
ABSTRACT
Protein kinase CK2 alpha (CK2α) is involved in the development of multiple malignancies. Overexpression of Y-box binding protein 1 (YBX1) is related to tumor proliferation, drug resistance, and poor prognosis. Studies have demonstrated that both CK2 and YBX1 could regulate the PI3K/AKT pathway. In addition, we predicted that CK2 might be the upstream kinase of YBX1 through the Human Protein Reference Database (HPRD). Herein, we hypothesize that CK2 may interact with YBX1 and they regulate the PI3K/AKT signaling pathway together. Expressions of CK2α and YBX1 in cancer cell lines were evaluated by immunoblotting. The results showed that CK2α could regulate the expression of YBX1 at the transcriptional level, which is dependent on its enzymatic activity. Synergistic effects of PI3K/AKT pathway inactivation could be observed through combined inhibition of CK2α and YBX1, and YBX1 was required for CK2α-induced PI3K/AKT pathway activation. Further results demonstrated that CK2α could interact with YBX1 and PI3K/AKT antagonist decreased cell resistance to doxorubicin induced by co-activation of CK2α and YBX1. These results indicated that combined inhibition of CK2α and YBX1 showed synergistic effects in inactivating the PI3K/AKT signaling pathway and may be one of the mechanisms involved in tumor growth and migration.
Introduction
CK2, a multifunctional serine/threonine kinase, plays important roles in cancer [Citation1]. CK2 is known to be transcribed from two genes CK2α and CK2α’, and CK2α is known to have higher and more widely distributed expression [Citation2]. CK2 can function as a monomeric kinase, or as a tetrameric complex composed of two CK2α kinase subunits and two CK2β regulatory subunits, and the CK2β subunits determine substrate specificity [Citation3]. Abnormal activation of CK2 occurs in many tumors, such as breast, prostate, lung, leukemia, brain tumors, and multiple myeloma [Citation4–Citation7]. Abnormal CK2 kinase activity leads to the change of cell life processes through the regulation of the JAK/STAT, NF-κB, PI3K/AKT, Wnt and Hedgehog signaling pathways [Citation8–Citation12]. Studies have shown that CK2 is abnormally activated in mice and plays functions as an oncogene in the development and progression of tumors [Citation13,Citation14]. CK2 inhibits apoptosis of cells by decreasing the expression of tumor suppressors p21, PTEN and p53 [Citation15]. CK2 can regulate cell survival by participating in the damage and repair of DNA. Recently, large-scale protein phosphorylation showed that radioactive analog drugs or ionizing radiation treatment of melanoma cells (G361) and lymphocyte (GM00130B) induced the collapse of DNA double strands. Many related proteins can be phosphorylated in the specific position by CK2 [Citation16]. Immunohistochemical analysis showed that CK2 exerts functions as a carcinogen in HCC and might be a major indicator for the treatment and prognosis of HCC. Functional analysis indicated that knockdown of CK2 inhibited cell proliferation, migration, invasion and promoted apoptosis by decreasing the expression of p-AKT (ser473) and Bcl-2 and upregulating p-p53, p53, Bax, and Caspase3 in HCC [Citation17].
CX4945, a specific inhibitor of CK2, can regulate the survival and angiogenesis of tumor cells. CX4945 combined with other chemotherapeutic drugs can reduce the occurrence of drug toxicity and resistance, and effectively improve the therapeutic effect of tumors. CK2 inhibitors are good candidates for clinical combinational therapy in cancer because of their important roles in regulating multiple cell pathways [Citation18]. The antitumor ability of CX4945 is dependent on the expression level of CK2α, the mutant states of CK2α catalytic subunit and the regulation subunit CK2β, PI3K/AKT, and PTEN. Treatment of cancer cells with CX4945 increased the expression of p21 and p27, and the activity of caspase 3/7 by decreasing phosphorylated p21 (T145) [Citation19].
YBX1, known as DNA binding protein, contains highly conserved cold stress domains (CSD); thus, it is classified as one of the members of the CSD family. The CSD domain of YBX1 could bind specifically to DNA/RNA to regulate gene transcription, replication and shear mRNA precursor [Citation20]. Overexpression of YBX1 is closely related to the progression and prognosis of tumors, including stromal tumors, pancreatic cancer, colon cancer, and other tumor tissues [Citation21,Citation22]. Approximately 40% of the breast cancers exhibit increased YBX1 transcription and translation levels and are closely related to patient survival rates [Citation23]. Overexpression of YBX1 in the breast cells of mice causes breast cancer, suggesting that YBX1 may function as a carcinogen in breast carcinogenesis [Citation24]. YBX1 (ser102) can be directly activated by AKT [Citation25] and ribosomal S6 kinase (RSK) and therefore plays an important role in the PI3K/AKT and MAPK signaling pathways [Citation26]. Studies have shown that YBX1 can influence the viability of cells by regulating the expression of multiple genes, such as CD44 [Citation27], PI3KCA [Citation28], and MET [Citation29]. Previous studies have shown that YBX1 increased drug resistance in cancer cells by inducing the expression of MDR-1 [Citation30,Citation31]. Consistent with this finding, the deregulation of YBX1 augmented the susceptibility of breast carcinoma cells to paclitaxel [Citation32]. YBX1 is highly expressed in colon cancer and is positively associated with the expression of DNA topoisomerase IIα independent of the multidrug resistance gene (MDR1) expression [Citation33]. In the treatment of colon cancer with the chemotherapeutic drug vinblastine, YBX1 can enhance drug resistance via accumulation in the nucleus [Citation34]. YBX1 can promote tumor development, cell proliferation, angiogenesis, migration, and invasion, which are involved in Hanahan and Weiberg effects [Citation35,Citation36]. In addition, evidences indicate that YBX1 knockdown by siRNA could reduce the tumorigenic ability in HER2 positive breast cancer cells, suggesting that YBX1 exerts an oncogenic function in breast cancer [Citation32]. Therefore, YBX1 is thought to be an oncogene. PI3K/AKT promotes the growth of breast cancer cells by phosphorylating YBX1 at serine102, leading to the entrance to the nucleus of YBX1 [Citation37]. YBX1 regulates breast cancer invasion by combining with the PIK3CA (PI3K catalytic subunit) promoter. Knockdown of YBX1 can reduce the expression of p-S6RP (ser235/236), p-RSK (ser380), p-AKT (ser473), P110 alpha and uPA [Citation28]. Immunohistochemical analysis of three colorectal cancer patients with clinical samples showed that YBX1 gathered in the nucleus, and the YBX1 expression level in the nucleus of colorectal cancer (CRC) is a useful prognostic biomarker that is related to the EGFR status [Citation38]. YBX1 combines to the CD44 and CD49f promoters to regulate self-renewal and microsphere formation in breast cancer. Paclitaxel induces the phosphorylation of YBX1 (ser102) and causes the accumulation of CD44 and YBX1 in the nucleus. Overexpression of wild-type YBX1 can enhance the resistance of breast cancer to Paclitaxel, while down-regulation of YBX1 increased the sensitivity of breast cancer to Paclitaxel [Citation27].
We hypothesized that CK2 might be the upstream protein of YBX1 through HPRD and CK2 could phosphorylate YBX1 to exert its kinase function. In addition, a previous study showed that CK2 participated in cell cycle and DNA repair by modulating the AKT signaling pathway [Citation39]. YBX1 has also been shown to regulate the AKT signaling pathway by binding to the promoter of many proteins related to it [Citation37].
Here, we assessed the expression of CK2α and YBX1 in several cancer cells. We explored the effects of CK2α and YBX1 inhibition on proliferation, survival, and migration, and examined the regulation mechanisms of YBX1 by CK2α. We investigated the interaction between CK2α and YBX1 through co-immunoprecipitation in different cancer cell lines. YBX1 inactivation increased CX4945 sensitivity in cancer cells, indicating that inhibition of the CK2α/YBX1 signaling pathway may be one of the mechanisms involved in tumor growth and migration, and might be a rational therapeutic strategy in drug-resistant cancers.
Materials and methods
Antibodies, plasmids, and reagents
Monoclonal antibodies to CK2α, cyclin A, cyclin D1, and GAPDH were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Monoclonal antibodies to p-AKT (Ser473), AKT, p-YBX1 (Ser102) and YBX1 were obtained from Cell Signaling Technology (Beverly, MA, USA). Polybrene was obtained from Sigma-Aldrich. CX4945 and doxorubicin were obtained from Selleck Chemicals. LY294002 was obtained from Cell Signaling Technology (Beverly, MA, USA). The inhibitors CX4945, doxorubicin and LY294002 were reconstituted in DMSO. Small interfering RNAs (siRNAs) and Lentiviral shRNA constructs were obtained from GenePharma (Suzhou, China) and included YBX1: AGAAGGUCAUCGCAACGAATT (siRNA or shRNA), CK2: GUGGAUUUAUAGUAGUUCATT (siRNA or shRNA), and Negative control: UUCUCCGAACGUGUCACGUTT (scramble siRNA or scramble shRNA). pZW6 (CK2alpha) and pGV15 (CK2alpha K68M) were gifts from David Litchfield (Addgene plasmid # 27086 and plasmid # 27089); pDESTmycYBX1 was a gift from Thomas Tuschl (Addgene plasmid # 19878).
Cell lines
The 293T, HeLa, SKOV3, OVCA429, ES2, Hep3B, HepG2, BGC823, MKN28, PC-9, and A549 cell lines were obtained from the Chinese Academy of Science (Shanghai, China) and were maintained in 10% FBS 1640 basic medium. GIST430, GIST882, and GIST-T1 were kind gifts from Dr. Wen-Bin Ou (College of Life Sciences and Zhejiang Sci-Tech University). GIST882 and GIST-T1 were maintained in 15% FBS 1640. GIST430 was maintained in DMEM/F-12 with 15% FBS. All cultures were supplemented with penicillin/streptomycin.
Protein lysate preparation and immunoblotting
The cell lysates were rocked in lysis buffer overnight at 4°C and spun down by centrifugation at 14,000 rpm for 30 min at 4°C. Cell lysates were standardized for the protein content with a Bio-Rad protein assay and resolved by SDS-PAGE. Electrophoresis and immunoblotting were performed as previously described [Citation37].
Ni-NTA pull-down
The cell lysates were rocked in lysis buffer (10 mM Tris-HCl, 200 mM NaCl, 8 M Urea, pH 7.4) overnight at 4°C and spun down by centrifugation at 14,000 rpm for 30 min at 4°C. After centrifugation, cell supernatant was conducted to Ni-NTA pull-down assay. The beads were then washed by washing buffer (10 mM Tris-HCl, pH 7.4, 50 mM imidazole) three times. Finally, the beads were eluted by 200 mM imidazole (pH 8.0)
RNA isolation and qRT-PCR
RNA extraction from cells was conducted with a TIANGEN RNA extraction kit (DP430). cDNAs synthesis were performed with an M-MLV (Moloney murine leukemia virus) system (Promega, M1705), using 1 μg portion of the total RNA and the reverse-transcribed DNA was subjected to PCR. The primers were as follows: CK2α forward, 5'-GCTGGGGGTAAGACCTTGTT-3', and CK2α reverse, 5'-TTGTCTGTGTGAGCAGAGGG-3'; YBX1 forward, 5'-CGCAGTGTAGGAGATGGAGAG-3', and YBX1 reverse, 5'-GAACACCACCAGGACCTGTAA-3'; 18S forward, 5'-TCGAGGCCCTGTAATTGGAA-3', and 18S reverse, 5'-CTTTAATATACGCTATTGGAGCTG-3'.
Luciferase reporter assay
The full-length sequence of YBX1 3'-UTR was amplified by PCR using the primer set: YBX1 3'UTR forward: 5'-ACTGCATTAGCCTCCTAACTG-3' and reverse: 5'-GGTTGCGGTGATGGTGACTGG-3' followed by cloned into pGL3-Basic Vector. For reporter assay, cells were co-transfected with WT or mutant of CK2α(K68M), pGL3-YBX1 and pRL-TK-Renilla as reference control. Forty-eight hours after post transfection, reporter assay was performed by Dual-Luciferase Reporter Assay system (Promega, Madison, WI, USA).
Immunoprecipitation
For immunoprecipitation analyses, 500 μg of lysates were incubated with the primary antibodies and rocked overnight at 4°C followed by the addition of protein A/G agarose beads (Invitrogen Life Technologies). SDS-PAGE was used to separate the immunoprecipitates, and proteins were transferred onto PVDF membranes (Millipore) for immunoblotting.
Transfection
CK2α siRNA (shRNA) or YBX1 siRNA (shRNA) and the co-transfection of CK2α and YBX1 expression vectors were performed with the transfection reagent LipofectamineTM 2000 (Invitrogen) according to the manufacturer’s instruction.
Preparation of shRNA lentiviruses
Co-transfection of 293T with LV3-pGLV-h1-GFP-puro plasmids that contained CK2α, YBX1 or CK2α plus YBX1 shRNAs and packaging plasmids Phr’∆R8.2 and pCMV-VSVG (ratio 9:8:1) was employed for lentivirus production. Transfections were conducted using LipofectamineTM 2000 (Invitrogen). Lentiviruses were collected at 72 h after transfection and frozen at −80°C. Lentiviruses were used for CK2α, YBX1 or CK2α plus YBX1 knockdowns.
Virus infection
Cells were seeded in 24-well plates and infected with lentiviral CK2α, YBX1 or CK2α plus YBX1 shRNA by the addition of 6 μg/mL of polybrene. SKOV3, ES2, PC9, and HepG2 cells were chosen for the stable deregulation of CK2α, YBX1 or CK2α plus YBX1 using 1 ~ 2 μg/ml of puromycin after transduction.
Cell migration assay
Cell migration assays were performed using a chamber system. Forty-five thousand cells were suspended in 250 μL of 1640 medium supplemented with 3% FBS and were seeded to the top chamber with 650 μL of 1640 medium supplement with 15% FBS added to the bottom chamber for 24 h. After incubating for 24 h, the cancer cell lines in the top chamber were detached with cotton swabs. Then, 75% methanol was inserted into the membrane for 15 min and stained with 0.5% crystal violet for 30 min.
Cell proliferation assay
Five thousand cells were seeded in 96-well plates cultured in basic 1640 medium with 10% FBS. The cell proliferation assay was carried out at 48 h or 72 h using a Cell Proliferation Assay Kit (Promega) according to the manufacturer’s instructions.
Colony formation assay
SKOV3, ES2, PC9, and HepG2 cells were plated at 5,000 cells/well in six-well plates and cultured in basic 1640 medium for 7 days before treatment with lentiviral CK2α shRNA and/or lentiviral YBX1 shRNA. After treatment with lentiviral for 14 days, the medium was removed, the cells were washed with PBS, and the cells were then stained with 0.5% Crystal violet in methanol for 15 min. Excess stain was removed by washing with PBS buffer. Colonies were photographed and counted. The experiments were performed in duplicate wells and repeated three times.
Cell-cycle analysis
SKOV3 cells in six-well plates were trypsinized and washed with PBS buffer at 72 h after incubation with 2.5 μM CX4945 for 48 h and transfection with YBX1 shRNA for 72 h. Nuclear staining was with a propidium iodide solution and the cell suspension was tested in a flow cytometer (C6 plus). Data processing was carried out by Flowjo VX10.
In vitro wound-healing assay
A wounding healing assay was carried out according to previously described methods [Citation40]. A scratch was produced in the cultures that were close to fusion by 200 μL pipetman at 48 h after treatment with the inhibitor CX4945 (2.5, 5 μM) or YBX1 siRNA for 48 or 72 h, respectively, or 96 h after shRNA transduction. The plates were photographed at 0, 12, 24, 36 and 48 h using a Leica DMI 3000B inverted microscope (Leica Microsystems, Germany). Experiments were performed in triplicate.
Xenotransplant murine models
Female adult athymic nude mice (4 to 6 weeks old) were raised in a designated sterile infrastructure. Mice in each group underwent hypodermic injection with 3 × 106 SKOV3 cells suspended in PBS expressing scramble shRNA (n = 5), CK2α-targeting shRNA (n = 5), YBX1-targeting shRNA (n = 5), or CK2α plus YBX1-targeting shRNA. After 4 weeks, the mice were sacrificed by CO2 inhalation after injection to evaluate the tumor volume and CK2α and YBX1 expressions. All animal experimental protocols were approved by the Independent Ethics Committee at Yangtze Delta Region Institute of Tsinghua University Zhejiang (Permit Number: 2014–0012), and all procedures were performed in accordance with the approved guidelines.
Immunohistochemistry (IHC) analysis
CK2α, YBX1, and Ki67 were detected in the specimens of xenotransplant tumor from female adult athymic nude mice. After fixed in 4% paraformaldehyde, the specimens of tumor were embedded in paraffin. After dewaxing and hydration, antigen retrieval and blocking, the tumor specimens were incubated with specific primary antibodies (CK2α, Santa Cruz, 1:300, sc12738; YBX1, Beyotime, 1:100, AF1690 and Ki67, Beyotime, 1:100, AF1738) overnight at 4°C, and followed by the observation using 3,3-diaminobenzidine color kit (Dako, Denmark).
Statistical analysis
Data between two groups were analyzed using independent-unpaired T-tests, and data among multiple groups were compared by one-way analysis of variance (ANOVA), followed by a Tukey’s multiple comparisons posttest. Significant differences were defined as *p < 0.05, **p < 0.01 and ***p < 0.001.
Results
Inhibition or knockdown of CK2α results in down-regulation of YBX1
The expression of CK2α and YBX1 was measured by immunoblotting in various cancer cells, including GIST, ovarian, non-small cell lung, liver, and gastric cancer cell lines ()). CK2α and YBX1 were expressed in all selected cell lines, without consideration of the type of cancer cell lines ()).
Figure 1. The regulatory role of CK2α on expression of YBX1. (a). Immunoblotting assessment of CK2α and YBX1 in cancer cell lysates, including gastrointestinal stromal tumors (GIST882, GIST-T1, and GIST430), ovarian cancer cell lines (SKOV3, OVCA429, and ES2), non-small cell lung cancer cell lines (PC9 and A549), liver cancer cell lines (HepG2 and Hep3B) and gastric cancer cell lines (AGS, MKN28, and BGC823).(b). Expression of YBX1 was assessed by immunoblotting after treatment with the CK2 inhibitor CX4945 for 48 h. (c). Expressions of YBX1, p-AKT, AKT, cyclin A, and cyclin D1 were evaluated by immunoblotting after the transfection with CK2α siRNA for 72 h in ovarian cancer cell lines (SKOV3 and OVCA429), non-small cell lung cancer cell lines (PC9) and liver cancer cell lines (HepG2). (d). Immunoblotting evaluation of CK2α, p-AKT, AKT, cyclin A, and cyclin D1 after the transfection with YBX1 siRNA for 72 h in ovarian cancer cell lines (SKOV3 and OVCA429) and liver cancer cell lines (HepG2). (e). Immunoblotting evaluation of YBX1 expression after the transfection with wild-type CK2α overexpression vector HA-CK2α or kinase-dead mutation HA-CK2α (K68M) for 72 h in ovarian cancer cell lines (SKOV3 and ES2), non-small cell lung cancer cell lines (PC9) and liver cancer cell lines (HepG2).
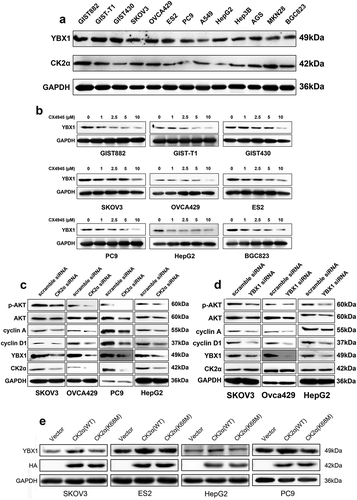
We assessed the effects of CK2α inhibitor CX4945 on YBX1 and p-YBX1 by immunoblotting in various cell lines, including GIST, ovarian, liver, and gastric cancer cell lines (); Supplementary Fig. S1a). CX4945 induced the down-regulation of YBX1 in all cell lines in a dose-dependent manner (); Supplementary Fig. S1a). We subsequently examined the YBX1 expression after CK2α was knockdown by CK2α siRNA. Three siRNA sequences were designed to knockdown CK2α expression in different cancer cell lines, and CK2α siRNA 2′ was the most efficient in silencing CK2α expression at both the protein and mRNA levels (data not shown). Therefore, we utilized CK2α siRNA 2′ to conduct subsequent experiments. Western blotting analysis showed that CK2α knockdown decreased the expression of YBX1 in SKOV3, HepG2, PC9 and OVCA429 cell lines ()); however, the deregulation of YBX1 had little influence on the expression of CK2α ()). As predicted, the overexpression of CK2α induced an increased expression of YBX1; however, the K68M (kinase dead) mutant of CK2α had no effect on the expression of YBX1 ()). Next, qRT-PCR analysis showed that CK2α knockdown decreased the expression of YBX1 in SKOV3, HepG2, PC9, and OVCA429 cell lines, however, the deregulation of YBX1 had little influence on the expression of CK2α (Supplementary Fig. S1b). Further results demonstrated that the overexpression of CK2α increased the expression of YBX1 in SKOV3, HepG2, and PC9 cell lines; however, the K68M (kinase dead) mutant of CK2α had no effect on the expression of YBX1 (Supplementary Fig. S1b).
As CK2 and YBX1 play important roles in PI3K/AKT pathway regulation, we investigated the effects of CK2α and YBX1 on PI3K/AKT pathway. The effects of CK2α or YBX1 silencing on PI3K/AKT pathway were assessed by immunoblotting in SKOV3, OVCA429, HepG2 and PC9 cells with the transfection of CK2α or YBX1 siRNA (,)). Three siRNA sequences were designed to knockdown YBX1 expression in different cancer cell lines, and YBX1 siRNA 1′ was the most efficient one at both the protein and mRNA levels (data not shown). Therefore, we utilized YBX1 siRNA 1′ as to conduct subsequent experiments. CK2α siRNA resulted in decreases of p-AKT, cyclin D1, cyclin A and YBX1 in the SKOV3, OVCA429, HepG2 and PC9 cell lines ()). YBX1 siRNA resulted in decreases of p-AKT, AKT, cyclin A and cyclin D1 expression in the SKOV3, OVCA429 and HepG2 cell lines; however, it had no effect on the expression of CK2α ()).
Ck2α regulated the expression of YBX1 at the transcriptional level
Due to the observation that inhibition of CK2α down-regulated the expression of YBX1 (), we examined how CK2α affected the expression of YBX1. As post-translational modification of genes is frequently related to the expression of proteins, we examined the effects of CK2α on the stability of YBX1. To further analyze if this degradation is associated with proteasome-dependent degradation, cells were treated with MG132 (proteasome inhibitor). MG132 did not result in an increase of YBX1 (data not shown), suggesting that down-regulation of YBX1 by CK2α inhibition is independent on proteasome-mediated YBX1 degradation. To investigate the regulation of YBX1 expression under steady-state conditions, SKOV3 cells were treated with cycloheximide (CHX) to inhibit protein synthesis after treatment of CK2α shRNA or CX4945, and the results showed that inhibition of CK2α failed to increase the half-life of YBX1 (Supplementary Fig. S2a). Further results demonstrated that knockdown of CK2α in SKOV3 cells did not increase the ubiquitination of YBX1 (Supplementary Fig. S2b). These results suggest that CK2α-induced YBX1 degradation is not regulated at the protein level, but at the level of transcription.
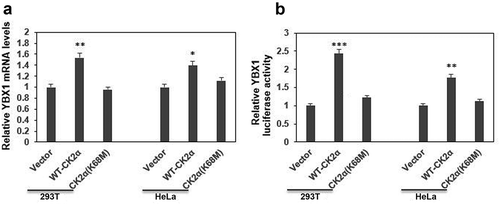
In order to test this hypothesis, expression of YBX1 was assessed by qRT-PCR after cells were transfected with WT-CK2α or K68M-CK2α plasmids. WT-CK2α, not K68M-CK2α, resulted in increase of YBX1 in 293T cells ()). Next, YBX1 luciferase assay was carried out to determine the effects of CK2α on YBX1 transcriptional activation in 293T cells. As shown in ), overexpression of WT-CK2α activated YBX1, while overexpression of K68M-CK2α mutant showed no effects on YBX1 activation ()). Impressively, similar results were found in HeLa cells (,)), suggesting that CK2α is an activator of YBX1, and K68 of CK2α (kinase activity site) plays a critical role in the regulation of YBX1 transcriptional activation.
Figure 2. CK2α regulated the expression of YBX1 at transcriptional level. (a). The YBX1 transcript levels determined by qRT-PCR after the transfection with wild-type CK2α overexpression vector HA-CK2α or kinase-dead mutation HA-CK2α (K68M) for 48 h in both 293T and HeLa cell lines. (b). The activities of the reporter genes were determined by a dual-luciferase reporter assay system after the transfection with wild-type CK2α overexpression vector HA-CK2α or kinase-dead mutation HA-CK2α (K68M), firefly reporter plasmid and pRL-TK-Renilla for 24 h in 293T and Hela cell lines. The data represent the mean values (±s.d.) of three independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001).
Synergistic effects of combined inhibition of CK2α and YBX1
Due to the observations that YBX1 down-regulation occurred under the treatment of CX4945 or CK2α siRNA and that they both regulated the PI3K/AKT pathway, we hypothesized that synergistic inhibitory effects of the PI3K/AKT pathway on cancer cells should be observed with YBX1 knockdown and CK2α inhibition. Immunoblotting evaluated p-AKT, cyclin A and cyclin D1 expression in cell lines (SKOV3, ES2, OVCA429, PC9, HepG2, and BGC823) after 72 h of YBX1 knockdown and 48 h of CX4945 treatment.
CX4945 treatment alone decreased the expression of p-AKT, cyclin A and cyclin D1 in a dose-dependent manner. The combination of YBX1 knockdown and CK2α inhibition by CX4945 led to decreased expressions of p-AKT, cyclin A and cyclin D1 to a larger degree than in the independent manipulations ()).
Figure 3. Additive effects were observed through coordinated inhibition of CK2α and YBX1. (a and c) Expression of p-AKT, AKT, cyclin A, cyclin D1, CK2α and YBX1 were assessed by immunoblotting after incubation with CX4945 for 48 h and transfection with YBX1 siRNA for 72 h (a) or 3 days after the transduction with lentiviral CK2α shRNA and/or lentiviral YBX1 shRNA in cancer cell lines (c). (b and d) Cell proliferation was assessed after incubation with CX4945 for 48 h and transfection with YBX1 siRNA for 72 h (b) or 4 days after transduction with lentiviral CK2α shRNA and/or lentiviral YBX1 shRNA in cancer cell lines (d). (e). Colony growth assays were performed after the transduction with lentiviral CK2α shRNA and/or lentiviral YBX1 shRNA for 14 days in cancer cell lines. (f). Cell-cycle analysis was performed after incubation with 2.5 μM CX4945 for 48 h and transfection with YBX1 shRNA for 72 h in SKOV3 cell lines. (g). Wound-healing assays were performed after treatment with CX4945 for 48 h and transfection with YBX1 siRNA for 72 h in cancer cell lines. Scale bar, 50 μm. (h). Transwell migration assays were performed after treatment with CX4945 for 48 h and transfection with YBX1 shRNA for 72 h in cancer cell lines. Scale bar, 50 μm. (i). Wound-healing assays were performed after transduction with lentiviral CK2α shRNA and/or lentiviral YBX1 shRNA for 2 days in cancer cell lines. Scale bar, 50 μm. (j). Transwell migration assays were performed after transduction with lentiviral CK2α shRNA and/or lentiviral YBX1 shRNA for 2 days in cancer cell lines. Scale bar, 50 μm. The data represent the mean values (±s.d.) of three independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001).
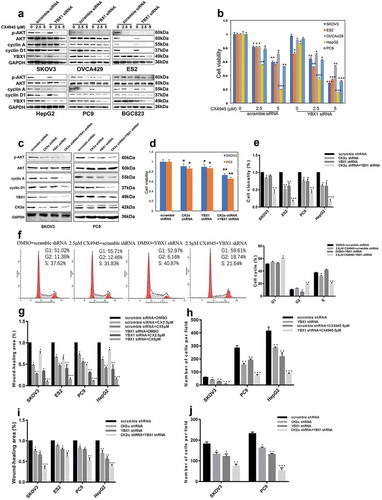
Additional inhibitory effects on the cell viability, colony formation, cell-cycle arrest, wound-healing, and Boyden chamber assay were obtained by the combined inhibition of CK2α and YBX1. YBX1 knockdown in cancer cell lines (SKOV3, ES2, OVCA429, PC9, and HepG2) resulted in a 10 ~ 20% decrease of cell viability 72 h after YBX1 siRNA treatment. The combination of YBX1 knockdown and the CK2α inhibitor CX4945 resulted in a 30 ~ 65% (2.5 μM of CX4945) or 45 ~ 90% (5 μM of CX4945) reduction of cell viability, respectively ()). Furthermore, we assessed the PI3K/AKT pathway and cell viability of the cancer cell lines after the knockdown of CK2α and YBX1 by shRNAs (,)). Synergistic treatment of SKOV3 and PC9 with CK2α shRNA and YBX1 shRNA resulted in a greater decrease of p-AKT, cyclin A and cyclin D1 compared with either intervention alone ()). Meanwhile, treatment with CK2α shRNA or YBX1 shRNA resulted in a 10 ~ 20% decrease in cell viability in SKOV3 and PC9. Nevertheless, co-targeting CK2α and YBX1 by shRNA achieved a 30 ~ 40% decrease compared with either intervention alone ()). SKOV3, ES2, PC9, and HepG2 cells were further evaluated by colony formation assays. Cells treated with CK2α shRNA or YBX1 shRNA formed fewer colonies compared with scramble shRNA. Combinational usage of CK2α shRNA and YBX1 shRNA further decreased the number of colonies in these cells, as compared with either intervention alone (); Supplementary Fig. S3a). CK2α and YBX1 inhibition in SKOV3 resulted in a minimal increase of G1 peaks, from 51% in cells treated with DMSO to 55% in cells treated with CX4945 and 53% in cells treated with YBX1 shRNA, respectively. Combinational treatment with CX4945 and YBX1 shRNA induced greater increase of G1 and G2 peaks than each intervention alone: G1 peaks account for 59.61% of the cells treated with CX4945/YBX1 shRNA; G2 peaks account for 18.74% of the cells treated with CX4945/YBX1 shRNA, 12.46% of those treated with CX4945, and in 6.16% of those treated with YBX1 shRNA ()).
Wound-healing assays in cancer cell lines (SKOV3, ES2, PC9, and HepG2) demonstrated that knockdown of YBX1 led to a slight suppression of wound healing at 72 h; however, intact wound healing was observed in the control cells. Addictive wound-closure assays were carried out at day 2 in these cells with the combined treatment of YBX1 siRNA and the CK2 inhibitor CX4945, resulting in a greater extent of wound-closure inhibition than that with either intervention alone in a dose-dependent manner (); Supplementary Fig. S3b). Similarly, Boyden chamber assays demonstrated that the combination of YBX1 knockdown and CK2 inhibitor CX4945 resulted in a greater inhibition of migration (); Supplementary Fig. S3c). Wound-healing assays and transwell migration assays were also evaluated after the knockdown of CK2α and YBX1 by shRNAs. Co-targeting CK2α and YBX1 by shRNAs achieved greater inhibition of the cell migration ability (,); Supplementary Fig. S3d and e).
YBX1 is required for CK2α-induced proliferation and migration
Next, we investigated the functions of YBX1 in CK2α-induced proliferation and migration. As shown in ), inhibition of CK2α by CX4945 decreased the expression of p-AKT, cyclin A and cyclin D1. Overexpression of YBX1 increased the expression of p-AKT, cyclin A and cyclin D1. However, overexpression of YBX1 partly compensated the decrease of p-AKT, cyclin A and cyclin D1 induced by CK2α inhibition ()). Furthermore, overexpression of WT-CK2α increased the expression of p-AKT, cyclin A and cyclin D1, however, K68M mutant of CK2α (kinase dead) had minor effects on it. Meanwhile, knockdown of YBX1 decreased the expression of p-AKT, cyclin A and cyclin D1. Intriguingly, after YBX1 knockdown, the induction of p-AKT, cyclin A and cyclin D1 by WT-CK2α-overexpression was abolished ()).
Figure 4. YBX1 is required for CK2α-induced proliferation and migration. (a). Immunoblotting evaluation of p-AKT, AKT, cyclin A, and cyclin D1 after treatment with 5 μM CX4945 for 48 h and transfection with YBX1 plasmids for 72 h in ovarian cancer cell lines (SKOV3). GAPDH staining is a loading control. (b). Cell viability was assessed after treatment with 5 μM CX4945 for 48 h and transfection with YBX1 plasmids for 72 h in ovarian cancer cell lines (SKOV3). Data were normalized to the vector and DMSO, and represent the mean values (± s.d.) from quadruplicate cultures. (c). Cell migration was assessed after treatment with 5 μM CX4945 for 48 h and transfection with YBX1 plasmids for 72 h in ovarian cancer cell lines (SKOV3). Data were normalized to the vector and DMSO, and represent the mean values (± s.d.) from quadruplicate cultures. Scale bar, 50 μm. (d). Immunoblotting evaluation of p-AKT, AKT, cyclin A, and cyclin D1 after treatment with CK2α plasmids and YBX1 siRNA for 48 h in ovarian cancer cell lines (SKOV3). GAPDH staining is a loading control. (e). Cell viability was assessed after treatment with CK2α plasmids and YBX1 siRNA for 48 h in ovarian cancer cell lines (SKOV3). Data were normalized to the vector and scramble siRNA, and represent the mean values (± s.d.) from quadruplicate cultures. (f). Cell migration was assessed after treatment with CK2α plasmids and YBX1 siRNA for 48 h in ovarian cancer cell lines (SKOV3). Scale bar, 50 μm. Data were normalized to the vector and scramble siRNA, and represent the mean values (±s.d.) of three independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001).
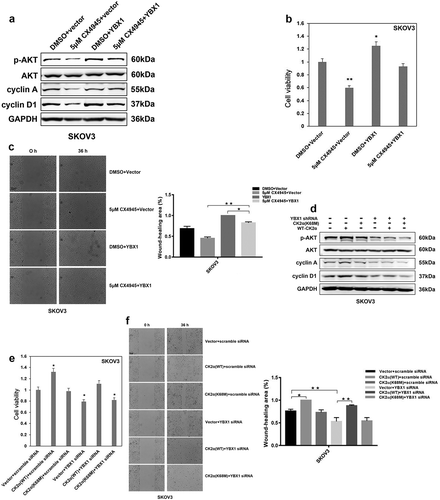
To investigate whether CK2α can promote proliferation and migration through YBX1, we performed cell viability and wound-healing assays in SKOV3 cells with YBX1 overexpression or YBX1 knockdown. CK2α inhibition in cancer cells (SKOV3) resulted in a ~ 40% decrease of cell viability after 5 μM CX4945 treatment ()). Cell viability was increased to ~1.3-fold in SKOV3 cells under YBX1 overexpression condition. Nevertheless, the combination of YBX1 overexpression and the CK2α inhibitor CX4945 did not result in an obvious reduction of cell viability compared to CX4945 treatment alone ()).
Furthermore, we assessed cell viability lines after CK2α overexpression under the condition of YBX1 knockdown ()). Upregulation of wild-type CK2α, not CK2α (K68M) resulted in a ~ 1.3-fold increase of cell viability in SKOV3 cells, while YBX1 knockdown resulted in a ~ 20% decrease of cell viability ()). Nevertheless, treatment with CK2α expression vector and YBX1 shRNA for 48 h resulted in a 1.1-fold increase in cell viability ()). Our results suggested that YBX1 overexpression restored the decreased cell viability induced by CK2α inhibitor CX4945.
Similar results were observed in SKOV3 cells with wound-healing assays. Wound-healing assays demonstrated that inhibition of CK2α by CX4945 led to an obvious suppression of wound healing; however, intact wound healing was observed in the group of YBX1 overexpression ()). Addictive wound-closure assays were carried out at 36 h in SKOV3 cells with combined treatment of YBX1 expression vector and the CK2α inhibitor CX4945, resulting in marked promotion of wound closure compared with CK2α inhibition, and a slight extent of wound-closure inhibition than that with YBX1 overexpression alone ()).
The results of wound healing after CK2α overexpression under the condition of YBX1 knockdown by siRNA were in line with previous assays ()). Upregulation of wild-type CK2α, not CK2α (K68M) resulted in an increase of wound healing in SKOV3 cells. YBX1 knockdown resulted in a decrease of wound healing ()). Addictive wound-closure assays were carried out at 36 h in these cells with the combined treatment of YBX1 siRNA and the CK2α vector, resulting in marked promotion of wound closure compared with YBX1 knockdown, and a slight extent of wound-closure inhibition than that with CK2α overexpression alone ()).
Inhibition of PI3K/AKT abolished the proliferation and migration induced by co-activation of CK2α and YBX1
Due to the observations that co-targeting CK2α and YBX1 achieved greater inhibition of cell proliferation and migration, we hypothesized that PI3K/AKT pathway should play an essential role in the process of these results. As shown in ), overexpression of CK2α or YBX1 increased the expression of p-AKT, cyclin A and cyclin D1. Inhibition of PI3K/AKT pathway by LY294002 decreased the expression of p-AKT, cyclin A and cyclin D1. Interestingly, when PI3K/AKT pathway was inhibited with LY294002, the induction of p-AKT, cyclin A and cyclin D1 by co-activation of CK2α and YBX1 overexpression was abolished.
Figure 5. Inhibition of PI3K/AKT abolished the proliferation and migration induced by CK2α and YBX1. (a). Immunoblotting evaluation of p-AKT, AKT, cyclin A, and cyclin D1 after treatment with 20 μM LY294002 for 48 h and transfection with CK2α and YBX1 plasmids for 72 h in ovarian cancer cell lines (SKOV3). GAPDH staining is a loading control. (b). Cell viability was assessed after treatment with 20 μM LY294002 for 48 h and transfection with CK2α and YBX1 plasmids for 72 h in ovarian cancer cell lines (SKOV3). (c). Cell migration was assessed after treatment with 20 μM LY294002 for 48 h and transfection with CK2α and YBX1 plasmids for 72 h in ovarian cancer cell lines (SKOV3). Scale bar, 50 μm. Data were normalized to the empty vector and DMSO, and represent the mean values (±s.d.) of three independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001).
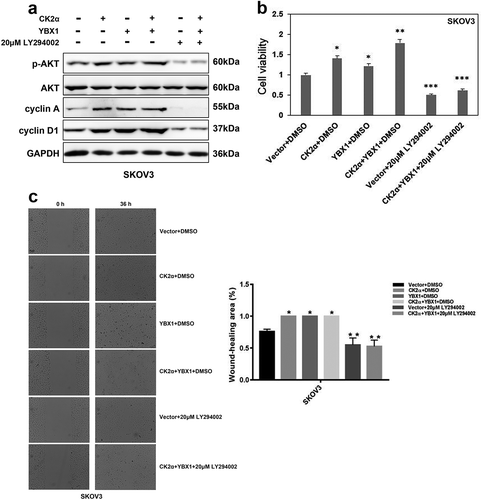
Next, we investigated the effects of PI3K/AKT pathway inhibition on co-overexpression of CK2α and YBX1-induced proliferation and migration. As shown in ,), overexpression of CK2α or YBX1 increased cell viability and wound-closure ability. Co-overexpression of CK2α and YBX1 achieved greater promotion of cell viability and wound-closure ability. Inhibition of PI3K/AKT pathway by LY294002 decreased cell viability and wound-closure ability. Furthermore, after PI3K/AKT pathway was inhibited by LY294002, the induction of cell viability and wound closure induced by CK2α and YBX1 expression was abolished.
CK2 interacts with YBX1
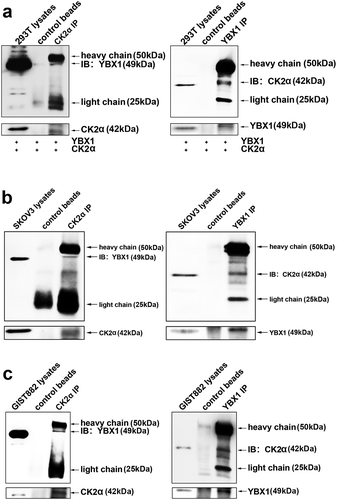
To investigate whether there is an interaction between CK2α and YBX1, we transfected 293T with CK2α and YBX1 expression vectors and conducted CK2α or YBX1 immunoprecipitation (co-IP) ()). CK2α immunoprecipitation displayed an obvious YBX1 49 kDa band, while YBX1 immunoprecipitation displayed an obvious CK2α 42 kDa band ()). We subsequently evaluated the CK2α and YBX1 interaction without extra transfection in GIST882 and SKOV3 cells (,)). CK2α immunoprecipitation indicated a dominant YBX1 49 kDa band in GIST882 ()) and SKOV3 ()) cells; this result was confirmed by CK2α immunoblotting in YBX1 immunoprecipitation (,)). Furthermore, the in vitro binding analysis indicated that CK2α directly bound to YBX1 (Supplementary Fig. S4). These results demonstrated that CK2α could interact with YBX1.
Figure 6. Interaction of CK2α and YBX1 in cancer cell lines. (a). CK2α-YBX1 complex was assessed after co-transfection of CK2α and YBX1 through immunoprecipitation followed by immunoblotting in 293T cell lines. (b and c) SKOV3 or GIST882 cell lysates were analyzed via immunoprecipitation and Western blot analysis as shown.
Combined inhibition of CK2α and YBX1 in vivo prevented ovarian cancer growth
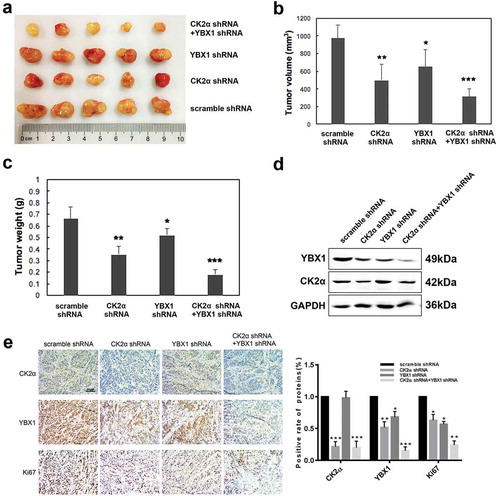
In a mouse xenograft model, the combination of CK2α and YBX1 knockdown in SKOV3 cells resulted in a greater decrease of tumor growth ()). At the end of the observation, the bulk of the tumor in CK2α and YBX1 knockdown group was obviously less than that with either intervention alone ()). Compared with cells with intervention alone, the combination of CK2α and YBX1 knockdown resulted in a lighter weight ()). CK2α and YBX1 knockdown were evaluated by immunoblot and IHC analysis after xenograft tissues were harvested. The shRNA-mediated knockdown resulted in the reduction of CK2α and YBX1 expression in the mouse xenograft tumor tissues, and the YBX1 expression was further inhibited in the xenograft tumors after treatment with CK2α shRNA. Co-targeting CK2α and YBX1 by shRNAs achieved greater inhibition of the expression of Ki67 than that with either intervention alone (,)).
Figure 7. Combination of CK2α knockdown and YBX1 knockdown inhibited the growth of SKOV3 xenografts in mice. (a). Tumors were resected and photographed, and decreased tumor growth was observed following treatment with a combination of CK2α shRNA and YBX1 shRNA. (b and c) Tumor volume and weight were measured after SKOV3 cells with stable shRNA expression were subcutaneously injected in mice for 4 weeks. (d). Western blot confirming CK2α and YBX1 knockdown in resected SKOV3 xenografts with expression of CK2α-targeting shRNA, YBX1-targeting shRNA, and CK2α plus YBX1-targeting shRNA. (e). Xenograft tumors were immunostained with CK2α, YBX1 or Ki67. Relative intensity of the immunostaining was quantified. Scale bar, 50 μm. The data represent the mean values (±s.d.) of three independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001).
PI3K/AKT antagonist inhibits the resistance of doxorubicin induced by co-activation of CK2α and YBX1
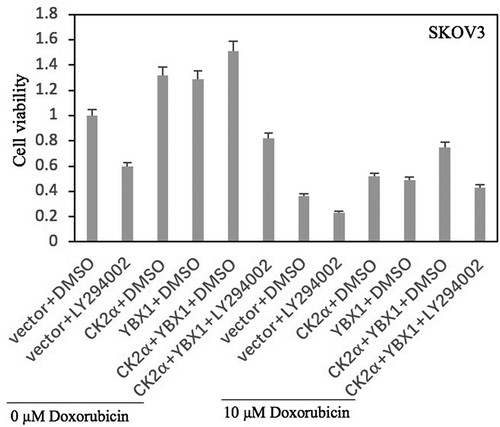
Additive effects on cell viability were obtained through coordinated doxorubicin treatment after CK2α and YBX1 activation (). Co-activation of CK2α and YBX1 expression in ovarian cell lines SKOV3 resulted in ~50% increase of cell viability at day 2 after 10 μM doxorubicin treatment for 24 h (). To determine whether PI3K/AKT pathway regulates chemotherapy sensitivity of SKOV3 cells to doxorubicin induced by co-activation of CK2α and YBX1, the PI3K/AKT pathway was inactivated using a small molecule inhibitor, LY294002. Treatment with LY294002 showed profound anti-proliferative effects at day 2 in SKOV3 cells. Co-overexpression of CK2α and YBX1 increased chemotherapeutic resistance of SKOV3 cells to doxorubicin (). However, even with CK2α and YBX1 overexpression, viability was decreased in SKOV3 cells after treatment with LY294002 ().
Figure 8. PI3K/AKT antagonist decreases the resistance of doxorubicin induced by co-activation of CK2α and YBX1. Cell proliferation was assessed in cell lines after incubation with doxorubicin for 24 h or/and transfection with CK2α or/and YBX1 plasmids for 72 h or/and 20 μM LY294002 for 48 h in ovarian cancer cell lines (SKOV3). Cell viability was assayed by Cell Proliferation Assay Kit (Promega) method. The data represent the mean values (±s.d.) of three independent experiments.
Discussion
The PI3K/AKT pathway plays a major role in normal and cancerous cytophysiology and is altered in many human cancers [Citation41–Citation44], which regulate cell cycle, cell survival and motility[Citation45]. Thus, targeting it has substantial therapeutic potential in the treatment of cancer. Studies have demonstrated that both CK2 and YBX1 could regulate PI3K/AKT pathway [Citation11,Citation37]. To date, however, no direct crosstalk between CK2 and YBX1 has been reported.
CK2 protein has been found to be overexpressed in numerous human neoplasms, and it has been suggested that this upregulation is closely related to the occurrence and progression of cancer [Citation4,Citation46]. The overexpression of YBX1 is related to the progression and prognosis of tumors, including stromal tumors, pancreatic cancer, colon cancer and other cancers [Citation21,Citation22]. In this study, immunoblotting evaluations of CK2α and YBX1 expression demonstrated that CK2α and YBX1 were ubiquitously expressed in cancer cell lines ()), indicating that the CK2α–YBX1 pathways may have a role in cancer cells.
CX4945, an orally bioavailable-specific inhibitor of CK2, restrains pro-survival activity and exerts sufficient anti-tumor activity. The positive association between CX4945 and anti-proliferation effects appeared to be dependent on CK2α, the catalytic subunit of CK2[Citation19]. Previous studies have shown that CK2 may be an upstream kinase of YBX1 with the phosphorylation site on serine 165[Citation47]. Our study suggested that CX4945 reduced YBX1 expression in cancer cell lines ()). Moreover, knockdown of CK2α decreased p-AKT, cyclin A, cyclin D1 and YBX1 expressions ()); this outcome is consistent with published articles showing that knockdown of CK2α resulted in the deregulation of cyclin A[Citation48] in T-ALL cells and cyclin D1 in NSCLC cells[Citation49]. To evaluate whether YBX1 was suppressed by CK2α, CK2α knockdown was carried out by siRNA or shRNA () and )). Knockdown of CK2α decreased YBX1 expression () and )). Furthermore, co-targeting CK2α (by CK2α shRNA or inhibitor CX4945) and YBX1 (by YBX1 shRNA) decreased p-AKT, cyclin A, cyclinD1 and YBX1 to a larger degree than that with the independent manipulations (,)). However, YBX1 knockdown had little effect on the CK2α expression () and )). The present data also indicated that overexpression of wild-type CK2α increased the YBX1 expression, whereas the mutant type CK2α (K68M, kinase dead) had no impact on YBX1 ()). Our results indicated that CK2α positively regulated the YBX1 expression which is dependent on CK2α kinase activity.
AKT, an important intermediate of RTK/PI3K signal transduction, has a major role in preventing cell proliferation, migration, and invasion [Citation50–Citation53]. Previous studies have shown that YBX1 can enhance the resistance of breast cancer to Paclitaxel[Citation27]. In the present study, we examined whether down-regulation of YBX1 improved the sensitivity of cancer cell lines to CX4945. Synergistic effects were obtained by combined treatment with CK2α inhibitor CX4945 or CK2α knockdown and YBX1 knockdown ( and ), which were shown by the decrease of p-AKT, cyclin A and cyclinD1 and the depression of proliferation, colony formation and cell-cycle arrest of cancer cells, the results of cell migration () and xenograft tumor growth (). These results suggest that co-targeting CK2α and YBX1 might be an effective therapeutic strategy for cancer patients.
Due to the observations that YBX1 down-regulation occurred with the treatment of CX4945 or CK2α siRNA (,)), we hypothesized that YBX1 could remedy the effects of CK2α on cancer cell proliferation and migration. Our study suggested that YBX1 overexpression weakened the phenotypes mediated by CK2α inhibitor CX4945 and YBX1 knockdown weakened the phenotypes induced by wild-type CK2α overexpression (). In conclusion, our findings for the first time demonstrated that YBX1 could be regulated through CK2α to promote proliferation and metastasis in cancer cells.
Although both CK2α and YBX1 are overexpressed in cancer cell lines and CK2α can positively regulate YBX1 expression, it remains unclear whether CK2α has interactions with YBX1. Through the immunoprecipitation assay, we demonstrated that CK2α interacted with YBX1 following the overexpression of CK2α and YBX1 ()). The results of the endogenous immunoprecipitation indicated that CK2α and YBX1 can interact with each other under physiological conditions in SKOV3 and GIST882 cells (,c)). In summary, our study showed that the CK2α and YBX1 expressions were correlated in cancer cells. The combination of CK2α and YBX1 inhibition appeared to exert synergistic effects in cancer cells in preventing cancer cell growth and cell migration compared with either intervention alone. Taken together, our results indicated that co-targeting CK2α and YBX1 may be one of the mechanisms involved in tumor growth and migration.
Furthermore, our results showed that CK2a regulates YBX1 abundance (), CK2a regulates p-AKT, and YBX1 also regulates p-AKT (,)). Combination of YBX1 knockdown and CK2α inhibition led to decreased expression of critical factors of PI3K/AKT pathway to a larger degree ()). Thus, we hypothesized that PI3K/AKT pathway should play an essential role in the process of these results. Our results showed that inhibition of PI3K/AKT pathway by LY294002 abolished the induction of p-AKT, cyclin A and cyclin D1, cell viability and migration by co-activation of CK2α and YBX1 (), suggesting that PI3K/AKT pathway activation is required for CK2α and YBX1-induced proliferation and migration.
Chemotherapy is one of the options available for ovarian cancer patients. The patients usually respond to chemotherapy initially, but most advanced ovarian cancer patients suffer from high rate of recurrence. Previous studies showed that CK2 inhibitor CX-4945 suppressed DNA repair response triggered by DNA-targeted anticancer drugs and augments efficacy, which is mechanistic rationale for drug combination therapy[Citation18]. Overexpression of YBX1 can enhance the resistance of breast cancer to Paclitaxel, while down-regulation of YBX1 increased the sensitivity of breast cancer to Paclitaxel[Citation27]. To demonstrate the role of co-targeting CK2α and YBX1 in the resistance of ovarian cancer cells against chemotherapeutic agents, we performed cell viability assay with the treatment of doxorubicin, a clinical chemotherapeutic agent for ovarian cancer patients (). Our results clearly showed that co-targeting CK2α and YBX1 decreased chemotherapeutic sensitivity of SKOV3. (). Intriguingly, anti-chemotherapy sensitivity induced by CK2α and YBX1 was abolished when cells were treated with LY294002, suggesting that CK2α and YBX1-induced chemotherapy resistance is at least partly dependent on PI3K/AKT pathway activity ().
To conclude, our studies demonstrated that CK2α could interact with YBX1, and CK2α could regulate the expression of YBX1. The inhibition of CK2α and YBX1 showed synergistic effects in inactivating PI3K/AKT signaling pathway. YBX1 is required for CK2α-induced proliferation and migration. Inhibition of PI3K/AKT abolished the proliferation, migration and chemotherapy resistance induced by CK2α and YBX1. These results indicate that combined inhibition of CK2α and YBX1 showed synergistic effects in inactivating PI3K/AKT signaling pathway and may be one of the mechanisms involved in tumor growth.
Author Contributions
WX, XF, and XL: designed the research, analyzed the data, and wrote the manuscript. WX, YM, and HM: performed experiments and prepared figs. LS, HM, WO, and JP: advised the design of the research and collected the tumor samples. TL, TQ, and HZ: reviewed the manuscript. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
All animal experimental protocols were approved by the Independent Ethics Committee at Yangtze Delta Region Institute of Tsinghua University Zhejiang (Permit Number: 2014–0012), and all procedures were performed in accordance with the approved guidelines.
Supplemental Material
Download Zip (8 MB)Acknowledgments
We thank David Litchfield and Thomas Tuschl for providing CK2α or YBX1 plasmids and Dr. Wen-Bin Ou for the gastrointestinal stromal tumor cell lines.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Pinna LA. The oncogenic potential of CK2. John Wiley & Sons, Inc. 2013; 10.
- Xu X, Landesman-Bolliag E, Channavajhala PL, et al. Murine protein kinase CK2: gene and oncogene. Mol Cell Biochem. 1999;191:65–74.
- Bibby AC, Litchfield DW. The multiple personalities of the regulatory subunit of protein kinase CK2: CK2 dependent and CK2 independent roles reveal a secret identity for CK2β. Int J Biol Sci. 2005;1:67–79.
- Trembley JH, Chen Z, Unger G, et al. Emergence of protein kinase CK2 as a key target in cancer therapy. Biofactors. 2010;36:187–195.
- Liu H, Wang H, Wang J, et al. Structural determinants of CX-4945 derivatives as protein kinase CK2 inhibitors: A computational study. Int J Mol Sci. 2011;12:7004–7021.
- Manni S, Carrino M, Piazza F. Role of protein kinases CK1α and CK2 in multiple myeloma: regulation of pivotal survival and stress-managing pathways. J Hematol Oncol. 2017;10:157.
- Chua M, Lee M, Dominguez I. Cancer-type dependent expression of CK2 transcripts. Plos One. 2017;12:e0188854.
- Zhang S, Wang Y, Mao JH, et al. Inhibition of CK2α down-regulates Hedgehog/Gli signaling leading to a reduction of a stem-like side population in human lung cancer cells. Plos One. 2012;7:e38996.
- Dominguez I, Sonenshein GE, Seldin DC. Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-kappaB signaling: linking development and cancer. Cell Mol Life Sci. 2009;66:1858–1867.
- Di MG, Salvi M, Arrigoni G, et al. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 2005;12:668–677.
- Park JH, Kim JJ, Bae YS. Involvement of PI3K-AKT-mTOR pathway in protein kinase CKII inhibition-mediated senescence in human colon cancer cells. Biochem Biophys Res Commun. 2013;433:420–425.
- Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;276:993–998.
- Seldin DC. New models of lymphoma in transgenic mice. Curr Opin Immunol. 1995;7:665–673.
- Landesmanbollag E, Song DH, Romieumourez R, et al. Protein kinase CK2: signaling and tumorigenesis in the mammary gland. Mol Cell Biochem. 2001;227:153–165.
- Agarwal M, Nitta RT, Li G. Casein Kinase 2: a novel player in glioblastoma therapy and cancer stem cells. J Mol Genet Med. 2014;8:pii:1000094.
- Bennetzen MV, Larsen DH, Bunkenborg J, et al. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol Cell Proteomics. 2010;9:1314–1323.
- Zhang HX, Jiang SS, Zhang XF, et al. Protein kinase CK2α catalytic subunit is overexpressed and serves as an unfavorable prognostic marker in primary hepatocellular carcinoma. Oncotarget. 2015;6:34800–34817.
- Siddiquijain A, Bliesath J, Macalino D, et al. CK2 inhibitor CX-4945 suppresses DNA repair response triggered by DNA-targeted anticancer drugs and augments efficacy: mechanistic rationale for drug combination therapy. Mol Cancer Ther. 2012;11:994–1005.
- Adam SJ, Denis D, Nicole S, et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010;70:10288–10298.
- Eliseeva IA, Kim ER, Guryanov SG, et al. YB-1 and its functions. Biochemistry (Mosc). 2011;76:1402–1433.
- Kohno K, Izumi HT, Ashizuka M, et al. The pleiotropic functions of the Y-box-binding protein, YB-1. Bio Essays. 2003;25:691–698.
- Kuwano M, Oda Y, Izumi H, et al. The role of nuclear Y-box binding protein 1 as a global marker in drug resistance. Mol Cancer Ther. 2004;3:1485–1492.
- Habibi G, Leung S, Law JH, et al. Redefining prognostic factors for breast cancer: YB-1 is a stronger predictor of relapse and disease-specific survival than estrogen receptor or HER-2 across all tumor subtypes. Breast Cancer Res. 2008;10:R86.
- Sperling O, Brosh S, De VA. YB-1 provokes breast cancer through the induction of chromosomal instability that emerges from mitotic failure and centrosome amplification. Cancer Res. 2005;65:4078–4087.
- Sutherland BW, Kucab J, Wu J, et al. Akt phosphorylates the Y-box binding protein 1 at Ser102 located in the cold shock domain and affects the anchorage-independent growth of breast cancer cells. Oncogene. 2005;24:4281–4292.
- Stratford AL, Fry CJ, Desilets C, et al. Y-box binding protein-1 serine 102 is a downstream target of p90 ribosomal S6 kinase in basal-like breast cancer cells. Breast Cancer Res. 2008;10:R99.
- To K, Fotovati A, Reipas KM, et al. Y-box binding protein-1 induces the expression of CD44 and CD49f leading to enhanced self-renewal, mammosphere growth, and drug resistance. Cancer Res. 2010;70:2840–2851.
- Astanehe A, Finkbeiner MR, Hojabrpour P, et al. The transcriptional induction of PIK3CA in tumor cells is dependent on the oncoprotein Y-box binding protein-1. Oncogene. 2009;28:2406–2418.
- Finkbeiner MR, Astanehe A, To K, et al. Profiling YB-1 target genes uncovers a new mechanism for MET receptor regulation in normal and malignant human mammary cells. Oncogene. 2009;28:1421–1431.
- Bargou RC, Jürchott K, Wagener C, et al. Nuclear localization and increased levels of transcription factor YB-1 in primary human breast cancers are associated with intrinsic MDR1 gene expression. Nat Med. 1997;26:447–450.
- Chatterjee M, Rancso C, Stühmer T, et al. The Y-box binding protein YB-1 is associated with progressive disease and mediates survival and drug resistance in multiple myeloma. Blood. 2008;111:3714–3722.
- Lee C, Dhillon J, Wang MY, et al. Targeting YB-1 in HER-2 overexpressing breast cancer cells induces apoptosis via the mTOR/STAT3 pathway and suppresses tumor growth in mice. Cancer Res. 2008;68:8661–8666.
- Shibao K, Takano HY, Okazaki K, et al. Enhanced coexpression of YB-1 and DNA topoisomerase II alpha genes in human colorectal carcinomas. Int J Cancer. 1999;83:732–737.
- Vaiman AV, Stromskaya TP, Rybalkina EY, et al. Development of drug resistance in the population of colon cancer cells under the effect of multifunctional protein YB-1. B Exp Biol Med. 2007;143:463–466.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674.
- Lasham A, Print CG, Woolley AG, et al. YB-1: oncoprotein, prognostic marker and therapeutic target? Biochem J. 2013;449:11–23.
- Evdokimova V, Ruzanov P, Anglesio MS, et al. Akt-mediated YB-1 phosphorylation activates translation of silent mRNA species. Cell Mol Biol Lett. 2006;26:277–292.
- Shiraiwa S, Kinugasa T, Kawahara A, et al. Nuclear Y-Box-binding protein-1 expression predicts poor clinical outcome in stage III colorectal cancer. Anticancer Res. 2016;36:3781–3788.
- Siddiquijain A, Drygin D, Streiner N, et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010;70:10288–10298.
- Shaw RJ, Paez JG, Curto M, et al. The Nf2 tumor suppressor, merlin, functions in Rac-dependent signaling. Dev Cell. 2001;1:63–72.
- Janus JM, Rfl OS, Harwood CA, et al. Phosphoinositide 3-kinase-dependent signalling pathways in cutaneous squamous cell carcinomas. Cancers (Basel). 2017;9:86–134.
- Mantamadiotis T. Towards targeting PI3K-dependent regulation of gene expression in brain cancer. Cancers (Basel). 2017;9:60.
- Crumbaker M, Khoja L, Joshua AM. AR signaling and the PI3K pathway in prostate cancer. Cancers (Basel). 2017;9:34.
- Piddock RE, Bowles KM, Rushworth SA. The role of PI3K isoforms in regulating bone marrow microenvironment signaling focusing on acute myeloid leukemia and multiple myeloma. Cancers (Basel). 2017;9:29.
- Mabuchi S, Kuroda H, Takahashi R, et al. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol Oncol. 2015;137:173–179.
- Pinna LA. 12. CK2 Suppression of apoptosis and its implication in cancer biology and therapy. John Wiley & Sons, Inc. 2013;12:319–343.
- Prabhu L, Mundade R, Wang B, et al. Critical role of phosphorylation of serine 165 of YBX1 on the activation of NF-κB in colon cancer. Oncotarget. 2015;6:29396–29412.
- Melão A, Spit M, Cardoso BA, et al. Optimal interleukin-7 receptor-mediated signaling, cell cycle progression and viability of T-cell acute lymphoblastic leukemia cells rely on casein kinase 2 activity. Haematologica. 2016;101:1368–1379.
- Cirigliano SM, Díaz Bessone MI, Berardi DE, et al. The synthetic peptide CIGB-300 modulates CK2-dependent signaling pathways affecting the survival and chemoresistance of non-small cell lung cancer cell lines. Cancer Cell Int. 2017;17:42.
- Sordella R, Bell DW, Haber DA, et al. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167.
- Stommel JM, Kimmelman AC, Ying H, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287–290.
- Harir N, Boudot C, Friedbichler K, et al. Oncogenic Kit controls neoplastic mast cell growth through a Stat5/PI3-kinase signaling cascade. Blood. 2008;112:2463–2473.
- Faber AC, Li D, Song Y, et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci U S A. 2009;106:19503–19508.