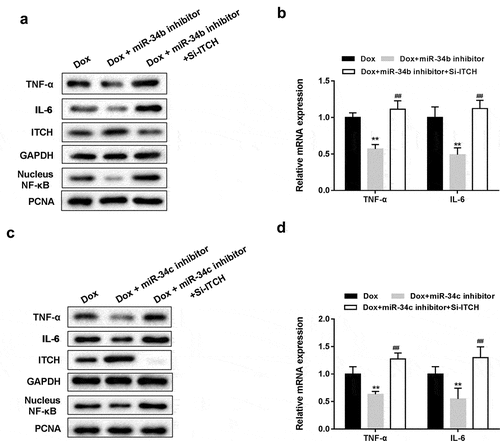
As shown in ,), miR-34b/c inhibitor reversed the Dox-induced decrease of ITCH protein level, promotion of NF-κB pathway and increase of TNF-α and IL-6 protein levels in HL-1 cells, whereas miR-34b/c inhibitor+si-ITCH could not reverse the effects induced by Dox. miR-34b/c inhibitor reversed the Dox-induced increase of mRNA expression of TNF-α and IL-6 in HL-1 cells, whereas miR-34b/c inhibitor+si-ITCH could not reverse the effects induced by Dox (,)). These findings indicated that silencing miR-34b/c inhibited proinflammatory cytokines and NF-κB pathway through ITCH.
Figure 4. Silencing miR-34 protected HL-1 cells through regulating ITCH. (a). Under the treatment of Dox, miR-34b inhibitor reversed the Dox-induced decrease of ITCH protein level and promotion of NF-κB pathway, whereas si-ITCH could not reverse the effects induced by Dox. (b). miR-34b inhibitor reversed the Dox-induced increase of mRNA expression of TNF-α and IL-6, whereas si-ITCH could not reverse the effects induced by Dox. (c). miR-34c inhibitor reversed the Dox-induced decrease of ITCH protein level and promotion of NF-κB pathway, whereas si-ITCH could not reverse the effects induced by Dox. (d). miR-34c inhibitor reversed the Dox-induced increase of mRNA expression of TNF-α and IL-6, whereas si-ITCH could not reverse the effects induced by Dox. **P < 0.01 vs Dox; ##P < 0.01 vs Dox+miR-34b/c inhibitor
miR-34 antagomir-protected myocardial cells in mouse model of cardiomyopathy
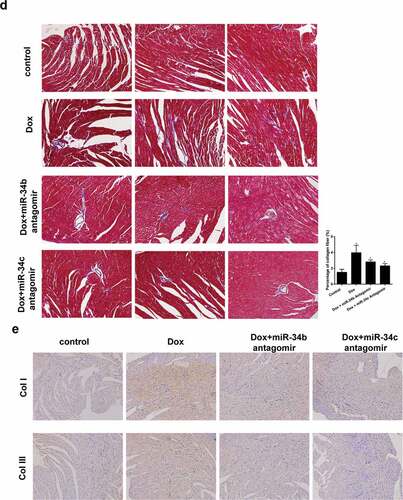
To investigate the role of miR-34b/c in cardiomyopathy, Dox-induced cardiomyopathy mice model was established, and miR-34b antagomir or miR-34c antagomir was intraperitoneally injected into mice. We found that protein levels and mRNA expressions of TNF-α and IL-6, protein levels of c-caspase3 and nucleus NF-κB were higher in heart tissue of Dox group than control group, and ITCH protein level was lower in heart tissue of Dox group ()). miR-34b/c antagomir treatment could reverse the Dox-induced effects ()). Immunohistochemistry results also proved that TNF-α and IL-6 levels were higher in heart tissue of Dox group than control group, miR-34b/c antagomir treatment decreased TNF-α and IL-6 levels in heart tissue ()). Besides, myofibrillar degeneration and disruption were observed in the Dox group by HE staining. More normal morphology and myofibrosis were observed in Dox+miR-34b/c antagomir groups than Dox group, and myofibrillar degeneration and disruption were relieved in Dox+miR-34b/c antagomir groups than Dox group ()). Finally, MASSON staining showed that myofibrosis was induced in the Dox group, and miR-34b/c antagomir treatment relieved myofibrosis ()). Immunohistochemistry for collagen type 1 and 3 further strengthened the results of MASSON staining ()).
Figure 5. miR-34 Antagomir protected myocardial cells in mouse model of cardiomyopathy. Mice were divided into control (n = 5), Dox-induced cardiomyopathy group (n = 6), Dox+miR-34b antagomir group (n = 6) and Dox+miR-34c antagomir group (n = 6). (a). In Dox group, TNF-α, IL-6, c-caspase3 and NF-κB protein levels were higher in heart tissue, and ITCH protein level was lower in heart tissue. miR-34b/c antagomir treatment could reverse these effects. (b). Immunohistochemistry showed that TNF-α and IL-6 levels were higher in heart tissue, miR-34b/c antagomir treatment decreased TNF-α and IL-6 levels. (c). HE staining showed that in Dox group, myofibrillar degeneration and disruption was observed. More normal morphology and myofibrosis were observed in Dox+miR-34b/c antagomir groups than Dox group, and myofibrillar degeneration and disruption were relieved in Dox+miR-34b/c antagomir groups than Dox group. (d). MASSON staining showed that myofibrosis was induced in the Dox group, and miR-34b/c antagomir treatment relieved myofibrosis. E. Collagen type 1 (col I) and collagen type 3 (col III) expressions were detected by Immunohistochemistry. *P < 0.05 vs control
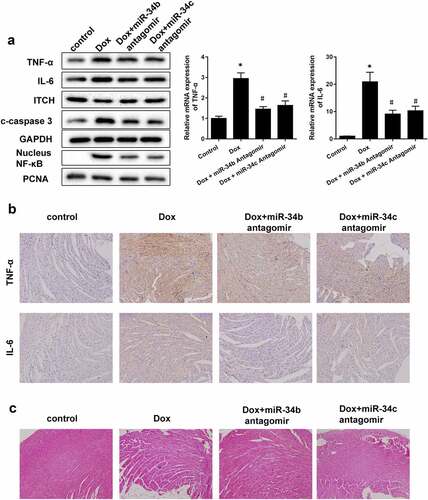
Figure 5. (continued)
Discussion
Dox is the most commonly used anti-cancer drug which belongs to anthracycline antibiotics [Citation22]. It can induce life-threatening cardiomyopathy and heart failure through the formation of reactive oxygen species (ROS), the damage to nuclear DNA, and the change of cellular contractility [Citation22]. So, revealing the underlying mechanism of Dox-induced heart failure is important for reducing the heart failure in clinical practice. Fortunately, clinicians and researchers have recognized the importance of basic research for reducing Dox-induced heart failure and obtained much advance. For example, loss of G protein inactivator RGS6 can reduce Dox-induced apoptosis of ventricular myocytes through modulating ROS generation to prevent heart failure [Citation23]. Bnip3 is involved in Dox-induced mitochondrial injury of myocardial cells, and knockdown of Bnip3 suppressed Dox-induced mitochondrial perturbations and ROS production [Citation24]. VEGF-B can protect Dox-induced cardiac endothelial cell damage and apoptosis, increased pERK1/2 signaling to promote angiogenesis, and reduced DNA damage, indicating VEGF-B gene therapy could suppress Dox-induced cardiotoxicity [Citation25].
Recent studies have shown that miRNAs play critical roles in the modulation of heart failure through different signaling pathways. miR-195 was up-regulated in failing myocardium of patients with heart failure which impaired energy metabolism through targeting mitochondrial deacetylase sirtuin 3 [Citation26]. miR-30 was down-regulated in Dox-treated myocardial cells and high miR-30 level could protect myocardial cells from Dox-induced apoptosis through regulating the β-adrenergic pathway and targeting BNIP3L/NIX [Citation27]. In failing human myocardium, ubiquitin proteasome system (UPS) activity was regulated by TWIST1/miR-199/214 cluster [Citation28]. In this study, we focused on the underlying mechanism mediated by miR-34b/c in myocardial cell apoptosis and inflammatory response. Previous reports have shown that miR-34 family acts as a tumor suppressor or tumor promoter in breast cancer cells, prostate cancer cells, esophageal squamous cell carcinoma cells, etc [Citation29–Citation32]. In addition, miR-34 family can be highly expressed in neuron to regulate neuronal differentiation [Citation33]. miR-34a can inhibit the neuroprotective effect of Schisandrin B on 6-OHDA-treated neuroblastoma SH-SY5Y cells [Citation34]. The role of miR-34a in myocardial cells under hypoxic stress has been proved [Citation19]; however, the role of miR-34b/c in Dox-induced heart failure is still not known. In the present study, we found miR-34b/c were up-regulated in Dox-induced myocardial cells, and miR-34b/c overexpression reduced myocardial cell viability and promoted the secretion of proinflammatory cytokines in myocardial cells.
ITCH is an E3 ligase that can be abnormally expressed in lung cancers, gastric cancer, pancreatic cancer to modulate the invasion and migration of these cancers [Citation13,Citation35,Citation36]. ITCH is also involved in the regulation of T helper cell differentiation and Treg differentiation [Citation37]. Studies have reported that miRNAs could target to the 3ʹ-UTR of ITCH mRNA thus to involve in the proliferation of human hepatocellular carcinoma cells or cardiac injury [Citation38,Citation39]. In this study, we found that ITCH mRNA and protein level were down-regulated in Dox-induced myocardial cells, and ITCH knockdown promoted the expression of NF-κB and the secretion of TNF-α and IL-6 in Dox-induced myocardial cells. Moreover, we proved ITCH was a target of miR-34b/c, and ITCH expression was negatively regulated by miR-34b/c. miR-34b/c knockdown protected myocardial cells from Dox through regulating ITCH. As far as we know, there were no other reports determined the regulation role of miR-34b/c in ITCH in Dox-induced myocardial cells and mouse model of cardiomyopathy, which will enrich the literature and provide target molecules for the treatment of Dox-induced heart failure. In this study, we mainly focused on Dox-induced myocardial injury mice, and conducted a series of in vitro experiments in HL-1 cells, as they may not fully represent human primary myocardial cells.
In conclusion, miR-34b and miR-34c were up-regulated in Dox-induced myocardial cells, and miR-34b/c decreased myocardial cell viability and promoted the secretion of proinflammatory cytokines in Dox-induced myocardial cells through ITCH/NF-κB pathway.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Ibrahim NE, Januzzi JL Jr. Established and emerging roles of biomarkers in heart failure. Circ Res. 2018;123(5):614–629.
- Uchakina ON, Ban H, Hostetler BJ, et al. Inhibition of hyaluronic acid formation sensitizes chronic myelogenous leukemia to treatment with doxorubicin. Glycobiology. 2016;26(11):1171–1179.
- Zheng S, Wang X, Weng Y-H, et al. siRNA knockdown of RRM2 effectively suppressed pancreatic tumor growth alone or synergistically with doxorubicin. Mol Ther Nucleic Acids. 2018;12:805–816.
- Wang Y, Cui X, Wang Y, et al. Protective effect of miR378* on doxorubicin-induced cardiomyocyte injury via calumenin. J Cell Physiol. 2018;233(10):6344–6351.
- Singh P, Sharma R, McElhanon K, et al. Sulforaphane protects the heart from doxorubicin-induced toxicity. Free Radic Biol Med. 2015;86:90–101.
- Ojha S, Al Taee H, Goyal S, et al. Cardioprotective potentials of plant-derived small molecules against doxorubicin associated cardiotoxicity. Oxid Med Cell Longev. 2016;2016:5724973.
- Wang Y, Lei T, Yuan J, et al. GCN2 deficiency ameliorates doxorubicin-induced cardiotoxicity by decreasing cardiomyocyte apoptosis and myocardial oxidative stress. Redox Biol. 2018;17:25–34.
- Kumar D, Kirshenbaum LA, Li T, et al. Apoptosis in adriamycin cardiomyopathy and its modulation by probucol. Antioxid Redox Signal. 2001;3(1):135–145.
- Zhang QL, Yang -J-J, Zhang H-S. Carvedilol (CAR) combined with carnosic acid (CAA) attenuates doxorubicin-induced cardiotoxicity by suppressing excessive oxidative stress, inflammation, apoptosis and autophagy. Biomed Pharmacothe. 2018;109:71–83.
- Deswal A, Petersen NJ, Feldman AM, et al. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the vesnarinone trial (VEST). Circulation. 2001;103(16):2055–2059.
- Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-κB in the heart: to be or not to NF-κB. Circ Res. 2011;108(9):1122–1132.
- Wang S, Wang Y, Zhang Z, et al. Cardioprotective effects of fibroblast growth factor 21 against doxorubicin-induced toxicity via the SIRT1/LKB1/AMPK pathway. Cell Death Dis. 2017;8(8):e3018.
- Li P-F, Zhang Q-G. Inhibition of ITCH suppresses proliferation and induces apoptosis of lung cancer cells. Cell Physiol Biochem. 2018;48(4):1703–1709.
- Ishihara T, Inoue J, Kozaki K-I, et al. HECT-type ubiquitin ligase ITCH targets lysosomal-associated protein multispanning transmembrane 5 (LAPTM5) and prevents LAPTM5-mediated cell death. J Biol Chem. 2011;286(51):44086–44094.
- Otaki Y, Takahashi H, Watanabe T, et al. HECT-type ubiquitin E3 ligase ITCH interacts with thioredoxin-interacting protein and ameliorates reactive oxygen species-induced cardiotoxicity. J Am Heart Assoc. 2016;5(1).e002485
- Liang L, Fan Y, Cheng J, et al. TAK1 ubiquitination regulates doxorubicin-induced NF-κB activation. Cell Signal. 2013;25(1):247–254.
- Fan J, Li H, Nie X, et al. MiR-665 aggravates heart failure via suppressing CD34-mediated coronary microvessel angiogenesis. Aging (Albany NY). 2018;10(9):2459–2479.
- Zhou Z-B, Niu Y-L, Huang G-X, et al. Silencing of circRNA.2837 plays a protective role in sciatic nerve injury by sponging the miR-34 family via regulating neuronal autophagy. Mol Ther Nucleic Acids. 2018;12:718–729.
- Zhang Y, Liu G, Gao X. Attenuation of miR-34a protects cardiomyocytes against hypoxic stress through maintenance of glycolysis. Biosci Rep. 2017;37(6):BSR20170925.
- Piegari E, Russo R, Cappetta D, et al. MicroRNA-34a regulates doxorubicin-induced cardiotoxicity in rat. Oncotarget. 2016;7(38):62312–62326.
- Holmgren G, Synnergren J, Andersson CX, et al. MicroRNAs as potential biomarkers for doxorubicin-induced cardiotoxicity. Toxicol In Vitro. 2016;34:26–34.
- Gianni L, Herman EH, Lipshultz SE, et al. Anthracycline cardiotoxicity: from bench to bedside. J Clin Oncol. 2008;26(22):3777–3784.
- Yang J, Maity B, Huang J, et al. G-protein inactivator RGS6 mediates myocardial cell apoptosis and cardiomyopathy caused by doxorubicin. Cancer Res. 2013;73(6):1662–1667.
- Dhingra R, Margulets V, Chowdhury SR, et al. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc Natl Acad Sci U S A. 2014;111(51):E5537–44.
- Räsänen M, Degerman J, Nissinen TA, et al. VEGF-B gene therapy inhibits doxorubicin-induced cardiotoxicity by endothelial protection. Proc Natl Acad Sci U S A. 2016;113(46):13144–13149.
- Zhang X, Ji R, Liao X, et al. MicroRNA-195 regulates metabolism in failing myocardium via alterations in sirtuin 3 expression and mitochondrial protein acetylation. Circulation. 2018;137(19):2052–2067.
- Roca-Alonso L, Castellano L, Mills A, et al. Myocardial miR-30 downregulation triggered by doxorubicin drives alterations in β-adrenergic signaling and enhances apoptosis. Cell Death Dis. 2015;6:e1754.
- Baumgarten A, Bang C, Tschirner A, et al. TWIST1 regulates the activity of ubiquitin proteasome system via the miR-199/214 cluster in human end-stage dilated cardiomyopathy. Int J Cardiol. 2013;168(2):1447–1452.
- Zhang L. MiR-34b/c-5p and the neurokinin-1 receptor regulate breast cancer cell proliferation and apoptosis. Cell Prolif. 2019;52(1): e12527.
- Bonetti P. Dual role for miR-34a in the control of early progenitor proliferation and commitment in the mammary gland and in breast cancer. Oncogene. 2019;38(3):360–374.
- Fang -L-L, Sun B-F, Huang L-R, et al. Potent inhibition of miR-34b on migration and invasion in metastatic prostate cancer cells by regulating the TGF-β pathway. Int J Mol Sci. 2017;18(12):2762.
- Yang L, Song X, Zhu J, et al. Tumor suppressor microRNA-34a inhibits cell migration and invasion by targeting MMP-2/MMP-9/FNDC3B in esophageal squamous cell carcinoma. Int J Oncol. 2017;51(1):378–388.
- Jauhari A, Singh T, Singh P, et al. Regulation of miR-34 family in neuronal development. Mol Neurobiol. 2018;55(2):936–945.
- Ba Q, Cui C, Wen L, et al. Schisandrin B shows neuroprotective effect in 6-OHDA-induced parkinson’s disease via inhibiting the negative modulation of miR-34a on Nrf2 pathway. Biomed Pharmacother. 2015;75:165–172.
- Gen Y, Yasui K, Kitaichi T, et al. ASPP2 suppresses invasion and TGF-β1-induced epithelial-mesenchymal transition by inhibiting Smad7 degradation mediated by E3 ubiquitin ligase ITCH in gastric cancer. Cancer Lett. 2017;398:52–61.
- Luo Z-L, Luo H-J, Fang C, et al. Negative correlation of ITCH E3 ubiquitin ligase and miRNA-106b dictates metastatic progression in pancreatic cancer. Oncotarget. 2016;7(2):1477–1485.
- Jin H-S, Park Y, Elly C, et al. Itch expression by Treg cells controls Th2 inflammatory responses. J Clin Invest. 2013;123(11):4923–4934.
- Chen Z-G, Liu H, Zhang J-B, et al. Upregulated microRNA-214 enhances cardiac injury by targeting ITCH during coxsackievirus infection. Mol Med Rep. 2015;12(1):1258–1264.
- Xia K, Zhang Y, Cao S, et al. miR-411 regulated ITCH expression and promoted cell proliferation in human hepatocellular carcinoma cells. Biomed Pharmacother. 2015;70:158–163.