
ABSTRACT
Spinocerebellar ataxias (SCA) are a genetically heterogeneous family of cerebellar neurodegenerative diseases characterized by abnormal firing of Purkinje neurons and degeneration. We recently demonstrated the slowed firing rates seen in several SCAs share a common etiology of hyper-activation of the Src family of non-receptor tyrosine kinases (SFKs). However, the lack of clinically available neuroactive SFK inhibitors lead us to investigate alternative mechanisms to modulate SFK activity. Previous studies demonstrate that SFK activity can be enhanced by the removal of inhibitory phospho-marks by receptor-protein-tyrosine phosphatases (RPTPs). In this Extra View we show that MTSS1 inhibits SFK activity through the binding and inhibition of a subset of the RPTP family members, and lowering RPTP activity in cerebellar slices with peptide inhibitors increases the suppressed Purkinje neuron basal firing rates seen in two different SCA models. Together these results identify RPTPs as novel effectors of Purkinje neuron basal firing, extending the MTSS1/SFK regulatory circuit we previously described and expanding the therapeutic targets for SCA patients.
Introduction
Cells organize kinase and phosphatase complexes within distinct micro-domains on their membranes to locally control responses to extrinsic signals. One organizer of these micro-domains is the I-BAR family of proteins who both sense, and induce, negative membrane curvature before assembling relevant regulatory complexes to control the kinase/phosphatase output. Mtss1, a founding member of the I-BAR family, acts as a docking site for receptor protein tyrosine phosphatases (RPTP) to locally control the activity of Src family non-receptor tyrosine kinases (SFKs) [Citation1-Citation4]. This local kinase regulation gives rise to a variety of cell biological phenotypes including local changes in the actin cytoskeleton [Citation5], as well as receptor internalization and recycling [Citation6]. In highly polarize cells, such as neurons, Mtss1 restraint of SFKs stabilizes dendritic spines [Citation7,Citation8].
The cerebellum is an evolutionarily conserved brain region with well-defined circuits that control movement and motor learning. A prominent feature of the cerebellum is that several distinct neuronal inputs are integrated by Purkinje neurons, who are the sole output. Spinocerebellar ataxias are a genetically heterogeneous family of cerebellar wasting disorders characterized by motor control problems, abnormal eye movements, and impairments in motor learning [Citation9]. Causative SCA loci are numbered in order of discovery and affect many compartments in the cell, ranging from ion channels at the cell membrane [Citation10] to transcription factors [Citation11] to proteolytic machinery [Citation12] or ER calcium flux [Citation13,Citation14]. Defects in any of these diverse pathways lead to a common endophenotype: Purkinje neuron dysfunction and degeneration. While the logic of the diverse array of loci appeared opaque, we recently demonstrated that multiple SCA models, originating from possible defects in transcription (SCA1), translation (SCA2) and cytoskeleton (SCA5, Mtss1), converge on preventing hyperactivity of the Src family of non-receptor tyrosine kinases [Citation7].
Mammals have 9 members of the Src family of non-receptor tyrosine kinases who have partially overlapping patterns of expression and activity, which may help mask identification of their individual functions (reviewed in [Citation15]). SFKs play essential roles in cell polarity and cytoskeletal organization as well fundamental cell processes including protein translation [Citation16,Citation17]. In the nervous system SFKs underlie the induction of long term potentiation(LTP) [Citation18], a synaptic correlate of learning, and are hyper-activated in a variety of neurodegenerative disorders associated with reduced synapse density including SCA1 and Alzheimer disease [Citation19,Citation20]. This suggests the localization and control of tyrosine kinase activity, within neurons, can help drive neurodegenerative disorders and represents a intriguing new therapeutic target. While we have shown that small molecular SFK inhibitors can ameliorate the clinical severity of the ataxia phenotype, clinically available drugs lack favorable CNS biodistribution [Citation21], and cause myeloproliferative defects preventing further clinical development to treat SCA.
By contrast, RPTPs that control SFK activation remain attractive therapeutic targets. The RPTP super family is divided into 8 subtypes [Citation22], where type IIa members LAR, PTPRS and PTPRD are strongly implicated in nervous system function, including synaptic density [Citation23], LTP [Citation24], spatial memory [Citation25] and neurofibrillary tangle formation for a variety of dementias [Citation26]. Type IIa RPTPs are characterized by dual catalytic domains where the membrane proximal D1 domain removes phosphates from substrates while the membrane-distal D2 domain has little activity alone, rather it controls dimerization and D1 catalytic function [Citation27,Citation28]. Exogenous expression of the PTPRS wedge domain that links the D1 and D2 catalytic sites is sufficient to reduce PTPRS function [Citation29], establishing a new class of specific RPTP inhibitor.
Here we provide proof of concept that RPTP inhibitors correct cerebellar Purkinje neuron firing rates, leading to the possibility of neuron survival, and amelioration of the SCA phenotype.
Materials and methods
Cell culture
MB55 mouse medulloblastoma cells [Citation30] were maintained in floating spheres in DMEM:F12 + B27 supplement. For ISP treatment cells were dissociated with accutase and plated on 1% matrigel. For ISP treatment cells were incubated in increasing concentrations overnight. COS-7 cells were maintained in DMEM+10% FBS and transfected with Fugene 6.
Co-IPs
GST-fusions encoding the intracellular D2 phosphatase domains of human CD45 (NM_002838 bp 2887–3831), LAR (AB177857 bp 5228–6014), PTPRS (NM_002850.3 bp 5290–6012), PTPRD (AB211400 bp 5667–6450), PTPRG (L09247 bp 3614–4300), PTPRN (BC070053 bp 2081–2926), were cloned into pGEX expression vectors and purified from bacteria using glutathion-sepharose beads then incubated with COS-7 cell lysate expressing Myc tagged human MTSS1. Protein complexes bound to beads were washed 3 times in PBS+0.5% NP40 before denaturing western blot, and probing with mouse anti-Myc (Sigma 5546) before stripping and reprobing with mouse anti-GST (Sigma G1160).
TAT-ISP peptide
TAT-ISP was synthesized by Genscript. Sequence: GRKKRRQRRRCDMAEHELADHIERLKANDNLKFSQEYE-amidation
Purkinje cell recordings from cerebellar slices
All experimental procedures were approved by Stanford University IACUC. Purkinje cell recordings from cerebellar slices was performed as in our previous publication [Citation7] with the following changes for TAT-ISP: Mice were injected with 50μg TAT-ISP the night before harvest, then cerebellar slices were bathed in ACSF supplemented with 2μM TAT-ISP during cutting and recording. MIMEX15 mice were characterized in [Citation7], and ATXN2Q127 mice were characterized in [Citation31].
Western blots
Cells or tissues were lysed in RIPA buffer supplemented with Roche complete-mini protease and Pierce phosphatase inhibitors. Samples were normalized with BCA assay (Pierce) and 30μg total protein was run on Novex 4–12% gels. Active SFK-Y416 (CST 6943), β-Actin (Sigma A1978) primary antibodies were detected by IR-dye conjugated secondaries and imaged with a LiCor scanner.
Results
MTSS1 binds specific RPTP family members
Because current SFK inhibitors lack favorable CNS biodistribution [Citation21] we explored the possibility that RPTP inhibitors would demonstrate efficacy in our MIMEX15 and ATXN2Q127 SCA models. The reciprocal interaction between MTSS1 and Type IIa-family member RPTPD controls membrane localization [Citation3] as well as the ability to remove inhibitory phospho-marks from SFKs [Citation1]. To determine whether MTSS1 specifically binds individual RPTPs expressed in the cerebellum, we performed co-precipitation experiments. We purified GST-fusion constructs of the intracellular domain of 5 of the 8 RPTP subtypes [Citation22]: CD45 (type 1/6), PTPRF (IIa), PTPRS (IIa), PTPRD (IIa), PTPRA (type 4), PTPRG (type 5), PTPRN (type 8), then incubated with recombinant MTSS1 from COS-7 cell-lysate. We observed specific interactions between MTSS1 and type IIa family members PTPRS and PTPRD with weak interaction with PTPRF (LAR) ()). Interestingly, these type II RPTP family members are also highly expressed in the cerebellum [Citation32–Citation35] and associated with synapse formation in other brain regions [Citation23,Citation24].
Figure 1. MTSS1 binds specific RPTP family members. (a) We purified GST-fusions of the intracellular domain of CD45 (type 1), LAR (2a), PTPRS (2a), PTPRD (2a), PTPRN (type 8), PTPRA (type 4), PTPRG (type 5), which represent 5 of the 8 RPTP subtypes, then incubated with COS-7 cell lysate expressing Myc- MTSS1. Samples were washed, subject to denaturing western blot, and probed with anti-Myc (MIM), or anti-GST. (b) We treated mouse medulloblastoma cells [Citation30], that express both PTPRS and PTPRD, with increasing concentrations of TAT-ISP for 24 hours then SFK activation through the abundance of the active phospho-mark SFK-Y416 by western blot. Intensity values are compared to β-Actin loading control. *p < 0.05, **p = 0.0057 1-way ANOVA with Tukey post-hoc. Error bars s.e.m
![Figure 1. MTSS1 binds specific RPTP family members. (a) We purified GST-fusions of the intracellular domain of CD45 (type 1), LAR (2a), PTPRS (2a), PTPRD (2a), PTPRN (type 8), PTPRA (type 4), PTPRG (type 5), which represent 5 of the 8 RPTP subtypes, then incubated with COS-7 cell lysate expressing Myc- MTSS1. Samples were washed, subject to denaturing western blot, and probed with anti-Myc (MIM), or anti-GST. (b) We treated mouse medulloblastoma cells [Citation30], that express both PTPRS and PTPRD, with increasing concentrations of TAT-ISP for 24 hours then SFK activation through the abundance of the active phospho-mark SFK-Y416 by western blot. Intensity values are compared to β-Actin loading control. *p < 0.05, **p = 0.0057 1-way ANOVA with Tukey post-hoc. Error bars s.e.m](/cms/asset/e6b1f4c2-fb5f-462d-86e2-e5f186d7f5b1/kccy_a_1695995_f0001_b.gif)
To determine whether RPTP inhibition reduced activation of SFKs, we took advantage of the recently described RPTPS wedge inhibitor for type IIa fused to the cell permeant TAT peptide for greater tissue distribution (TAT-ISP) [Citation29]. We treated mouse medulloblastoma cells [Citation30], that express type IIa RPTPs (PTPRD,PTPRF,PTPRS), with TAT-ISP inhibitory peptide and assayed SFK activation through the abundance of the active phospho-mark SFK-Y416. We found increasing levels of ISP reduced SFK-Y416 suggesting RPTP inhibition is sufficient to reduce SFK activity ()).
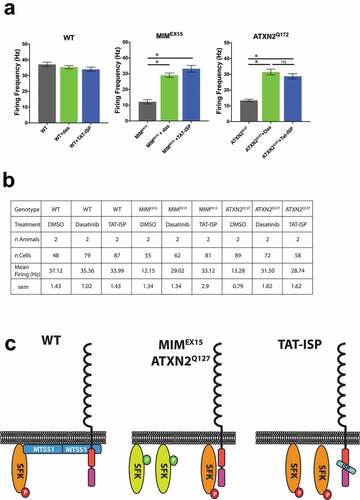
We recently demonstrated that Purkinje neuron basal firing rates are highly dependent on SFK activity, and that reducing SFK activity is sufficient to boost the suppressed firing rates and improve behavior seen in many spinocerebellar ataxias [Citation7]. To determine whether RPTP inhibition impacts basal firing rates we treated cerebellar slices from both MIMEX15 and ATXN2Q127 mice with TAT-ISP. We found 2μM TAT-ISP treatment boosted firing rates in both models to a level comparable to the small molecule SFK inhibitor dasatinib (), documenting the efficacy of the TAT-ISP approach.
Figure 2. RPTP inhibition rescues firing defects from multiple ataxias. (a) Mean Purkinje neuron basal firing rates for cerebellar slices incubated with DMSO (black), 200nM dasatinib (green), or 2μM TAT-ISP (blue). TAT-ISP treatment boosted firing rates in both MIMEX15 and ATXN2Q127 models to a level not statistically different from treating with the small molecule SFK inhibitor dasatinib. *p < 0.0001,ns p = 0.38 1-way ANOVA with Tukey post-hoc. Error bars s.e.m. (b) Summary table of firing data shown in A. C. Model where MTSS1 interacts with RPTPs and SFKs to restrain activity. When MTSS1 is lost (center) RPTPs remove inhibitory phospho marks from SFKs allowing activation. TAT-ISP blocks RPTP activity preventing the activation of SFKs. (c) Summary comparing how MTSS1 functions with SFKs and RPTPs
Discussion
We previously demonstrated SFK hyper-activation suppresses Purkinje neuron basal firing rates and leads to SCA by 1 month of age. Here we demonstrate that inhibition of RPTP family members known to interact with MTSS1 and control SFK activity also rescues SCA-dependent suppression of firing rates. These data suggest that aberrant regulation of RPTPs underlie one of the earliest SCA phenotypes: altered Purkinje neuron firing rates. RPTPs have large extracellular domains sufficient for dimerization and activation either through contact with other cells [Citation36] or with the extracellular matrix (ECM) [Citation37,Citation38]. The ability of RPTPs to respond to extracellular signals suggest some aspects of SCA disease progression may be driven in a non-cell autonomous manner, possibly by altered interaction with the ECM. Supporting the idea that ECM contributes to neurologic disease, conditionally removing HSPGs a major component of the extracellular matrix leads to neurologic deficits ranging from cerebellar agenesis [Citation39] to socio-communicative deficits [Citation40], while ablating CSPGs, an alternative ECM polysaccharide family, improves re-innervation after spinal injury [Citation41].
While the majority of studies using PTPRS and PTPRD mutant mice have focused on the hippocampus, the observed phenotypes contrast the cerebellar phenotypes seen in many SCA models. For example, PTPRS and PTPRD mutant mice have been shown to have increased hippocampal dendritic spine density [Citation23], and enhanced synaptic transmission associated with learning defects [Citation24], contrasting the reduced Purkinje neuron spine density [Citation7,Citation31,Citation42] and attenuated synaptic strength [Citation43] seen in some SCA models. Since SFKs and RPTPs likely work through a variety of mechanisms to regulate synapse stability, additional studies are required to elucidate which events result in the reduced synapse stability seen in SCA. In the hippocampus, SFK activity enhances LTP [Citation18,Citation44] by controlling glutamate receptor presentation and activity [Citation45], and helps control the formation and stability dendritic spines [Citation46] possibly through regulating the activity of the actin bundling protein Cortactin [Citation47]. While SFKs and protein tyrosine phosphatases have been shown to modulate mGluR1 activity at the Purkinje neuron/parallel fiber synapse to control LTD [Citation48]. Additionally, SFKs are a key link between RPTPs and TrkB to potentiate BDNF signaling and increase synaptic function [Citation49]. These findings suggest that one function for RPTPs in the nervous system is local control of SFK activity to stabilize the synapse, likely in response to multiple signaling pathways.
Our data expands the MTSS1/SFK regulatory circuit and identifies RPTPs as novel effectors of Purkinje neuron firing. Given the pleiotropic effects of SFK inhibition, RPTPs may prove a more idea candidate for SCA treatment.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Chaudhary F, Lucito R, Tonks NK. Missing-in-Metastasis regulates cell motility and invasion via PTPδ-mediated changes in SRC activity. Biochem J [Internet]. 2015;465;89–101. Available from http://biochemj.org/lookup/doi/10.1042/BJ20140573
- Zheng XM, Resnick RJ, Shalloway D. A phosphotyrosine displacement mechanism for activation of Src by PTPalpha. Embo J. 2000;19:964–978.
- Gonzalez-Quevedo R, Shoffer M, Horng L, et al. Receptor tyrosine phosphatase-dependent cytoskeletal remodeling by the hedgehog-responsive gene MIM/BEG4. J Cell Biol. 2005;168:453–463.
- Woodings JA, Sharp SJ, Machesky LM. MIM-B, a putative metastasis suppressor protein, binds to actin and to protein tyrosine phosphatase delta. Biochem J. 2003;371:463–471.
- Saarikangas J, Mattila PK, Varjosalo M, et al. Missing-in-metastasis MIM/MTSS1 promotes actin assembly at intercellular junctions and is required for integrity of kidney epithelia. J Cell Sci. 2011;124:1245–1255.
- Quinones GA, Jin J, Oro AE. I-BAR protein antagonism of endocytosis mediates directional sensing during guided cell migration. J Cell Biol. 2010;189:353–367.
- Brown AS, Meera P, Altindag B, et al. MTSS1/Src family kinase dysregulation underlies multiple inherited ataxias. Proc Nat Acad Sci. 2018;85: 16177–10.
- Saarikangas J, Kourdougli N, Senju Y, et al. MIM-induced membrane bending promotes dendritic spine initiation. Dev Cell. 2015;33(6):644–659.
- Paulson HL. The spinocerebellar ataxias. J Neuroophthalmol. 2009;29:227–237.
- Zhuchenko O, Bailey J, Bonnen P, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15:62–69.
- Nakamura K, Jeong SY, Uchihara T, et al. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet. 2001;10:1441–1448.
- Burnett B. The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum Mol Genet. 2003;12:3195–3205.
- Iwaki A, Kawano Y, Miura S, et al. Heterozygous deletion of ITPR1, but not SUMF1, in spinocerebellar ataxia type 16. J Med Genet. 2007;45:32–35.
- Hara K, Shiga A, Nozaki H, et al. Total deletion and a missense mutation of ITPR1 in Japanese SCA15 families. Neurology. 2008;71:547–551.
- Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609.
- Salmond RJ, Emery J, Okkenhaug K, et al. MAPK, phosphatidylinositol 3-kinase, and mammalian target of rapamycin pathways converge at the level of ribosomal protein S6 phosphorylation to control metabolic signaling in CD8 T cells. J Immunol. 2009;183:7388–7397.
- Li C, Götz J. Somatodendritic accumulation of Tau in Alzheimer’s disease is promoted by Fyn‐mediated local protein translation. Embo J. 2017;36:3120–3138.
- Grant SG, O’Dell TJ, Karl KA, et al. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science. 1992;258:1903–1910.
- Kaufman AC, Salazar SV, Haas LT, et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol. 2015;77:953–971.
- Chin J, Palop JJ, Puoliväli J, et al. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2005;25:9694–9703.
- Porkka K, Koskenvesa P, Lundan T, et al. Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood. 2008;112:1005–1012.
- Andersen JN, Mortensen OH, Peters GH, et al. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol. 2001;21:7117–7136.
- Horn KE, Xu B, Gobert D, et al. Receptor protein tyrosine phosphatase sigma regulates synapse structure, function and plasticity. J Neurochem. 2012;122:147–161.
- Uetani N, Kato K, Ogura H, et al. Impaired learning with enhanced hippocampal long‐term potentiation in PTPδ‐deficient mice. Embo J. 2000;19:2775–2785.
- Kolkman MJM, Streijger F, Linkels M, et al. Mice lacking leukocyte common antigen-related (LAR) protein tyrosine phosphatase domains demonstrate spatial learning impairment in the two-trial water maze and hyperactivity in multiple behavioural tests. Behav Brain Res. 2004;154:171–182.
- Chibnik LB, White CC, Mukherjee S, et al. Susceptibility to neurofibrillary tangles: role of the PTPRD locus and limited pleiotropy with other neuropathologies. Nat Publishing Group. 2017;23:1521–1529.
- Streuli M, Krueger NX, Thai T, et al. Distinct functional roles of the two intracellular phosphatase like domains of the receptor-linked protein tyrosine phosphatases LCA and LAR. Embo J. 1990;9:2399–2407.
- Wallace MJ, Fladd C, Batt J, et al. The second catalytic domain of protein tyrosine phosphatase delta (PTP delta) binds to and inhibits the first catalytic domain of PTP sigma. Mol Cell Biol. 1998;18:2608–2616.
- Lang BT, Cregg JM, DePaul MA, et al. Modulation of the proteoglycan receptor PTPσ promotes recovery after spinal cord injury. Nature. 2015;518:404–408.
- Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res. 2015;75(17):3623–3635.
- Hansen ST, Meera P, Otis TS, et al. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet. 2013;22:271–283.
- Schaapveld R, Schepens J, Bächner D. Developmental expression of the cell adhesion molecule-like protein tyrosine phosphatases LAR, RPTPδ and RPTPσ in the mouse. Mech Dev. 1998;77:59–62.
- Shishikura M, Nakamura F, Yamashita N, et al. Expression of receptor protein tyrosine phosphatase δ, PTPδ, in mouse central nervous system. Brain Res. 2016;1642:244–254.
- Meathrel K, Adamek T, Batt J, et al. Protein tyrosine phosphatase σ -deficient mice show aberrant cytoarchitecture and structural abnormalities in the central nervous system. J Neurosci Res. 2002;70:24–35.
- Longo FM, Martignetti JA, Le Beau JM, et al. Leukocyte common antigen-related receptor-linked tyrosine phosphatase. Regulation of mRNA expression. J Biol Chem. 1993;268:26503–26511.
- Wang J, Bixby JL. Receptor tyrosine phosphatase-δ Is a homophilic, neurite-promoting cell adhesion molecule for CNS neurons. Mol Cell Neurosci. 1999;14:370–384.
- Aricescu AR, McKinnell IW, Halfter W, et al. Heparan sulfate proteoglycans are ligands for receptor protein tyrosine phosphatase sigma. Mol Cell Biol. 2002;22:1881–1892.
- Fox AN, Zinn K. The heparan sulfate proteoglycan syndecan is an in vivo ligand for the drosophila LAR receptor tyrosine phosphatase. Curr Biol. 2005;15:1701–1711.
- Inatani M, Irie F, Plump AS, et al. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science. 2003;302:1044–1046.
- Irie F, Badie-Mahdavi H, Yamaguchi Y. Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate. Proc Natl Acad Sci U S A. 2012;109:5052–5056.
- Bradbury EJ, Moon LDF, Popat RJ, et al. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640.
- Clark HB, Burright EN, Yunis WS, et al. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. 1997;17:7385–7395.
- Hourez R, Servais L, Orduz D, et al. Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci. 2011;31:11795–11807.
- Lu YM, Roder JC, Davidow J, et al. Src activation in the induction of long-term potentiation in CA1 hippocampal neurons. Science. 1998;279:1363–1367.
- Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369:233–235.
- Morita A. Regulation of dendritic branching and spine maturation by semaphorin3A-fyn signaling. J Neurosci. 2006;26:2971–2980.
- Hering H, Sheng M. Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci. 2003;23:11759–11769.
- Canepari M, Ogden D. Evidence for protein tyrosine phosphatase, tyrosine kinase, and G-protein regulation of the parallel fiber metabotropic slow EPSC of rat cerebellar Purkinje neurons. J Neurosci. 2003;23:4066–4071.
- Yang T, Massa SM, Longo FM. LAR protein tyrosine phosphatase receptor associates with TrkB and modulates neurotrophic signaling pathways. J Neurobiol. 2006;66:1420–1436.