
ABSTRACT
Background Cerebral stroke refers to an acute onset of neurological deficit syndrome. In this research, we attempted to probe into the underlying mechanisms by which chrysophanol (CP) performed its regulatory roles in cerebral stroke. Methods OGD inducement was conducted in PC12 cells to construct a cerebral stroke model. Subsequently, CCK-8 assay, western blot, flow cytometry were utilized to determine cell viability, proliferation, and apoptosis, respectively. qRT-PCR was employed for detecting miR-216a expression level. Afterward, cell transfection was performed to alter miR-216a expression. Further, experiments were conducted to determine the expression of crucial factors participated in PI3 K/AKT and JAK2/STAT3 pathways for exploring the underlying mechanisms. Results OGD inducement suppressed cell viability, while promoted cell apoptosis. Besides, it enhanced the expression of proliferation-associated p53, p21, and apoptosis-associated Bax, and Cleaved-caspase-3, while suppressed the expression of Bcl-2. Furthermore, CHR exposure ameliorated the effects that OGD-evoked, and elevated the expression of miR-216a, as well as the expression of crucial factors participated in PI3 K/AKT and JAK2/STAT3 pathways. However, miR-216a silencing markedly reversed the effects triggered by CHR exposure. Conclusion CHR exposure relieved OGD-evoked PC12 cell damage by elevating miR-216a expression and thereby activating of PI3 K/AKT and JAK2/STAT3 pathways.
Introduction
Cerebral stroke refers to an acute onset of neurological deficit syndrome caused by local blood circulation disorders characterized by an instant deprivation of oxygen and glucose which resulting in disability and death to a large extent [Citation1]. Cerebral stroke causes neurological dysfunction via influencing the process of apoptotic or necrotic cell death [Citation2]. Besides, cerebral stroke induces series of pathophysiological changes, such as generation of free radical, activation of inflammatory response, and stimulation of oxidative phosphorylation, and so on [Citation3]. These pathophysiological changes contribute to serious brain damage which in turn affects human survival. However, the effective drugs used for cerebral stroke treatment are still limited until now.
Chrysophanol (CHR) is the main active ingredient of Chinese herbal medicine Rhubarb plant. Accumulating studies have reported that CHR possesses variety of activities, such as lipid-lowering and anti-diabetic, anti-tumor [Citation4], anti-apoptotic [Citation5], antifungal [Citation6], anti-allergic [Citation7] properties. It was once documented that CHR effectively reduced the death of ischemic apoptotic cells [Citation8]. Besides, Zhao et al. reported that CHR manifested neuroprotective properties in ischemia-triggered brain damage [Citation9]. Moreover, Zhang et al. verified that CHR could perform protective effects toward hippocampal neurons against lead neurotoxicity [Citation10]. Whereas, the underlying mechanisms by which CHR performs its neuroprotective roles in cerebral stroke remain unclear.
Latest researches have demonstrated that microRNAs (miRNAs) have a tight correlation with the development of cerebral ischemic stroke, and could be used as latent targets for diagnosis of cerebral ischemic stroke [Citation11,Citation12]. miR-216a is one of the miRNAs mentioned above. It was once documented that ischemia/reperfusion (I/R) treatment notably decreased the expression of miR-216a-5p [Citation13]. Besides, Koturbash et al. verified that miR-216a was involved in the cerebellar response to I/R treatment exhibiting as an elevated expression in male cerebellum [Citation14]. Moreover, Tian et al. specified that up-regulating the expression of miR-216a performed neuroprotective effects against ischemic damage both in vivo and in vitro [Citation15]. Although studies have demonstrated the association of miR-216a and I/R, but the potential mechanism of how miR-216a functions in cerebral stroke are still not fully understood.
Considering the characteristics of cerebral stroke, we conducted oxygen glucose deprivation (OGD) treatment on PC12 cells to simulate the conditions of cerebral stroke in vivo. This investigation made an attempt to explore the regulating mechanism of how CHR works in OGD-evoked PC12 cells. Firstly, we performed OGD treatment on PC12 cells to construct an in vitro cerebral stroke model. Subsequently, cell viability, proliferation, and apoptosis were examined in OGD + CHR evoked PC12 cells. Afterward, the expression of miR-216a was detected and then was altered by utilizing cell transfection. The expression of crucial factors participated in PI3 K/AKT and JAK2/STAT3 pathways were ultimately examined for exploring the underlying mechanisms.
Methods and materials
Cell culture and OGD treatment
The PC12 cells (American Type Culture Collection, ATCC, Rockville, MD, USA) were maintained in an incubator supplied with 5% CO2 + 95% air at 37°C in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, California, USA), which was replenished with 10% fetal bovine serum (FBS, BBI Solution, Crumlin, UK), 1% GlutaMAX (Thermo Fisher Scientific, Massachusetts, USA), and 1% streptomycin/penicillin (100 mg/ml: 100 U/ml). The culture medium was replaced 3 times per week. For inducing OGD, the culture medium was replaced with glucose-free DMEM (Invitrogen) and maintained in an anaerobic chamber which containing 5% CO2 + 95% N2 for 4 h at 37°C. Afterward, the culture medium was changed with normal DMEM medium 2 h before completing OGD inducement. Thereafter, the cells were maintained in the incubator for 24 h.
CHR (Sigma-Aldrich, St. Louis, Missouri, USA) was firstly made into stock solution and then diluted into series concentrations (20, 40, 60, 80, 100 μM) to treat PC12 cells for 24 h.
Cell counting kit assay (CCK-8 assay)
The PC12 cells were inoculated into a 96-well plate (5 × 103), and placed in an incubator supplied with 5% CO2 at 37°C. After treatment, 10 μl of CCK-8 reagent was added and the mixture was maintianed in the incubator for 40 min. Finally, the absorbance values of each group were measured at a wavelength of 450 nm by utilizing a Microplate Reader (Thermo Fisher, Massachusetts, USA).
Apoptosis assay
Cell apoptosis was analyzed by utilizing Annexin V-FITC/propidium iodide (PI) apoptosis detection kit (BBI Solution, Crumlin, UK). The cells were stimulated when grown to about 70% in a culture flask, and then were trypsinized, centrifuged, and collected. Afterward, cells were rinsed twice with pre-cooled phosphate-buffered saline (PBS), and suspended in binding buffer to a concentration of 2–5 × 105/ml. Subsequently, 10 μl of Annexin V-FITC was added to the cell suspension, and 10 μl of PI (20 μg/ml) was added 3 min later. The mixture was sustained at 25°C in dark condition for 15 min and then was analyzed by using a flow cytometry (BD Biosciences, San Jose, CA).
Cell transfection
miR-216a inhibitor (IN) and negative control (NC) inhibitor were produced by Sangon Biotech (Shanghai, China). Afterward, these sequences (150 nM) were respectively transfected into cells by utilizing Lipofectamine 3000 (Invitrogen) according to operating instructions. Cell collection was performed at 48 h after transfection.
Quantitative reverse-transcription polymerase chain reaction (qRT-PCR)
Total RNA was taken out by utilizing Trizol (TAKARA, Beijing, China), and the quality of extracted RNA was determined by using Nanodrop 2000 (Thermo Scientific, Massachusetts, USA) and gel electrophoresis. Afterward, the first strand of cDNA was synthesized by utilizing a reverse transcription kit (TAKARA, Beijing, China). Thereafter, qRT-PCR was performed by using SYBR Premix Ex Taq (UNIBIOTEST, Wuhan, China). U6 was used as the internal reference and the relative expression of miR-216a was calculated by utilizing 2−ΔΔCt method.
Western blot
The proteins were taken out by utilizing RIPA lysate (Sangon Biotech, Shanghai, China) complemented with protease inhibitor treatment (Roche, Basel, Switzerland). The concentration of proteins extracted was tested by using the BCA method (Solarbio, Beijing, China). After separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), the proteins were transferred onto polyvinylidene fluoride (PVDF) membranes (BBI Solution, Crumlin, UK). After that, the membranes carrying blots were blocked in 5% bull serum albumin (BSA, BBI Solution, Crumlin, UK) for 1 h at 25°C. Afterward, the membranes were co-incubated with primary antibodies directly against p53 (ab131442, Abcam, Cambridge, United Kingdom), p21 (ab109199, Abcam), Bcl-2 (ab196495, Abcam), Bax (ab32503, Abcam), Cleaved caspase-3 (ab49822, Abcam), t-PI3 K (ab151549, Abcam), p-PI3 K (ab182651, Abcam), t-AKT (ab8805, Abcam), p-AKT (ab38449, Abcam), t-JAK-2 (ab39636, Abcam), p-JAK2 (ab32101, Abcam), t-STAT3 (ab68153, Abcam), p-STAT3 (ab76315, Abcam), β-actin (ab8227, Abcam) for 16 h at 4°C. After rinsed with Tris-Buffered Saline Tween (TBST) for three times, the HRP-labeled secondary antibody (ab205718, Abcam) was added and sustained for 1 h. Thereafter, the developing solution ECL (Sangon Biotech, Shanghai, China) was supplied and reacted for 1 min. Finally, the image was scanned by utilizing the GS-800 imaging system (Bio-Rad, California, USA).
Statistical analysis
All trails were repeated for three times. Data analysis was performed by utilizing SPSS 19.0 statistical software (SPSS, Inc., Chicago, IL, USA). Data were displayed as mean ± standard deviation (SD) and analyzed by one-way analysis of variance (ANOVA). Multiple comparison between the groups was performed by using S-N-K method. P < 0.05 was recognized as statistically significant.
Results
OGD inducement restrained cell proliferation while augmented cell apoptosis
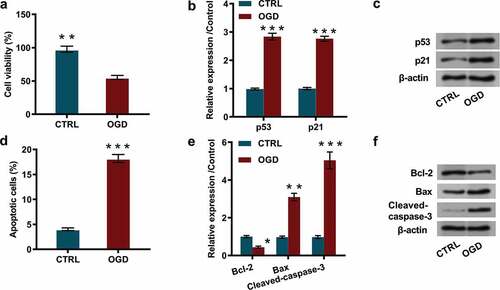
The PC12 cells were stimulated by OGD inducement, and cell characteristics including cell viability, cell proliferation, and the percentage of apoptotic cells, as well as the expression of proliferation and apoptosis-associated factors were subsequently detected. The results showed that OGD inducement significantly reduced cell viability (P < 0.01, ), and notably restrained cell proliferation, exhibiting as dramatically enhanced the expression of proliferation-associated proteins including p53 (P < 0.001) and p21 (P < 0.001, and ). Besides, flow cytometry results demonstrated that OGD inducement remarkably increased the percentage of apoptotic cells (P < 0.001, ), and western blot results revealed that the expression levels of apoptosis-associated proteins including Bax (P < 0.01) and Cleaved-caspase-3 (P < 0.001) were dramatically elevated, while Bcl-2 (P < 0.05, and ) was notably declined. These outcomes indicated that OGD inducement restrained cell proliferation while augmented cell apoptosis.
Figure 1. OGD inducement restrained cell proliferation while augmented cell apoptosis.
CHR exposure relieved OGD-evoked cell injury
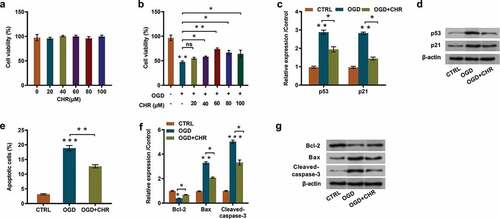
To investigate the influences of CHR on OGD-evoked PC12 cells, cells were firstly pretreated with CHR and then cell characteristics and the expression of proliferation/apoptosis-associated proteins were determined. Outcomes displayed in illustrated that CHR exposure (20–100 μM) had no significant influences on cell viability. Whereas, it had notably weakened OGD-evoked inhibitory effects on cell viability and exhibited in a dose-dependent way (P < 0.05 or P < 0.01, ). Considering that the most effective concentration was 60 μM, so we chose this concentration to conduct subsequent experiments. Besides, CHR exposure remarkably relieved OGD-triggered promoting effects that on the expression of proliferation-associated proteins-p53 (P < 0.05) and p21 (P < 0.05, and ). Moreover, results of flow cytometry demonstrated that CHR exposure effectively declined the proportion of apoptotic cells compared with OGD-induced group (P < 0.01, ), but was still higher than control group. These results were further verified by the outcomes of western blot, which revealed that CHR exposure markedly ameliorated the repressive effects on Bcl-2 (P < 0.05) expression and the enhancing effects on Bax (P < 0.05) and Cleaved-caspase-3 (P < 0.05, and ) expression. These outcomes illustrated that CHR exposure effectively ameliorated OGD-evoked cell injury in PC12 cells.
Figure 2. CHR exposure relieved OGD-evoked cell injury.
CHR exposure stimulated miR-216a expression
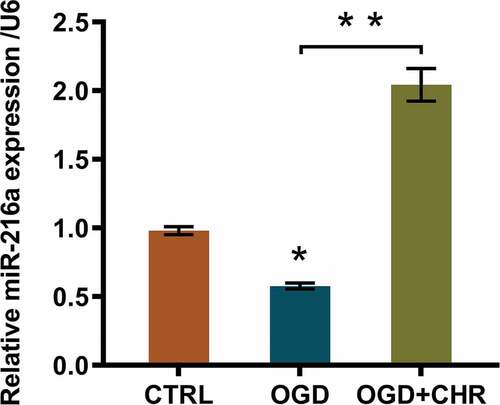
A previous study once clarified the correlation between miR-216a expression and ischemic injury [Citation15]. Therefore, we suspected that whether miR-216a was involved in the process of CHR ameliorating OGD-evoked cell injury. Thus, we detected the miR-216a expression both in OGD-induced PC12 cells and in OGD + CHR exposed cells. Data suggested that OGD inducement notably repressed miR-216a expression (P < 0.05, ), while CHR exposure markedly reversed the suppressive effects triggered by OGD inducement (P < 0.01, ). In other words, CHR exposure enhanced miR-216a expression.
Figure 3. CHR exposure stimulated miR-216a expression.
CHR exposure ameliorated OGD-evoked cell injury through increasing miR-216a expression
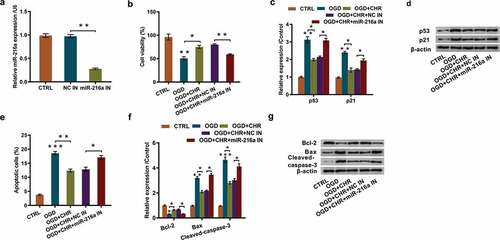
To probe into how miR-216a participated in the regulation of CHR ameliorating OGD-evoked cell injury, we silenced miR-216a expression by using cell transfection. Data displayed in showed that miR-216a expression was remarkably inhibited after cell transfection (P < 0.01). Besides, CCK-8 assay revealed that the increased cell viability stimulated by CHR exposure was remarkably decreased by miR-216a inhibition (P < 0.01, ). Moreover, miR-216a inhibition effectively alleviated the inhibitory effects triggered by CHR exposure on cell proliferation, exhibiting as partially reversed the inhibited expression of proliferation-associated proteins-p53 (P < 0.05) and p21 (P < 0.05, and ). As for cell apoptosis, outcomes of flow cytometry showed that miR-216a inhibition markedly increased the number of apoptotic cells (P < 0.05, ) which was previously reduced by CHR exposure. These results were further proved by western blot outcomes which demonstrated that miR-216a inhibition notably reversed the elevated expression of apoptosis-related Bcl-2 (P < 0.05) and the declined expression of Bax (P < 0.05) and Cleaved-caspase-3 (P < 0.05, and ). The aforementioned results indicated that CHR exposure might ameliorate OGD-evoked cell injury via enhancing miR-216a expression.
Figure 4. CHR exposure ameliorated OGD-evoked cell injury through increasing miR-216a expression.
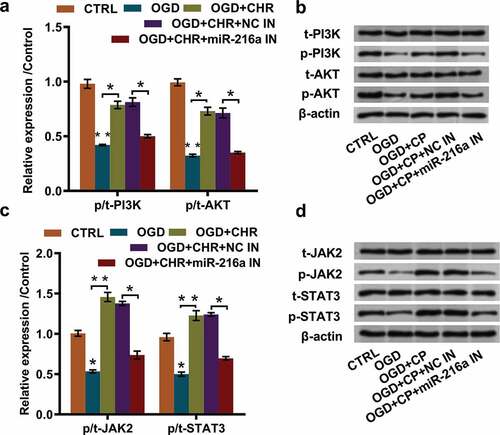
CHR exposure accelerated activation of PI3 K/AKT and JAK2/STAT3 pathways via elevating miR-216a expression
Substantial previous investigations have recorded that PI3 K/AKT and JAK2/STAT3 pathways participated in the neuro-protection after stroke [Citation16–Citation18]. To probe into the potential regulating mechanisms, we investigated the expression of pivotal factors participated in PI3 K/AKT and JAK2/STAT3 pathways. Western blot results specified that OGD inducement notably repressed the expression of p/t-PI3 K and p/t-AKT (both P < 0.01), while CHR exposure attenuated these effects (both P < 0.05). Moreover, miR-216a inhibition partially reversed the effects that CHR exposure had induced in OGD + CHR exposed PC12 cells (both P < 0.05, and ). In addition, the expression of crucial proteins in JAK2/STAT3 pathway exhibited the same variation trend, showing that expression of p/t-JAK2 and p/t-STAT3 were firstly reduced by OGD inducement (both P < 0.05), and then elevated by CHR exposure (both P < 0.01). Afterward, CHR exposure triggered impacts were dramatically reversed by miR-216a inhibition (both P < 0.05). These data hinted that CHR exposure might accelerate activation of PI3 K/AKT and JAK2/STAT3 pathways through elevating miR-216a expression.
Figure 5. CHR exposure accelerated activation of PI3 K/AKT and JAK2/STAT3 pathways via elevating miR-216a expression.
Discussion
In this investigation, we demonstrated that OGD inducement notably suppressed cell viability and proliferation, while enhanced cell apoptosis. However, CHR exposure effectively diminished the effects OGD evoked. Besides, it had accelerated miR-216a expression. Moreover, silencing miR-216a expression remarkably reversed the effects that CHR exposure induced on cell characteristics, and the expression of key players participated in the PI3 K/AKT and JAK2/STAT3 pathways. These outcomes hinted that CHR might ameliorate OGD-evoked cell injury through elevating miR-216a expression and thereby regulating PI3 K/AKT and JAK2/STAT3 pathways.
It has been widely acknowledged that cerebral stroke is characterized by an instant deprivation of oxygen and glucose, therefore, we dealt PC12 cells with OGD inducement in this investigation. The outcomes specified that OGD inducement remarkably suppressed cell viability and proliferation, while enhanced cell apoptosis. These results were subsequently verified by the elevated expression of proliferation-associated p53 and its down-stream factor p21, as well as the declined expression of apoptosis-associated Bcl-2, and elevated expression of Bax and Cleaved-caspase-3. These outcomes described above were consistent with earlier studies [Citation19–Citation22].
An increasing number of literatures have demonstrated the neuroprotective properties of CHR. For instance, Zhao et al. previously clarified that CHR ameliorated focal I/R triggered brain injury through anti–inflammatory actions [Citation9] and effectively relieved I/R triggered nitrosative/oxidative stress damage [Citation23]. Besides, Chae et al. elucidated that CHR reduced the death rate of hippocampal neuronal cells induced by glutamate exposure through regulating mitochondrial fission [Citation24]. Moreover, Yan et al. revealed that CHR liposome performed its neuroprotective properties toward cerebral I/R injury under middle cerebral artery occlusion circumstance [Citation25]. However, fewer investigations were conducted toward the neuroprotective mechanisms of CHR in cerebral stroke. Only a study performed by Zhang et al. elucidated that CHR could play protection roles in cerebral stroke through suppressing NALP3 inflammasome activation [Citation26]. Therefore, we exposed PC12 cells to CHR to quest the potential influences of CHR on OGD-evoked cells in this investigation. The results specified that CHR exposure increased OGD previously decreased cell viability in a dose-dependent way. Besides, it had attenuated the inhibitory effects on cell proliferation and the promoting effects on cell apoptosis exhibiting as notably declining the expression of proliferation-associated p53 and p21, and reducing the expression of apoptosis-associated Bax and Cleaved-caspase-3, while elevated the expression of Bcl-2. These outcomes were in line with earlier studies stated above [Citation8,Citation23], which hinted that CHR exactly possesses neuroprotective properties toward cerebral stroke and this properties would make great sense for future treatment of stroke.
Earlier studies elucidated that miR-216a dysregulation participated in the neuroprotective effects against I/R injury. For instance, a study performed by Ding et al. demonstrated I/R treatment notably reduced the expression of miR-216a-5p in neuronal cells [Citation13]. More importantly, an investigation performed in primary cortical neuronal cells confirmed that elevating the expression of miR-216a could stimulate the neuroprotective effects on ischemic damages via targeting JAK2 both in vivo and in vitro [Citation15]. Furthermore, a previous study specified that Ginsenoside Rg1, the primary active composition extracted from ginseng, exerted its protective roles against H2O2-triggered damages through elevating miR-216a-5p expression in PC12 cells [Citation27]. Therefore, we suspected that CHR might exert its regulatory roles via mediating the expression of miR-216a as well. Interestingly, we found that OGD exposure significantly decreased miR-216a expression, while CHR completely unregulated miR-216a. To further ensure this speculation, we inhibited miR-216a expression by utilizing cell transfection. Succeeding results uncovered that miR-216a inhibition observably reversed the elevating effects on cell viability and the suppressive effects on cell proliferation and apoptosis that CHR exposure triggered in OGD-evoked PC12 cells. Besides, it also had dramatically counteracted the suppressive effects on the expression of cell proliferation-associated proteins p53, p21, and apoptosis-associated proteins Bax and Cleaved-caspase-3. Combination of our results and previous findings hinted that CHR attenuated OGD-evoked cell damage, which might be achieved by elevating miR-216a expression.
Accumulating researches have reported the participation of PI3 K/AKT and JAK2/STAT3 pathways in the regulatory process of CHR and miR-216a. For instance, Xia et al. disclosed that overexpressing the expression of miR-216a/217 accelerated the activation of PI3 K/AKT pathway in hepatocellular carcinoma [Citation28]. Besides, it was reported that PI3 K/AKT pathway was also involved in emodin-mediated hepatoblastoma [Citation29]. Moreover, an earlier research about gastric cancer elucidated that miR-216a repressed cancer metastasis through targeting the epithelial–mesenchymal transition process which was mediated by JAK2/STAT3 pathway [Citation30]. What's more, researches conducted in cardiomyocyte, pancreatic tumor and airway smooth muscle cells commonly demonstrated that miR-216a functioned via targeting JAK2/STAT3 pathway [Citation31–Citation33]. Yuan et al. once clarified that CHR also ameliorated isoproterenol-evoked cardiac hypertrophy through regulating JAK2/STAT3 pathway [Citation34]. In our investigation, the results illustrated that CHR exposure notably elevated the phosphorylation levels of crucial factors participated in PI3 K/AKT and JAK2/STAT3 pathways, such as PI3 K, AKT, JAK2, and STAT3. In other words, CHR exposure accelerated the activation of PI3 K/AKT and JAK2/STAT3 pathways. However, the following trails showed that miR-216a inhibition dramatically reversed the promoting effects on the variations of protein expression triggered by CHR exposure. These results intimated that CHR might accelerate the activation of PI3 K/AKT and JAK2/STAT3 pathways via elevating the expression of miR-216a in OGD-evoked PC12 cells.
Overall, CHR exposure relieved OGD-evoked PC12 cell damage by elevating miR-216a expression and activating PI3 K/AKT and JAK2/STAT3 pathways. This investigation might have an important significance for clinical use of CHR and future treatment of stroke.
Authors’ Contributions
Conceived and designed the experiments: Xipeng Yan
Performed the experiments and analyzed the data: Yuanyuan Liu, Chuanqian Liu, Xueting Zhang, Zhenzhen Liu
Manuscript writing and revision: Yuanyuan Liu, Chuanqian Liu, Xueting Zhang, Xipeng Yan
All authors read and approved the final manuscript.
Availability of Data and Materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Disclosure Statement
The authors declare that they have no competing interests.
Additional information
Funding
References
- Siniscalchi A, Gallelli L, Malferrari G, et al. Cerebral stroke injury: the role of cytokines and brain inflammation. J Basic Clin Physiol Pharmacol. 2014;25:131–137.
- Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev. 2007;54:34–66.
- Gupta YK, Briyal S, Gulati A. Therapeutic potential of herbal drugs in cerebral ischemia. Indian J Physiol Pharmacol. 2010;54:99–122.
- Ni CH, Yu CS, Lu HF, et al. Chrysophanol‐induced cell death (necrosis) in human lung cancer A549 cells is mediated through increasing reactive oxygen species and decreasing the level of mitochondrial membrane potential. Environ Toxicol. 2014;29:740–749.
- Chen YC, Shen SC, Lee WR, et al. Emodin induces apoptosis in human promyeloleukemic HL-60 cells accompanied by activation of caspase 3 cascade but independent of reactive oxygen species production. Biochem Pharmacol. 2002;64:1713–1724.
- Agarwal SK, Singh SS, Verma S, et al. Antifungal activity of anthraquinone derivatives from Rheum emodi. J Ethnopharmacol. 2000;72:43–46.
- Kim DH, Park EK, Bae EA, et al. Metabolism of rhaponticin and chrysophanol 8-o-beta-D-glucopyranoside from the rhizome of rheum undulatum by human intestinal bacteria and their anti-allergic actions. Biol Pharm Bull. 2000;23:830–833.
- Zhao Y, Fang Y, Zhao H, et al. Chrysophanol inhibits endoplasmic reticulum stress in cerebral ischemia and reperfusion mice. Eur J Pharmacol. 2018;818:1–9.
- Zhao Y, Fang Y, Li J, et al. Neuroprotective effects of Chrysophanol against inflammation in middle cerebral artery occlusion mice. Neurosci Lett. 2016;630:16–22.
- Zhang J, Yan C, Wang S, et al. Chrysophanol attenuates lead exposure-induced injury to hippocampal neurons in neonatal mice. Neural Regen Res. 2014;9:924–930.
- Yao S, Tang B, Li G, et al. miR-455 inhibits neuronal cell death by targeting TRAF3 in cerebral ischemic stroke. Neuropsychiatr Dis Treat. 2016;12:3083–3092.
- Guo D, Ma J, Yan L, et al. Down-regulation of Lncrna MALAT1 attenuates neuronal cell death through suppressing beclin1-dependent autophagy by regulating Mir-30a in cerebral ischemic stroke. Cell Physiol Biochem. 2017;43:182–194.
- Ding H, Gao S, Wang L, et al. Overexpression of miR-582-5p inhibits the apoptosis of neuronal cells after cerebral ischemic stroke through regulating PAR-1/Rho/Rho Axis. J Stroke Cerebrovasc Dis. 2019;28:149–155.
- Koturbash I, Zemp F, Kolb B, et al. Sex-specific radiation-induced microRNAome responses in the hippocampus, cerebellum and frontal cortex in a mouse model. Mutat Res Genet Toxicol Environ Mutagen . 2011;722:114–118.
- Tian YS, Zhong D, Liu QQ, et al. Upregulation of miR-216a exerts neuroprotective effects against ischemic injury through negatively regulating JAK2/STAT3-involved apoptosis and inflammatory pathways. J Neurosurg. 2018;130:977–988.
- Hou Y, Wang K, Wan W, et al. Resveratrol provides neuroprotection by regulating the JAK2/STAT3/PI3K/AKT/mTOR pathway after stroke in rats. Genes Dis. 2018;5:245–255.
- Xu X, Chua CC, Gao J, et al. Neuroprotective effect of humanin on cerebral ischemia/reperfusion injury is mediated by a PI3K/Akt pathway. Brain Res. 2008;1227:12–18.
- Kretz A, Happold CJ, Marticke JK, et al. Erythropoietin promotes regeneration of adult CNS neurons via Jak2/Stat3 and PI3K/AKT pathway activation. Mol Cell Neurosci. 2005;29:569–579.
- Chiu BY, Chang CP, Lin JW, et al. Beneficial effect of astragalosides on stroke condition using PC12 cells under oxygen glucose deprivation and reperfusion. Cell Mol Neurobiol. 2014;34:825–837.
- Guo H, Kong S, Chen W, et al. Apigenin mediated protection of OGD-evoked neuron-like injury in differentiated PC12 cells. Neurochem Res. 2014;39:2197–2210.
- Gaire BP, Jamarkattel-Pandit N, Lee D, et al. Terminalia chebula extract protects OGD-R induced PC12 cell death and inhibits lps induced microglia activation. Molecules. 2013;18:3529–3542.
- Chang R, Zhou R, Qi X, et al. Protective effects of aloin on oxygen and glucose deprivation-induced injury in PC12 cells. Brain Res Bull. 2016;121:75–83.
- Zhao Y, Huang Y, Fang Y, et al. Chrysophanol attenuates nitrosative/oxidative stress injury in a mouse model of focal cerebral ischemia/reperfusion. J Pharmacol Sci. 2018;138:16–22.
- Chae U, Min JS, Leem HH, et al. Chrysophanol suppressed glutamate-induced hippocampal neuronal cell death via regulation of dynamin-related protein 1-dependent mitochondrial fission. Pharmacology. 2017;100:153–160.
- Yan J, Zheng M, Zhang D. Chrysophanol liposome preconditioning protects against cerebral ischemia-reperfusion injury by inhibiting oxidative stress and apoptosis in mice. Int J Pharmacol. 2014;10:55–68.
- Zhang N, Zhang X. Chrysophanol inhibits NALP3 inflammasome activation and ameliorates cerebral ischemia/reperfusion in mice. Mediators Inflammation. 2014;2014:370530.
- Yi G, Liu L, Lv C, et al. Ginsenoside Rg1 defenses PC-12 cells against hydrogen peroxide-caused damage via up-regulation of miR-216a-5p. Life Sci. 2019;236:116948.
- Xia H, Ooi LL, Hui KM. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology. 2013;58:629–641.
- Cui Y, Lu P, Song G, et al. Involvement of PI3K/Akt, ERK and p38 signaling pathways in emodin-mediated extrinsic and intrinsic human hepatoblastoma cell apoptosis. Food Chem Toxicol. 2016;92:26–37.
- Tao Y, Yang S, Wu Y, et al. MicroRNA-216a inhibits the metastasis of gastric cancer cells by targeting JAK2/STAT3-mediated EMT process. Oncotarget. 2017;8:88870–88881.
- Wang J, Chen X, Shen D, et al. A long noncoding RNA NR_045363 controls cardiomyocyte proliferation and cardiac repair. J Mol Cell Cardiol. 2019;127:105–114.
- Hou BH, Jian ZX, Cui P, et al. miR-216a may inhibit pancreatic tumor growth by targeting JAK2. FEBS Lett. 2015;589:2224–2232.
- Yan YR, Luo Y, Zhong M, et al. MiR-216a inhibits proliferation and promotes apoptosis of human airway smooth muscle cells by targeting JAK2. J Asthma. 2019;56:938–946.
- Yuan J, Hong H, Zhang Y, et al. Chrysophanol attenuated isoproterenol-induced cardiac hypertrophy by inhibiting Janus kinase 2/signal transducer and activator of transcription 3 signaling pathway. Cell Biol Int. 2019;43:695–705.