
ABSTRACT
Secondary brain damage plays an important role in Traumatic brain injury (TBI) and inhibition of this damage has benefit for TBI treatment. However, the pathogenesis of secondary brain damage remains largely unknown. Here, we tried to explore the influence of microRNAs (miRNAs) on neuron apoptosis and inflammatory response after TBI. Firstly, the miRNA expression profiles were analyzed in the cerebral cortex tissues from the TBI mice model (controlled cortical impact) using miRNA microarray. miR-23a-3p (miR-23a) attracted our attention as its suppressive effects on apoptosis and inflammation. The further results showed that miR-23a upregulation improved long-term neurological function, the neuron apoptosis, and inhibited neuroinflammation, whereas knockdown of miR-23a had an opposite result. Using etoposide-induced primary cortical neurons injury model, we found that miR-23a was decreased in this cell model and miR-23a overexpression-suppressed etoposide induced the activity of caspase 3 and the releases of inflammatory mediators in primary cortical neurons. Phosphatase and tensin homolog (PTEN), a well‑known regulator of the AKT/mTOR pathway, was found to be a direct target of miR‑23a in the primary cortical neurons. Most importantly, it was found that miR-23a overexpression reactivated the AKT/mTOR pathway in TBI mice model, as demonstrated by the upregulation of phosphorylated (p‑)AKT and p‑mTOR. Taken together, these data indicate that miR-23a may serve as a therapeutic target for the treatment of TBI.
Introduction
Traumatic brain injury (TBI) is a major cause of injury-induced mortality and disability, worldwide [Citation1,Citation2]. The annual TBI incidence is approximately 1.7 million reported injuries in the USA and one third of survivors experiencing long-term disability [Citation1,Citation3,Citation4], despite advancements in understanding the underlying mechanisms of TBI. A number of studies have reported that secondary brain damage is critical in the pathological progression of TBI, with marked effects on cell apoptosis and inflammatory response which can lead to further neurological dysfunction [Citation5,Citation6]. However, few studies conducted on the mechanisms of secondary brain damage. MicroRNAs (miRNAs) are single-stranded non-coding RNAs that bind to the target mRNAs and interfere in the translation process [Citation7,Citation8]. Notably, aberrantly expressed miRNAs have been found in the cerebral cortex and hippocampus of rats and mice after TBI, suggesting miRNAs have important roles in the development of TBI [Citation9,Citation10]. For example, Ge et al. showed that miR-21 was found to be upregulated in the traumatic foci after TBI and upregulation of miR-21inhibited apoptosis and promoted angiogenesis, and contributed to antagonizing TBI [Citation11]. Another study showed that miR-155 inhibition protected TBI mice from post-traumatic neuro-inflammatory responses [Citation12]. However, limited studies have focused on the role of miRNAs in the pathophysiology of secondary damage after TBI.
In the present study, the miRNA profiles in TBI mice model were determined by a miRNA microarray. Moreover, the potential roles of miR-23aon apoptosis and inflammatory response, and the underlying mechanisms were examined in the TBI mice model and cell model. Our findings suggest that miR-23a may be a therapeutic target for TBI treatment.
Materials and methods
Animals
Male C57BL/6 mice (3-month-old, 22–26 g) were housed under standard conditions (12 h light-dark cycle, 25–27ºC, ~40% humidity) with free access to food and water throughout the duration of the experiments. Animal procedures were approved by the Institutional Animal Care and Use Committee of TongRen Hospital, Shanghai Jiao Tong University School of Medicine.
Controlled cortical impact (CCI) injury
C57BL/6 mice were randomly divided into four groups, a sham group, an injury group, an injury + agomir-23 group, and an injury + antagomir-NC group. In the injury group (n = 6/group), the mice were subjected to controlled cortical impact (CCI) injury as previously described [Citation13]. Mice in the sham group underwent all surgical procedures without being exposed to CCI. In the injury + agomir-23 group/antagomir-23/agomir-NC group (n = 6/group), the mice were treated with agomir-23/antagomir-23/agomir-NC (8 mg/kg) via intracerebroventricular infusion approach at 30 minas previously reported [Citation11]. After 2 weeks, all mice were sacrificed following 10% chloride hydrate (3.0 mL/kg) anesthesia, and the cerebral cortex tissues were collected for further experiments.
miRNA microarray assay
Total RNA was isolated from cortical tissues using anmiRNeasy Mini kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s protocol. The samples were assessed using the miRCURY LNA™ Array v. 16.0 (Agilent Technologies, Santa Clara, CA, USA). The procedure and imaging processes were as described previously[Citation14].
RNA isolation and quantitative RT-PCR
Total RNA was extracted from cortical tissues and cells with the TRIzol reagent (Invitrogen, Carlsbad, CA). Reverse transcription of miR-23a and PTENwas synthesized using the miScript II RT kit and the reverse transcription kit (Invitrogen, Carlsbad, CA), respectively. miR-23a and PTENexpressions were measured using the Exiqon SYBR Green Master Mix (Exiqon, Vedbaek, Denmark) on a Light Cycler instrument (Bio-Rad, Hercules, CA, USA). The primers used for were as follows: miR-23a F 5´-CCGCCGGGATCCACGGCCGGCTGGGGTTCC-3´, R: 5´-CCGCCGAAGCTTCAGAGCTCAGGGTCGGTTGG-3´; U6 F: 5´-GCTTCGGCAGCACATATACTAAAAT-3´, R: 5´-CGCTTCACGAATTTGCGTGTCAT-3´; PTEN F: 5´-AATTTTTAAAGGCACAAGAGG-3´, R: 5´-TCCAGGAAGAGGAAAGGA-3´;GAPDH F: 5´-GTGGTGAAGACGCCAGTGGA-3´; and GAPDH R:5´-CGAGCCACATCGCTCAGACA-3´. The relative expression of each gene was calculated using the 2−∆∆Ct method[Citation15].
TUNEL staining
The Situ Cell Death Detection Kit (cat no.11684817910, Roche, Basel, Switzerland) was used to measure the apoptosis in cerebral cortex tissues as previously described[Citation16]. The cerebral cortex sections were deparaffinized and rehydrated in an alcohol gradient. After washing with PBS, the sections were stained with the TUNEL reaction mixture, followed counterstaining with DAPI (1 µg/ml). The images were captured by the fluorescence microscope (Olympus Inc., Tokyo, Japan) at 200 × magnification. The TUNEL-positive cells in 10 random fields of each section were counted for analysis.
Immunocytochemistry (Ihc)
Cerebral cortex sections were blocked with 5% BSA for 30 min at 37°C, followed by incubation overnight at 4°C with cleaved-caspase-3 antibody (1:500, cat no. #9664) and PTEN antibody (1:500, cat no. #9188). All antibodies were obtained from Cell Signaling Technology, Inc. After washing with PBS, the sections were incubated with horseradish peroxidase-conjugated secondary antibodies (cat. no. 7074; 1:5,000; Cell Signaling Technology, Inc, Danvers, MA, USA) for 1.5 h at room temperature and then with DAB for 5 min. Stained sections were photographed with a fluorescence microscope (Olympus Inc., Tokyo, Japan) at 200 × magnification. Images were analyzed using Image J software (Version1.46r, Wayne Rasband, NationalInstitute of Mental Health, USA).
Animal experiment
Brain water content measurement
The brain water content was measured using the wet-dry weight method as previously reported[Citation17]. Briefly, mice were sacrificed to remove the brain at 7 days post-injury. The brain tissues were immediately weighted to obtain its wet weight. And then, the brain tissues were dried at 104°C for 24 h to obtain the dry weight. The brain water content was calculated as follows: [(wet weight – dry weight)/wet weight] ×100%.
Lesion volume quantification
Cerebral cortex sections were stained with hematoxylin and eosin (H&E) staining kit (Beyotime Company, Jiangsu, China) according to the kit instructions. Photographs of cerebral cortex tissues were taken under a light microscopy (Olympus Inc., Tokyo, Japan) at 200 × magnification. The results were presented as a percentage of the lesion volume as compared to the contralateral hemisphere as previously reported[Citation18].
mNSS test
The modified neurological severity score (mNSS) was evaluated before and at days 1, 3, 7, and 14 after CCI as previously reported[Citation19].
Morris Water Maze (MWM) test
The spatial acquisition trial was assessed using the MWM task from 14 d to 18 d following TBI as described previously[Citation20]. The entire procedure in the pool was monitored by a tracking system (HVS Imaging, Hampton, UK) and the latency-time to reach the platform and the time spent in the goal quadrant and the average swim speed were record and calculate.
Primary cortical neurons and treatment
Mice cortical neurons (MCNs) were purchased from Thermo Fisher (cat no.A15586) and maintained in Neurobasal® Medium (cat. no. 21,103) supplemented with GlutaMAX™-I supplement (cat. no. 35,050) and B-27® Supplement (cat. no. 17,504) in 5% CO2 at 37°C. All reagents were obtained from Thermo Fisher Scientific, USA. Four hours after incubation, media was replaced with normal condition media and cells were treated with etoposide (50 μM, cat no. #2200, Cell Signaling Technology) as previously described [Citation21,Citation22].
Transfection
After mice cortical neurons cells in six-well plate grown for 6 d, miR-23a-3p mimics (50 nmol/L) and miR-23a-3p inhibitor (50 nmol/L) were transfected into cells at 37°C for 24 h, using Lipofectamine® 2000 (Invitrogen, Carlsbad, CA). Subsequently, cells were cultured in Neurobasal® Medium supplemented with GlutaMAX™-I supplement and B-27® Supplement for 24 h. Then, the cells were treated with etoposide (50μM) for 24 h and utilized in subsequent experiments.
Caspase-3 activity
After treatment, the caspase-3 activity was evaluated using a caspase-3 activity kit (Beyotime Institute of Biotechnology). The OD was then measured using a microplate reader (Model 680, Bio-Rad, Hercules, CA, USA) at an absorbance of 405 nm.
Immunofluorescence assay (IFA)
The mice cortical neurons were screened by IFA for the secretion of the desired antibodies. In brief, the cells were fixed in absolute ethyl alcohol for 30 min at room temperature (RT). After washing twice with PBS, the fixed cells were stained with cleaved-caspase-3 antibody (1:1000, cat no. #9664, Cell Signaling Technology, Inc., Danvers, MA, USA) for 1 h at RT. Then, the secondary antibody conjugated with FITC (1:100, Sigma-Aldrich, St Louis, MO) was added for 2 h in the dark. Immunostained samples were imaged by fluorescence microscopy (Olympus, Japan) at 200× magnification.
ELISA assay
IFN-α (cat no.96–416), IL-1β (cat no.96–403), IL-6 (cat no.96–407), IL-10 (cat no.96–408), and MCP1 (cat no.96–413) were measured by ELISA kit (all from Merck-Millpore, Billerica, MA, USA) according to manufacturers.
Luciferase reporter assay
pGL3-PTEN wide type (Wt) or pGL3-PTEN mutant type (mut) plasmids were co-transfected with 20 nMmiR-23a-3p mimics, 20 nM miR-23a-3p inhibitor or 20 nMmiR-NC into HEK 293T cells in 24-well plates (2 × 105/well) using Lipofectamine 2000 (Invitrogen). At 24 h post-transfection, the double luciferase activities were analyzed using the Dual-Luciferase Reporter Assay system (Promega Corporation, Madison, WI, USA). Luciferase activity was normalized to Renilla luciferase activity.
Western blot analysis
Western blot was performed as previously described[Citation23]. Briefly, 40 μg extracted protein samples were transferred onto a polyvinylidene difluoride (PVDF, MilliporeBillerica, MA, USA) membrane and then this membrane was blocked with 5% skim milk for 2 h at room temperature, followed by incubation with primary antibodies against cleaved-caspase-3 (cat. no. #9664, 1:1,000), total-caspase-3 (cat. no.#9665, 1:1,000), anti-PTEN (cat. no.#9188), AKT (cat. no.#9272, 1:1,000), phospho-AKT (Ser473; cat. no.#4060, 1:1,000), mammalian target of rapamycin (mTOR; cat. no.#2972, 1:1,000), phospho-mTOR (Ser2448; cat. no.#2971, 1:1,000) and β-actin (cat. no.#4970, 1:1,000) at 4°C overnight. All antibodies were obtained from Cell Signaling Technology, Inc. Subsequently, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies (1:2,000; cat. no. #7074; Cell Signaling Technology, Inc.) for 1 h at room temperature. The protein bands were developed using ECL kit (GE Healthcare) and blot bands were quantified with ImageJ version 1.46 (Rawak Software, Inc. Munich, Germany).
Statistical analysis
Statistical analysis was performed using GraphPad Prism (version 5.0, GraphPad Software, Inc., La Jolla, CA, USA). Data were recorded as means ± SD. Differences between groups were analyzed using one way ANOVA or Student’s t-test. A p value < 0.05 was considered significant.
Results
miR-23a was downregulated in mice post-traumatic brain injury (TBI)
To identify miRNAs associated with TBI, we performed an miRNA microarray in injured cerebral cortex tissues from injury group (n = 3) and cerebral cortex tissues from sham group (n = 3). As demonstrated in ), 23 miRNAs were up-regulated and 13 miRNAs were down-regulated in injured cerebral cortex tissues, compared with sham group. Among them, miR-23a-3p was selected for further investigation as its expression level was identified as the lowest in the injury group. Consistent with our results, a previous study also reported the downregulation of miR-23a after TBI in rat [Citation24]. Another study showed that miR-23a upregulation in mouse neuronsalleviatedhypoxia induced neuronal apoptosis [Citation25]. Additionally, two previous studies reported that miR-23a has a protective effect on several types of cells injury models [Citation26,Citation27]. However, whether miR-23a has a protective effect against the secondary injury of neurons in TBI remains to be elucidated. Therefore, the present study focused on miR-23a-3pfor further investigation.
Figure 1. TBI induces miRNAs aberrant expression in mice TBI model. (a) Heat map shows significant expressional changes of 36 miRNAs levels in mice after TBI. The color green indicates downregulation and red upregulation. (b) Based on these microarray data, the miR-23a was selected and verified by qRT-PCR in cerebral cortex tissues at different time points after TBI. Data were represented as the mean ± SD of three independent experiments. *p < 0.01, **p < 0.01 vs sham group.
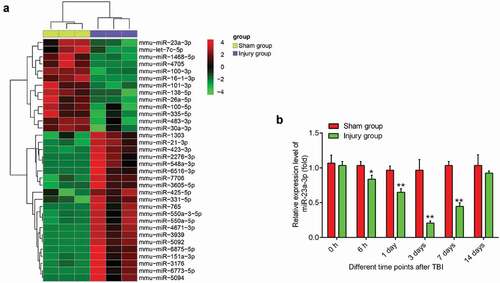
Next, we detected the expression levels of miR-23a-3p in injured cerebral cortex tissues at different time-points from 0 h to 14 d post-injury by qRT-PCR. As shown in), the expression of miR-23a-3p in the injury group was reduced 6 h post-injury, reached a plateau at 3 d post-injury, and then gradually increased at 3–14 d post-injury. These data indicated that miR-23a-3p may be important in the pathogenesis of TBI.
Upregulation of miR-23a level in cerebral cortex improves the neurons apoptosis after TBI
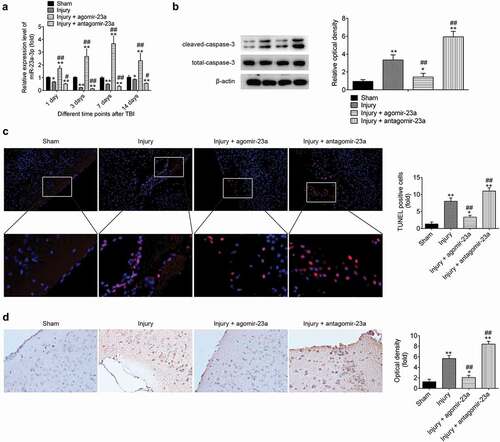
As we know, neuronal cell apoptosis has a crucial role in secondary brain damage[Citation28]. Following CCI for 15 min, miR-23a-3p agomir or antagomir was injected into mice through a single intracerebroventricular injection (CVI). As shown in ), the expression of miR-23a-3p was significantly increased (or decreased) in cerebral cortex tissues 1 d post-injury and reached the peak (or plateau) at 7 or 3 d post-injury after agomir-23a (or antagomir-23a) injection. Subsequently, the expressions of apoptosis-associated protein, cleaved-caspase-3 were measured by Western Blot, and the result showed that compared with that in sham group, the cleaved caspase-3 level was increased in injury group. However, cleaved caspase-3 expression was significantly reduced by agomir-23a and markedly enhanced by the antagomir-23a, compared with that in the injury group ()). Moreover, TUNEL staining showed that the positive number of TUNEL staining cells was significantly up-regulated compared to sham group. However, compared with the injury group, the number of TUNEL-positive-stained cells was significantly lower (or higher) in the agomir-23a (or the antagomir-23a) group ()).In addition, in the immunohistochemistry staining results, the level of cleaved caspase-3 was significantly decreased (or increased) in the agomir-23a (the antagomir-23a) group, compared to injury group ()). These findings indicated that the upregulation of miR-23a-3p in the cerebral cortex can improve TBI-induced neurons apoptosis.
Figure 2. Upregulation of miR-23a level in cerebral cortex improves the neurons apoptosis after TBI. At 15 min after injury, mice received a single intracerebroventricular injection (CVI) of miR-23aagomir or antagomir with a final concentration of 50 μM. At the scheduled time points, mice were euthanized with an overdose of 10% chloral hydrate (3 mg/kg) and cerebral cortex tissues were collected for subsequent experiments. (a) The expression of miR-23a was measured by qRT-PCR at different time points after TBI. (b) The expression levels of apoptosis-related proteins including cleaved caspase-3 and total caspase-3were detected by Western Blot. (c) TUNEL staining was used to analyze neuronal apoptosis (200× magnification). (d) Immunocytochemistry was performed to assess the expression of cleaved caspase-3 (200× magnification). Data were represented as the mean ± SD of three independent experiments. *p < 0.01, **p < 0.01 vs sham group, ##p < 0.01 vs injury group.
Upregulation of miR-23a level in cerebral cortex improves the inflammatory response after TBI
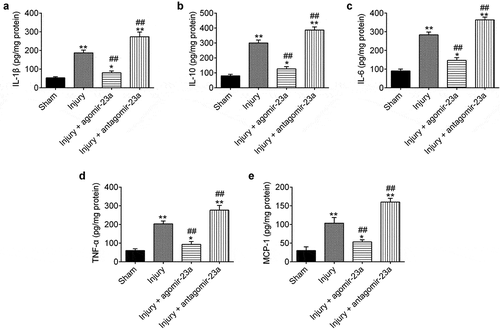
It is now well accepted that inhibition of inflammation can improve the prognosis of TBI [Citation29–Citation31]. Of note, miR-23a-3p has been reported to possess anti-inflammatory activity in different injuries [Citation27,Citation32]. To determine the impact of miR-23a-3p on the inflammatory response in TBI mice, we detected the levels of pro-inflammatory cytokines, including IL-1β, IL-10, IL-6, TNF-α, and MCP1. As shown in –), the levels of pro-inflammatory cytokines were significantly higher in the injury group of mice compared to the sham group, indicating that TBI leads to inflammation in the cerebral cortex. Meanwhile, compared with the injury group, we observed that the expressions of these pro-inflammatory cytokines induced by TBI were markedly reduced in the agomir-23a group, while their expressions were evidently enhanced in the antagomir-23a group. The results suggest that the upregulation of miR-23a-3p in the cerebral cortex can alleviate TBI-induced inflammatory response.
Figure 3. Upregulation of miR-23a level in cerebral cortex improves the neurons apoptosis after TBI. At 15 min after injury, mice received a single intracerebroventricular injection (CVI) of miR-23a agomir or antagomir with a final concentration of 50 μM. At the scheduled time points, mice were euthanized with an overdose of 10% chloral hydrate (3 mg/kg). Brain tissue in the cerebral cortex around the injured area was collected and homogenized. The homogenates were centrifuged at 4°C at 12,000 × g for 15 min, and supernatants were collected carefully. IFN-α (a), IL-1β (b), IL-6 (c), IL-10 (d) and MCP1 (e) in the supernatant were measured by ELISA kit. Data were represented as the mean ± SD of three independent experiments. *p < 0.01, **p < 0.01 vs sham group; ##p < 0.01 vs injury group.
Upregulation of miR-23a level in cerebral cortex improved the neurological outcome after TBI
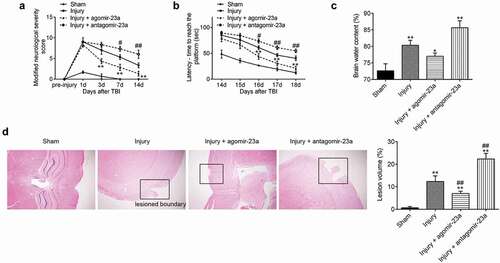
As mentioned above, miR-23a plays a suppressive role in neurons apoptosis and inflammatory response in TBI mice. Therefore, we asked whether exogenous miR-23a-3p could improve neurological dysfunctions after TBI. As shown in ), neurological function was gradually improved in four groups during the evaluation period, as demonstrated by mNSS assay. Notably, compared with the injury group, the better functional recovery after 3 days was observed in the agomir-23a group, while antagomir-23a exacerbated TBI induced the functional deficit. Additionally, the MWM test was performed to assess mice spatial cognition in each group. Compared with the injury group, escape latency was improved after agomir-23a treatment, while impaired by antagomir-23a ()) (day 16, p < 0.05; days 17 and 18, p < 0.01). It was also observed that the brain edema and lesion volume were notably increased in TBI mice, as determined by brain water content and HE staining. However, the brain edema and lesion volume were attenuated by agomir-23a, whereas antagomir-23a treatment aggravated brain edema and lesion volume as compared to the injury group (,)). Collectively, these results suggested that miR-23a upregulation protected rat neuroprotection against brain injury.
Figure 4. Upregulation of miR-23a level in cerebral cortex improved the neurological outcome after TBI. At 15 min after injury, mice received a single intracerebroventricular injection (CVI) of miR-23a agomir or antagomir with a final concentration of 50 μM. The long-term neurological function evaluated by the mNSS test (a) (n = 6) and Morris Water Mass test (b) (n = 6). (c) The quantitative data of brain water content measurement for lesioned cerebral hemisphere at 7d post-injury (d) (n = 6). (d) The H&E staining and the quantitative data of lesion volume at 7 d post-injury (n = 6)(200× magnification). Data were represented as the mean ± SD of three independent experiments. *p < 0.01, **p < 0.01 vs sham group; #p < 0.05, ##p < 0.01 vs injury group.
Upregulation of miR-23a level in primary cortical neurons suppresses etoposide induced the apoptosis and inflammatory response
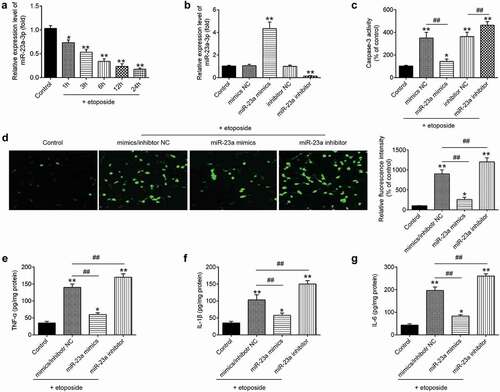
It is well known that etoposide-treated cortical neurons model is widely used to mimic pathological conditions of TBIin vitro[Citation33]. Following the treatment of the cortical neurons with etoposide (50μM), levels of miR-23a were analyzed at different time points by qRT-PCR. The expression of miR-23a was significantly reduced compared to the control group (untreated cells), and this inhibitory effect was time-dependent ()). To examine the role of miR-23a in TBI, miR-23a mimics/inhibitor was transfected into the primary cortical neuron, followed by 50 μM etoposide treatment. The transfection efficiency of miR-23a was detected by qRT-PCR. Compared with mimics NC/inhibitor NC, miR-23a obviously increased/decreased in neurons transfected with miR-23a mimics/miR-23a inhibitor ()). The biological consequences of miR-23a in the regulation of neuron apoptosis and inflammation were evaluated. As expected, miR-23a overexpression attenuatedetoposide-induced caspase-3 activity and the protein expression of cleaved caspase-3 in primary cortical neurons. Conversely, treatment with miR-23a inhibitor enhanced etoposide-induced neuronal apoptosis ()). In addition, it was also observed that overexpression of miR-23a inhibited etoposide-induced the secretion of TNF-α, IL-1β, and IL-6, while knockdown of miR-23a enhanced the secretion of these pro-inflammatory cytokines. Together, these data demonstrate that miR-23a upregulation exerts anti-apoptotic and anti-inflammatory effects in the TBI cell model.
Figure 5. Upregulation of miR-23a level in primary cortical neurons suppresses etoposide induced the apoptosis and inflammatory response. (a) Levels of miR-23a were analyzed by qRT-PCR at different time points after etoposide treatment. (b) Primary cortical neurons were transfected with miR-23a mimics, miR-23a inhibitor, and 4 h later treated with 50 μM etoposide. The expression levels of miR-23a were analyzed by qRT-PCR. (c) The caspase-3 activity was measured by caspase-3 activity kit. (d) The expression of cleaved caspase-3 was detected by immunofluorescence assay(200× magnification). (e-g) IFN-α (e), IL-1β (f) and IL-6 (g) in cell supernatant were measured by the ELISA kit. Data were represented as the mean ± SD of three independent experiments. *p < 0.01, **p < 0.01 vs control group; ##p < 0.01 vs etoposide group.
PTEN is a direct target of miR-23a in primary cortical neurons
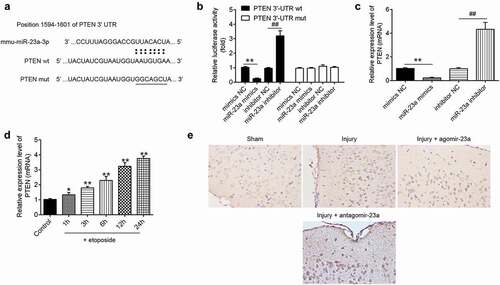
To determine the mechanism function of miR-23a in TBI, we predicted the target of miR-23a through bioinformatics prediction (TargetScan 7.0 and miRanda). As shown in ), PTEN was identified as a potential target gene of miR-23a. To validate the targeting interaction between PTEN and miR-23a, luciferase reporter assay was performed. It was found that the miR-23a mimics significantly inhibited the luciferase activity of the PTEN-3´UTR wt and that the miR-23a inhibitor transfection resulted in a significant increase in luciferase activity; however, no changes were observed in the cells co-transfection of PTEN 3ʹ-UTR-mut with either miR-23a mimics or inhibitor ()). In addition, qRT-PCR analyses showed that miR-23a negatively regulated the mRNA expression levels of PTEN in primary cortical neurons ()). It was also found that the mRNA expression levels of PTEN were significantly increased in etoposide-treated primary cortical neurons, and this effect was time-dependent ()). Consistent with the results in vitro, the protein expression level of PTEN was significantly decreased (or increased) after the agomir-23a (the antagomir-23a) injection, compared with the injury group in TBI mice ()). These findings indicate that miR-23a directly targeted PTEN and suppresses its expression in vitro and in vivo.
Figure 6. PTEN is a direct target of miR-23a in primary cortical neurons. A putative miR-23a-binding site exists in the 3ʹ-UTR of PTEN mRNA and 7-point mutations were generated in the binding site. (b) The relative luciferase activity of PTEN wild type or mutant 3ʹ-UTR in HEK 293 cells following transfection with the miR-23a mimics and miR-23a inhibitor. Data were represented as the mean ± SD of three independent experiments. **p < 0.01 vs mimics NC group; ##p < 0.01 vs inhibitor NC group. (c) Relative PTEN mRNA level was detected using qRT-PCR after transfection with miR-23a mimics and miR-23a inhibitor. Data were represented as the mean ± SD of three independent experiments. **p < 0.01 vs mimics NC group; ##p < 0.01 vs inhibitor NC group. (d) Primary cortical neurons were transfected with miR-23a mimics, miR-23a inhibitor, and 4 h later treated with 50 μM etoposide. The expression levels of PTEN were analyzed by qRT-PCR. Data were represented as the mean ± SD of three independent experiments. *p < 0.05, **p < 0.01 vs control group. (e) In the TBI mice model, at 15 min after injury, mice received a single intracerebroventricular injection (CVI) of miR-23a agomir or antagomir with a final concentration of 50 μM. The expression of PTEN in cerebral cortex tissues was measured by Immunocytochemistry after TBI (200× magnification).
miR-23a upregulation reactivated the PTEN/AKT/mTOR signaling pathway
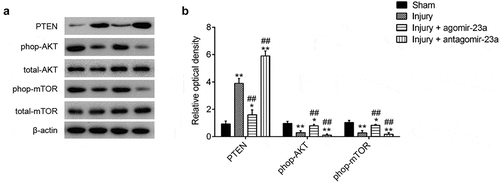
The previous study has proven reactivation of the AKT/mTOR signaling pathway contributes to the improvement of cognitive outcome in brain damage in vivo [Citation34–Citation36]. Given that PTEN is a negative regulator of the AKT/mTOR pathway [Citation37,Citation38], we speculated whether miR-23a improved brain injury via controlling the activation of PTEN/AKT/mTOR pathway in TBImice model. In the injury group, the expression level of PTEN was significantly increased, but phop-AKT and phop-mTOR were markedly decreased compared to the sham group. No alterations in total Akt and total mTOR were observed between sham and TBI group, suggest that TBI can block the activation of PTEN/AKT/mTOR pathway. Following agomir-23a and antagomir-23a treatment in TBI mice, we found that the overexpression of miR-23a reactivated this pathway inhibited by TBI, whereas miR-23a inhibition had an opposite effect ()). These findings suggested that the upregulation of miR-23a reactivates this pathway through downregulating PTEN in the TBI mice model.
Figure 7. miR-23a upregulation reactivated the PTEN/AKT/mTOR signaling pathway. At 15 min after injury, mice received a single intracerebroventricular injection (CVI) of miR-23a agomir or antagomir with a final concentration of 50 μM. At 7d post injection, mice were euthanized with an overdose of 10% chloral hydrate (3 mg/kg) and cerebral cortex tissues were collected for Western Blot. (a) The immunoblotting and (b) quantitative data of PTEN/AKT/mTOR signaling pathway-related molecules (PTEN, p-AKT, and p-mTOR) acquired from the cerebral cortex tissues. Data were represented as the mean ± SD of three independent experiments. *p < 0.01, **p < 0.01 vs sham group; ##p < 0.01 vs injury group.
Discussion
In the present study, miR-23a was downregulated in the cerebral cortex tissues from TBI mice and etoposide-induced primary cortical neurons. Upregulation of miR-23a level improves the neurological function recovery of TBI mice through attenuating cell apoptosis and inflammatory response. Moreover, we demonstrate that PTEN/Akt/mTOR signaling mediated the protection of miR-23a against TBI in mice. Our findings suggest that miR-23a may serve as a potential therapeutic target for TBI.
Growing evidence has been reported that recently discovered miRNAs are essential mediators in neural development and diverse neurological diseases including TBI [Citation39–Citation42]. Redell et al. reported that miR-21 was significantly increased in the hippocampus after TBI and elevated miR-21 expression improved brain injury via targeting programmed cell death 4 (PDCD4)[Citation43]. The roles of miR-21 in the pathogenesis of TBI have also reviewed by Ji et al. [Citation44]. Chen et al. found that elevated expression of miR-302 improved TBI by inhibiting phosphorylation of connexin43 via ERK signaling[Citation45]. Sabirzhanov et al. showed that miR-711 knockdown protected neurons against TBI-induced celldeath [Citation46]. Sun et al. found that elevated miR-144 promoted cognitive impairments after TBI through suppressing of ADAM10 expression[Citation47]. Although identification of a number of miRNAs involved in TBI has been reported, the functions and mechanisms of miRNAs in TBI remain to be elucidated. In this study, using a miRNA microarray, it was found that miR-23a was the most significantly downregulated microRNA post-injury, and the reduction lasted for up to 14 days post-injury. These data suggest that miR-23a may play an important role in the pathogenesis of secondary brain damage after TBI.
It is well known that cellular apoptosis and inflammation in the brain are induced within hours to days after the secondary brain damage [Citation11,Citation48]. Previous studies reported that if not properly controlled, apoptosis and inflammatory response can aggravate tissue damage and result in neurological deficiency [Citation49,Citation50]. To explore the miR-23a effects on inflammatory response and apoptosis in TBI, the mice treated intracerebroventricularly with agomir-23a and antagomir-23a in the TBI model. Our findings showed that upregulation miR-23a improved the neurological function, inhibited apoptotic cell death and suppressed inflammatory response induced by TBI in mice. Furthermore, we performed in vitro experiments to explore the therapeutic effect and mechanism of miR-23a upregulation on TBI. Using an etoposide-induced TBI cell model, we found that miR-23a was markedly downregulated in etoposide-induced TBI cell model. Moreover, overexpression of miR-23a significantly reduced etoposide-induced neuronal cell apoptosis and inflammatory response, whereas knockdown of miR-23a has an opposite result. These data indicated that miR-23a protects cellular apoptosis and inflammation in the TBI model.
PTEN is a negative regulator of the AKT/mTOR signal pathway and inhibition of PTEN activity has become a potential target for the improvement of neurological functional recovery following an injury to the nervous system [Citation51,Citation52]. In TBI, inhibition of PTEN promoted the recovery of neurological function by decreasing apoptosis [Citation53]. Ding et al. also reported thatPTEN inhibition reduced neuron apoptosis and improved neurological function recovery in TBI rats [Citation54].In addition, Liu et al. found evidence of bpV(pic), a commercially available inhibitor of PTEN, regulated neuroinflammation through AKT activation and NF-κB p65 inhibition in TBI rats [Citation55]. Interestingly, previous studies indicated that miR-23a enhanced the osteosarcoma cell migration and invasion by targeting PTEN [Citation56]. In this study, PTEN was identified as a target of miR-23a and its expression was negatively regulated by miR-23a in vitro and in vivo. Additionally, it was observed that PTEN was significantly increased the cerebral cortex of TBI mice model and etoposide-induced primary cortical neurons model. All data suggested that miR-23a may have the protection effect in TBI via targeting PTEN expression.
It is commonly known that the AKT/mTOR signaling pathway exerts powerful effects on the apoptosis and inflammatory response after central nervous system injury [Citation57,Citation58], in addition to its role in tumor cells. For example, Xu et al. found that the AKT/mTOR activation has been demonstrated to mediate a protective effect in a mouse model of TBI through suppressing neurons apoptosis[Citation59]. Ma et al. showed that miR-22 attenuated neuronal cell injury and apoptosis in a cell model of TBI through the PTEN/PI3K/AKT signaling pathway[Citation60]. In addition, Xu et al. reported that LY294002, an inhibitor of the PI3K/AKT pathway attenuated the inhibitory effect of histone deacetylase 1 (HDAC1) on the neuroinflammation of TBI mice[Citation61]. Zhang et al. showed that the PI3K/AKT pathway mediated the anti-inflammatory effect of Isoliquiritigenin (ILG) for TBI in mice[Citation62]. Notably, a recent study uncovered that miR-23a modulated PTEN/AKT/mTOR pathway to promote hepatic fibrosis[Citation63]. Given the association between PTEN and miR-23a, it was hypothesized that miR-23a improved brain injury by mediating the PTEN/AKT/mTOR pathway. Our results showed that the levels of phop-AKT and phop-mTOR were significantly decreased, while the expression of PTEN was increased after TBI, suggesting that TBI leads to the inactivation ofPTEN/AKT/mTOR pathway. However, agomir-23a or antagomir-23a treatment significantly increased (or decreased) the levels of phop-AKT and phop-mTOR in injured cerebral cortex compared with injury group but decreased (or increased) PTEN level. These data indicated that upregulation of miR-23a reactivates AKT/mTOR signaling pathway blocked by TBI in mice.
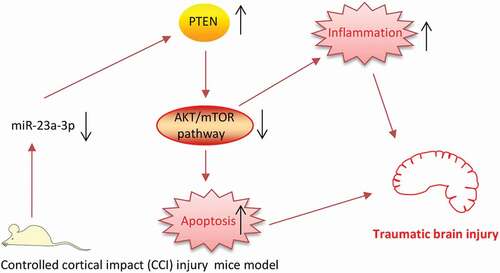
In conclusion, we demonstrated that the upregulation of miR-23a can improve the neurological outcome after TBI in mice by inhibiting neurons apoptosis and inflammatory response via reactivating PTEN/AKT/mTOR signaling pathway (). Our results indicated that the upregulation of miR-23a may be a promising strategy for TBI therapies.
Figure 8. Schematic diagram of the signaling pathway in which miR-23a protects against TBI.miR-23a was downregulated in the TBI mice model and downregulated miR-23a inactivated PTEN/AKT/mTOR signaling pathway through upregulating PTEN, thus promoting apoptosis and inflammatory response, finally leading to TBI.
However, there are still some limitations in the current study. For example, PTEN may not be the only target for the miR-23a. Using bioinformatics prediction, we also found other potential targets such as: apoptosis-related proteins including caspase 7 and X-linked inhibitor of apoptosis (XIAP) and inflammation-related protein including leucine-rich repeats and immunoglobulin-like domains 2 (LRIG2) and immunoglobulin superfamily, member 10 (IGSF10). These potential targets may also participate in the role of MicroRNA-23a-3p on apoptosis and inflammatory response in TBI mice. This limitation will be resolved in future experiments.
Authors’ contribution
Wei Xu was responsible for the main conceive of the study and the draft of the manuscript. Zhikun Li, Ruijun Xu, Xiaodong Zhu, Yifan Li, and Yi Wang helped to design the study and performed the statistical analysis. Zhikun Li and Ruijun Xu helped to revise the manuscript and participated in its design. All authors have read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–378.
- León-Carrión J, Del Rosario Domínguez-Morales M,Y, Martín JMB, et al. Epidemiology of traumatic brain injury and subarachnoid hemorrhage. Pituitary. 2005;8:197–202.
- Daneshvar DH, Nowinski CJ, McKee AC, et al. The epidemiology of sport-related concussion. Clin Sports Med. 2011;30: 1–17. vii.
- Taylor CA, Bell JM, Breiding MJ, et al. Traumatic brain injury-related emergency department visits, hospitalizations, and deaths - United States, 2007 and 2013. Morbidity and mortality weekly report Surveillance summaries 2017; 66:1–16.
- Liang J, Wu S, Xie W, et al. Ketamine ameliorates oxidative stress-induced apoptosis in experimental traumatic brain injury via the Nrf2 pathway. Drug Des Devel Ther. 2018;12:845–853.
- Jassam YN, Izzy S, Whalen M, et al. Neuroimmunology of traumatic brain injury: time for a paradigm shift. Neuron. 2017;95:1246–1265.
- Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–714.
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233.
- Lei P, Li Y, Chen X, et al. Microarray based analysis of microRNA expression in rat cerebral cortex after traumatic brain injury. Brain Res. 2009;1284:191–201.
- Redell JB, Liu Y, Dash PK. Traumatic brain injury alters expression of hippocampal microRNAs: potential regulators of multiple pathophysiological processes. J Neurosci Res. 2009;87:1435–1448.
- Ge XT, Lei P, Wang HC, et al. miR-21 improves the neurological outcome after traumatic brain injury in rats. Sci Rep. 2014;4:6718.
- Henry RJ, Doran SJ, Barrett JP, et al. Inhibition of miR-155 limits neuroinflammation and improves functional recovery after experimental traumatic brain injury in mice. Neurotherapeutics. 2019;16:216–230.
- Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999;160:226–234.
- Hu JZ, Huang JH, Zeng L, et al. Anti-apoptotic effect of microRNA-21 after contusion spinal cord injury in rats. J Neurotrauma. 2013;30:1349–1360.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408.
- Cuello-Carrion FD, Ciocca DR. Improved detection of apoptotic cells using a modified in situ TUNEL technique. J Histochem Cytochem. 1999;47:837–839.
- Yan EB, Hellewell SC, Bellander BM, et al. Post-traumatic hypoxia exacerbates neurological deficit, neuroinflammation and cerebral metabolism in rats with diffuse traumatic brain injury. J Neuroinflammation. 2011;8:147.
- Xiong Y, Zhang Y, Mahmood A, et al. Neuroprotective and neurorestorative effects of thymosin beta4 treatment initiated 6 hours after traumatic brain injury in rats. J Neurosurg. 2012;116:1081–1092.
- Chen J, Sanberg PR, Li Y, et al. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke. 2001;32:2682–2688.
- Chen X, Lin YP, Wang D, et al. Dexamethasone exacerbates spatial acquisition deficits after traumatic brain injury in rats. Neurol Res. 2010;32:1097–1102.
- Di Giovanni S, Movsesyan V, Ahmed F, et al. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci U.S.A 2005; 102:8333–8338.
- Yakovlev AG, Di X, Movsesyan V, et al. Presence of DNA fragmentation and lack of neuroprotective effect in DFF45 knockout mice subjected to traumatic brain injury. Mol Med. 2001;7:205–216.
- Han Z, Zhou X, Li S, et al. Inhibition of miR-23a increases the sensitivity of lung cancer stem cells to erlotinib through PTEN/PI3K/Akt pathway. Oncol Rep. 2017;38:3064–3070.
- Sabirzhanov B, Zhao Z, Stoica BA, et al. Downregulation of miR-23a and miR-27a following experimental traumatic brain injury induces neuronal cell death through activation of proapoptotic Bcl-2 proteins. J Neurosci. 2014;34:10055–10071.
- Chen Q, Xu J, Li L, et al. MicroRNA-23a/b and microRNA-27a/b suppress Apaf-1 protein and alleviate hypoxia-induced neuronal apoptosis. Cell Death Dis. 2014;5:e1132.
- Lin H, Qian J, Castillo AC, et al. Effect of miR-23 on oxidant-induced injury in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2011;52:6308–6314.
- Kou Y, Zheng WT, Zhang YR. Inhibition of miR-23 protects myocardial function from ischemia-reperfusion injury through restoration of glutamine metabolism. Eur Rev Med Pharmacol Sci. 2016;20:4286–4293.
- Li Z, Wang B, Kan Z, et al. Progesterone increases circulating endothelial progenitor cells and induces neural regeneration after traumatic brain injury in aged rats. J Neurotrauma. 2012;29:343–353.
- Wei HL, Ma SQ, Li CX. Deficiency of unc-51 like kinase 1 (Ulk1) protects against mice traumatic brain injury (TBI) by suppression of p38 and JNK pathway. Biochem Biophys Res Commun. 2018;503:467–473.
- Eme R. Neurobehavioral outcomes of mild traumatic brain injury: a mini review. Brain Sci. 2017. 7.
- Brogan RJ, Kontojannis V, Garara B, et al. Near-infrared spectroscopy (NIRS) to detect traumatic intracranial haematoma: A systematic review and meta-analysis. Brain Inj. 2017;31:581–588.
- Zhao H, Tao Z, Wang R, et al. MicroRNA-23a-3p attenuates oxidative stress injury in a mouse model of focal cerebral ischemia-reperfusion. Brain Res. 2014;1592:65–72.
- Ma Y, Liu W, Wang Y, et al. VEGF protects rat cortical neurons from mechanical trauma injury induced apoptosis via the MEK/ERK pathway. Brain Res Bull. 2011;86:441–446.
- Chen A, Xiong LJ, Tong Y, et al. Neuroprotective effect of brain-derived neurotrophic factor mediated by autophagy through the PI3K/Akt/mTOR pathway. Mol Med Rep. 2013;8:1011–1016.
- Zhu X, Park J, Golinski J, et al. Role of Akt and mammalian target of rapamycin in functional outcome after concussive brain injury in mice. J Cereb Blood Flow and Metab. 2014;34:1531–1539.
- Park J, Zhang J, Qiu J, et al. Combination therapy targeting Akt and mammalian target of rapamycin improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow and Metab. 2012;32:330–340.
- Blanco-Aparicio C, Renner O, Leal JF, et al. PTEN, more than the AKT pathway. Carcinogenesis. 2007;28:1379–1386.
- Hill R, Wu H. PTEN, stem cells, and cancer stem cells. J Biol Chem. 2009;284:11755–11759.
- Fiore R, Siegel G, Schratt G. MicroRNA function in neuronal development, plasticity and disease. Biochim Biophys Acta. 2008;1779:471–478.
- Wang WX, Rajeev BW, Stromberg AJ, et al. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J Neurosci. 2008;28:1213–1223.
- Wang WX, Visavadiya NP, Pandya JD, et al. Mitochondria-associated microRNAs in rat hippocampus following traumatic brain injury. Exp Neurol. 2015;265:84–93.
- Pan YB, Sun ZL, Feng DF. The role of MicroRNA in traumatic brain injury. Neuroscience. 2017;367:189–199.
- Redell JB, Zhao J, Dash PK. Altered expression of miRNA-21 and its targets in the hippocampus after traumatic brain injury. J Neurosci Res. 2011;89:212–221.
- Ji W, Jiao J, Cheng C, et al. MicroRNA-21 in the pathogenesis of traumatic brain injury. Neurochem Res. 2018;43:1863–1868.
- Chen W, Zhao L, Zhang J, et al. Elevated expression of miR-302 cluster improves traumatic brain injury by inhibiting phosphorylation of connexin43 via ERK signaling. J Chem Neuroanat. 2019;99:1–8.
- Sabirzhanov B, Stoica BA, Zhao Z, et al. miR-711 upregulation induces neuronal cell death after traumatic brain injury. Cell Death Differ. 2016;23:654–668.
- Sun L, Zhao M, Zhang J, et al. MiR-144 promotes beta-amyloid accumulation-induced cognitive impairments by targeting ADAM10 following traumatic brain injury. Oncotarget. 2017;8:59181–59203.
- Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7:22–30.
- Chen X, Zhang KL, Yang SY, et al. Glucocorticoids aggravate retrograde memory deficiency associated with traumatic brain injury in rats. J Neurotrauma. 2009;26:253–260.
- Blennow K, Hardy J, Zetterberg H. The neuropathology and neurobiology of traumatic brain injury. Neuron. 2012;76:886–899.
- Backman SA, Stambolic V, Suzuki A, et al. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. 2001;29:396–403.
- Park KK, Liu K, Hu Y, et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966.
- Wang G, Shi Y, Jiang X, et al. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3beta/PTEN/Akt axis. Proc Natl Acad Sci U.S.A 2015; 112:2853–2858.
- Ding J, Guo J, Yuan Q, et al. Inhibition of phosphatase and tensin homolog deleted on chromosome 10 decreases rat cortical neuron injury and blood-brain barrier permeability, and improves neurological functional recovery in traumatic brain injury model. PloS One. 2013;8:e80429.
- Liu R, Liao XY, Tang JC, et al. BpV(pic) confers neuroprotection by inhibiting M1 microglial polarization and MCP-1 expression in rat traumatic brain injury. Mol Immunol. 2019;112:30–39.
- Tian K, Di R, Wang L. MicroRNA-23a enhances migration and invasion through PTEN in osteosarcoma. Cancer Gene Ther. 2015;22:351–359.
- Park KK, Liu K, Hu Y, et al. PTEN/mTOR and axon regeneration. Exp Neurol. 2010;223:45–50.
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318.
- Xu K, Wu F, Xu K, et al. NaHS restores mitochondrial function and inhibits autophagy by activating the PI3K/Akt/mTOR signalling pathway to improve functional recovery after traumatic brain injury. Chem Biol Interact. 2018;286:96–105.
- Ma J, Shui S, Han X, et al. microRNA-22 attenuates neuronal cell apoptosis in a cell model of traumatic brain injury. Am J Transl Res. 2016;8:1895–1902.
- Xu L, Xing Q, Huang T, et al. HDAC1 silence promotes neuroprotective effects of human umbilical cord-derived mesenchymal stem cells in a mouse model of traumatic brain injury via PI3K/AKT pathway. Front Cell Neurosci. 2018;12:498.
- Zhang M, Wu Y, Xie L, et al. Isoliquiritigenin protects against bloodbrain barrier damage and inhibits the secretion of pro-inflammatory cytokines in mice after traumatic brain injury. Int Immunopharmacol. 2018;65:64–75.
- Dong Z, Li S, Wang X, et al. lncRNA GAS5 restrains CCl4-induced hepatic fibrosis by targeting miR-23a through the PTEN/PI3K/Akt signaling pathway. Am J Physiol Gastrointest Liver Physiol. 2019;316:G539–G50.