
ABSTRACT
Pyroptosis is a form of programmed cell death initiated by inflammasomes and is critical for immunity. SIRT1, a NAD+-dependent deacetylase, plays multiple roles in inflammatory response and immunity. Metformin can activate SIRT1 to participate in different biological processes and exert its anticancer effects. However, the mechanism by which metformin activates SIRT1 to drive cancer cell pyroptosis has not been reported. In this study, we treated cancer cells with metformin for diverse periods of time (0–24 h) and found that cell viability was decreased obviously. Interestingly, pyroptosis occurred when cancer cells were treated with metformin for the indicated time (4, 8 and 12 h), which was elucidated by the cell swelling and bubbles blowing in the membrane. Metformin also increased the release of lactate dehydrogenase (LDH, an indication of pyroptotic cell cytotoxicity) remarkably. The underlying mechanisms were that metformin enhanced AMPK/SIRT1 pathway and further increased NF-κB p65 expression to stimulate Bax activation and cytochrome c release, triggering caspase3 cleavage of GSDME, which is a characteristic pyroptotic marker. Depletion of SIRT1 inhibited metformin-induced these protein expression, revealing that metformin promotes AMPK/SIRT1/NF-κB signaling to drive cancer cell pyroptosis. Meantime, metformin induced mitochondrial dysfunction to trigger activation of caspase3 and generation of GSDME-N. Moreover, mitochondrial dysfunction activated AMPK/SIRT1 pathway to cause pyroptotic death upon metformin treatment. This research firstly reveals that metformin as a sensitizer amplifies AMPK/SIRT1/NF-κB signaling to induce caspase3/GSDME-mediated cancer cell pyroptosis. Induction of cellular pyroptosis by metformin is considered as a novel therapeutic option against various cancers.
Graphical abstract
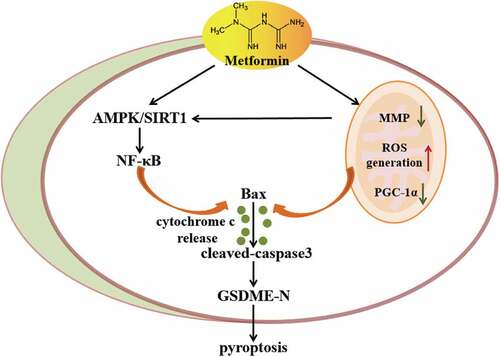
1. Introduction
With increasing incidence and mortality, cancer has been the leading cause of death with about 9.6 million deaths occurred in 2018 worldwide [Citation1]. Cancer cells facing their death can actively choose their cell fate determination following exposure to ionizing radiation and chemotherapeutic drugs. Programmed cell fate decisions mostly focus on classical types of cell death (apoptosis and necroptosis) and therapy-induced cellular senescence [Citation2–Citation5]. There is necessary to seek out an alternative pathway when these methods of cell death are compromised.
Pyroptotic death is distinguished from other forms of cell death partly due to its unique morphological and biochemical characteristics, which is lytic, featuring cellular swelling, cell membrane pore formation and large bubbles blowing from the plasma membrane [Citation6,Citation7]. It is regarded as a critical host defense mechanism against pathogenic bacteria by releasing them into the extracellular environment where they can be killed by immune cells [Citation8–Citation10]. Pyroptosis is originally measured in immune cells with inflammatory stimulations which induce inflammasome and caspase1/4/5/11 activation. These caspases, in turn, cleave gasdermin D (GSDMD) to generate the N-terminal fragment (GSDMD-NT), which forms nonselective pores in the plasma membrane [Citation11–Citation13]. Despite the well-known anti-infection effect of pyroptosis is induced by GSDMD in immune cells, a few reports about the functions and mechanisms of other gasdermin activation have been identified. Gasdermin E (GSDME) is specifically cleaved by caspase3 and releases its N-terminal pore-forming domain for pore formation when treating with certain apoptotic stimuli, eventually causing pyroptotic death in gastric and lung cancer cells [Citation6,Citation14]. Activation of the mitochondrial apoptotic pathway in the GSDME-deficient 293 T cells leads to induction of apoptosis, only when GSDME is stably expressed in these cells they progress to pyroptosis [Citation15]. Thus, GSDME is identified as a key mediator of pyroptosis and a downstream of apoptotic caspase3. Carbonyl cyanide m-chlorophenyl hydrazone (CCCP) induces mitochondrial dysfunction via the mitophagy pathway and causes ROS generation to activate caspase3, consequently driving pyroptotic death by induction of GSDME cleavage in melanoma cells [Citation3]. Recently, chemotherapeutic cisplatin, doxorubicin and paclitaxel have been reported to activate caspase3/GSDME and exert their cytotoxicity on lung cancer and macrophage cells by inducing pyroptosis. Concurrent with their therapeutic effects on cancer cells, these anticancer chemotherapy administration cause adverse toxicity to normal tissues of patients, leading to severe side effect [Citation14,Citation16]. Metformin is the most widely prescribed drug for the treatment of type 2 diabetes owing to its minimal side effects. It prevents the onset of human cancers in diabetic patients. As a complex I inhibitor, metformin directly induces mitochondrial dysfunction in epithelial cancer cell compartment [Citation17,Citation18]. It also activates AMP-activated protein kinase (AMPK), a major sensor of cellular energy status in multiple types of cancer and human fibroblasts [Citation19,Citation20]. AMPK activation raises the intracellular NAD+ concentrations and activates SIRT1 [Citation20,Citation21]. Our previous results show that SIRT1 increases nuclear factor-kappa B (NF-κB) expression significantly upon resveratrol treatment in MCF-7 and H1299 cells [Citation22]. NF-κB is comprised by several subunits including p50, p52, p65 (RelA), RelB and c-Rel, of which p65 is a major subunit of NF-κB well known for its role in the innate immune response [Citation23–Citation25]. It increases IL-1β production to trigger Bax accumulation, release of cytochrome c and caspase3 activation, which contribute to neuroblastoma cell death [Citation26]. Up to now, the exact molecular mechanism underlying for metformin to activate AMPK/SIRT1/NF-κB pathway and induce mitochondrial dysfunction to drive cancer cell pyroptosis has not been reported yet.
In the current study, we report that metformin treatment stimulates AMPK/SIRT1/NF-κB signaling to drive caspase3/GSDME-mediated cancer cell pyroptosis. Furthermore, metformin-induced mitochondrial dysfunction can promote AMPK/SIRT1 pathway to trigger cancer cell pyroptosis. These data reveals that metformin induces cancer cell pyroptosis by AMPK/SIRT1/NF-κB pathway and mitochondrial dysfunction to exhibit anticancer effects.
2. Materials and methods
2.1. Cell culture and reagents
The human hepatoblastoma cells HepG2, breast cancer cells MCF-7, colon cancer cell lines HCT-15 and HT-29 were obtained from the Cell Bank of Type Culture Collection of Chinese Academy of Science (Shanghai, China). HepG2 and MCF-7 cells were cultured in DMEM and other cells were in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin sulfate (Solarbio, Beijing, China). The cells were cultured at 37°C in a humidified 5% CO2 incubator. Cells were treated with 20 mM metformin (MET) (Sigma-Aldrich, St. Louis, USA) for diverse periods of time, 40 μM CCCP (Solarbio, Beijing, China) for 24 h, 10 μM EX527 (Selleck Chemicals, Houston, TX, USA) for 24 h, 10 μM BAY 11–7082 (Beyotime, Shanghai, China) for 1 h, and 3 μM Compound C (Selleck Chemicals, Houston, TX, USA) for 24 h supplemented with 10% FBS medium.
2.2. MTT assay
MTT assay was used to assess cell viability. Briefly, cells were seeded in 96-well plates at a density of 6 × 103 cells per well overnight. Cells were treated with or without 20 mM metformin for diverse periods of time (2, 4, 8, 12, 16 and 24 h). Then MTT solution (5 mg/mL) was added (20 uL per well). After incubation at 37°C for 4 h, the formazan crystals were dissolved by 150 µl DMSO, the solution was absorbed at 492 nm using enzyme-linked immunosorbent assay reader (Awareness, Palm City, FL, USA).
2.3. Microscopy imaging
To examine the morphology of pyroptotic cells, cells were treated in 6-well plates. Static bright-field images were captured using an Olympus IX73 microscope.
2.4. Hoechst/propidium iodide nuclear staining and fluorescence microscopy
HepG2 cells were treated with or without 20 mM metformin for 4, 8, 12 h. The staining was performed using Apoptosis and Necrosis Assay Kit (Beyotime Institute of Biotechnology) according to the manufacturer’s instructions. Cells were stained with 10 ng/mL Hoechst 33342 and 10 ng/mL propidium iodide (PI) for 20 min at 0°C in dark. Then, the nuclei of apoptotic cells were measured by fluorescence microscope. The method differentiates the normal live cells (stained light blue), apoptotic cells (stained bright blue) and necrotic cells (stained red). A minimum of 600 cells were counted.
2.5. LDH release assay
Pyroptosis was analyzed by detecting the activity of LDH released into cell culture supernatants using the CytoTox 96 Non-Radioactive Cytotoxicity Assay Kit (Beyotime, Shanghai, China) according to the manufacturer’s protocol.
2.6. Western blotting and antibodies
The cells were lysed by RIPA (Beyotime, Shanghai, China), and then centrifuged for 5 min at 12000 rpm at 4°C. Proteins were separated by SDS-PAGE (10-12% gels) and transferred on to PVDF membranes. Blots were blocked with 5% nonfat milk for 1.5 h and then probed with primary antibodies against AMPK, p-AMPK, cleaved-caspase3 and NF-κB p65 (CST, Danvers, MA, USA), SIRT1 (Millipore Corp, Billerica, MA), GSDME-N (Abcam, Cambridge, UK), PGC-1α and cytochrome c (Santa Cruz, CA, USA), Bax and Actin (Affinity, Cincinnati, OH, USA) overnight at 4°C, followed by incubation with HRP-conjugated rabbit/mouse secondary antibodies (ZSGB-BIO, Beijing, China) for 2 h. Protein expression was visualized by ECL detection (Millipore, Billerica, MA, USA).
2.7. siRNA experiments
Control siRNA and specific siRNA directed against of SIRT1 were from Guangzhou RiboBio. HepG2 and MCF-7 cells were transfected with (50 nM) SIRT1 knockdown siRNA (GGAAATATATCCTGGACAA) or control siRNA. Transfection was performed by using lipofectamine 2000 (Invitrogen, Waltham, MA, USA) according to the manufacturer’s instruction. The transfection medium was replaced with fresh 10% FBS medium 6 h post-transfection. Cells were incubated for 24–36 h, and then collected for subsequent experiments.
2.8. Mitochondrial membrane potential detection
We indirectly assessed mitochondrial function by tracking relative mitochondrial transmembrane potential through detecting JC-1 fluorescence according to the manufacturer’s instruction (JC-1, Beyotime, Shanghai, China). Briefly, cells in 6-well plates were treated with 20 mM metformin for 8 h in the absence or presence of Compound C (AMPK inhibitor). Then, we monitored the mitochondrial membrane potentials by determining the relative amounts or dual emissions from both mitochondrial JC-1 monomers (green) and aggregates (red) by using an Olympus fluorescent microscope. Mitochondrial depolarization was expressed by an increase in the intensity ratio of green/red fluorescence.
2.9. ROS assay
ROS production was detected by ROS specific fluorescent probe 2ʹ, 7ʹ-Dichlorofluorescin diacetate (DCFH.DA, Beyotime, Shanghai, China). Cells were treated with or without 20 mM metformin for different time, and then washed, incubated with 10 μM DCFH.DA for 30 min at 37°C in dark. Then, cells were washed and examined using SpectraMax M5 microplate reader (Molecular Devices) and Olympus fluorescence microscopy.
2.10. Statistical analysis
All of the assays were repeated at least three times independently. GraphPad Prism 6 and Adobe Illustrator CS6 were used for data analysis. Statistical significance was assessed by Student’s t-test and one-way analysis of variance (ANOVA) for experiments with more than two groups. P < 0.05 was considered statistically significant.
3. Results
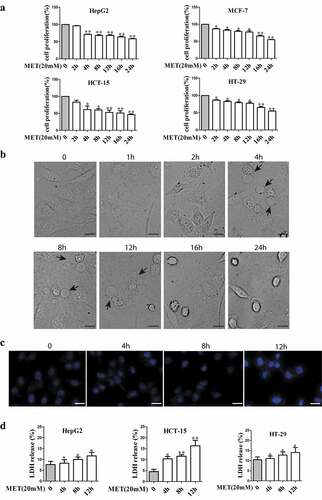
3.1. Metformin inhibited cell proliferation and induced pyroptosis in cancer cells
To examine proliferative effect of metformin on cancer cell lines, HepG2, MCF-7, HCT-15 and HT-29 cells were treated with metformin for various time (2, 4, 8, 12, 16 and 24 h) and MTT assay was performed (). The result showed that cell proliferation was inhibited after metformin treatment. Then, HepG2 cells were exposed to metformin for 1, 2, 4, 8, 12, 16 and 24 h. The phase-contrast images displayed that metformin did not induce typical pyroptotic morphology for 1 and 2 h, and the dying cells blew characteristic large bubbles from the plasma membrane, causing typical swelling when metformin treatment for 4, 8 and 12 h, then pyroptotic morphology disappeared upon metformin treatment for 16 and 24 h (). So 4, 8 and 12 h were selected for detecting molecular mechanism of pyroptosis with 20 mM metformin treatment. Hoechst staining assay further confirmed that there was no obvious cell apoptosis when HepG2 cells incubated with metformin for 4, 8 and 12 h (). As an indication of pyroptotic cell cytotoxicity, the release of lactate dehydrogenase (LDH) was detected in HepG2, MCF-7, HCT-15 and HT-29 cells with the same treatment, and the results showed that metformin enhanced LDH release (), which was consistent with the result in . These data together suggest that metformin triggered pyroptosis in cancer cells.
Figure 1. Metformin inhibited cell proliferation and induced pyroptosis in cancer cells. (a) Cell viability of cancer cells treated with 20 mM metformin at indicated time (2, 4, 8, 12, 16 and 24 h). (b) Representative microscopic images of HepG2 cells treated with 20 mM metformin for 1, 2, 4, 8, 12, 16 and 24 h. Black arrows indicate the characteristic balloon in the cell membrane. Scale bar is 50 μm. (c) Cell apoptosis rates were determined by using Hoechst and propidium iodide nuclear staining assay after HepG2 cells treated with 20 mM metformin for 4, 8 and 12 h. Scale bar is 50 μm. (d) Cytotoxicity of cancer cells was measured by LDH release in the culture supernatants; *P < 0.05, **P < 0.01 compared with control
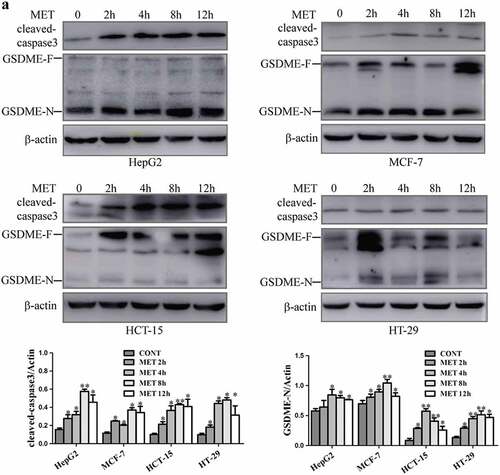
3.2. Metformin activated pyroptosis-associated proteins caspase3 and GSDME in cancer cells
Pyroptosis is triggered primarily by activation of the inflammatory caspases, which cleave a cellular substrate called GSDMD, generating a necrotic N-terminal fragment capable of inducing pyroptosis [Citation12]. Recently, shao et al have found that apoptotic caspases (caspase3) can cleave GSDME specifically to induce pyroptotic death in GSDME-expressing cells [Citation7]. In the present study, metformin time-dependently elevated the cleavage of caspase3 expression. The protein level of GSDME was detected and it constitutively expressed in HepG2, MCF-7, HCT-15 and HT-29 cells. When metformin was added to these cancer cells, GSDME-N protein level was increased for diverse periods of time (2, 4, 8 and 12 h) (), indicating that metformin effectively activated caspase3 cleavage to induce GSDME-mediated cancer cell pyroptosis.
Figure 2. Metformin increased expression of cleaved-caspase3 and GSDME-N in cancer cells. (a) Expression of cleaved-caspase3, full-length GSDME (GSDME-F) and GSDME-N terminal by western blotting analysis after 20 mM metformin treatment at indicated time (2, 4, 8, 12 h) in HepG2, MCF-7, HCT-15 and HT-29 cells. Quantitative results of cleaved-caspase3 and GSDME-N in the cells. *P < 0.05, **P < 0.01 compared with control
3.3. Metformin activated AMPK/SIRT1/NF-κB pathway to induce cancer cell pyroptosis
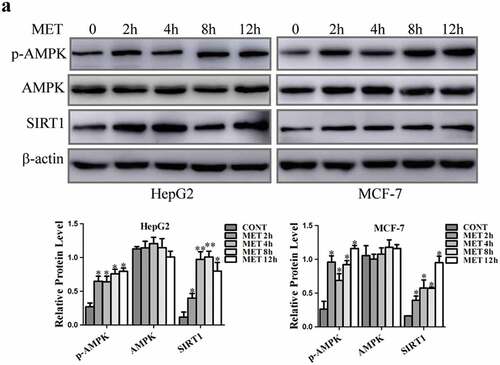
3.3.1. Metformin stimulated AMPK/SIRT1 signaling to cause pyroptotic death
Metformin activates AMPK, and AMPK further enhances SIRT1 activity by increasing cellular NAD+ levels to modulate downstream targets of SIRT1 [Citation20,Citation21]. When HepG2 and MCF-7 cells were treated with metformin for 2, 4, 8 and 12 h, our results showed that metformin led to a significant increase in p-AMPK and SIRT1 protein levels compared with control cells, but had no obvious modulatory function on AMPK expression (). The enhancement of SIRT1 expression was accorded with the increase of cleaved-caspase3 and GSDME-N protein levels maintained in metformin treatment with the extension of treating time.
Figure 3. Metformin induced cancer cell pyroptosis in a SIRT1-dependent manner. (a) AMPK, p-AMPK and SIRT1 expression were detected by western blotting after 20 mM MET treatment at indicated time (2, 4, 8 and 12 h) in cells and their quantification. *P < 0.05, **P < 0.01 compared with control
3.3.2. SIRT1 up-regulated NF-κB p65 expression to trigger caspase3/GSDME activation upon metformin treatment
SIRT1 plays a critical role in regulation of NF-κB expression to involve in inflammation process, cellular senescence and apoptosis induction [Citation27–Citation31]. NF-κB also induces caspases to contribute to apoptosis in neuroblastoma and colorectal cancer cells [Citation26,Citation31]. The expression of SIRT1 and its downstream targets were determined in HepG2 and MCF-7 cells. The results showed that metformin increased SIRT1 protein level and enhanced expression of NF-κB p65 and Bax to release cytochrome c, finally activating cleavage of caspase3 and GSDME. SIRT1 inhibitor EX527 treatment reversed metformin-induced these protein expression, and did not alter the metformin-induced decrease of PGC-1α level (). Knocking down SIRT1 in HepG2 by siRNA significantly reversed the increase of NF-κB p65 and pyroptosis-associated protein expression induced by metformin, and had no modulation of PGC-1α expression (), suggesting that NF-κB, Bax, cytochrome c, caspase3 and GSDME worked downstream of SIRT1 except for PGC-1α. Next, we also explored the effect of NF-κB on metformin-induced cancer cell pyroptosis. Results showed that metformin increased expression of NF-κB p65 and Bax to activate caspase3 and GSDME (). When NF-κB inhibitor BAY 11–7082 was added to HepG2 and MCF-7 cells, the metformin-induced these protein expression were suppressed. Above results indicate that metformin activated AMPK/SIRT1/NF-κB pathway to trigger caspase3/GSDME-mediated cancer cell pyroptosis.
Figure 4. SIRT1 increased protein level of NF-κB p65 and its downstream targets upon metformin treatment in cancer cells. (a) SIRT1, PGC-1α, NF-κB p65, Bax, cytochrome c, cleaved-caspase3, GSDME-F and GSDME-N protein levels were detected in cells treated with 20 mM MET for 8 h in the absence or presence of 10 μM EX527 for 24 h. (b) Protein levels of SIRT1, PGC-1α, NF-κB p65, cleaved-caspase3 and GSDME-N after MCF-7 cells were transfected with SIRT1 siRNA, incubated for 36 h and further treated with or without 20 mM MET for 8 h. *P < 0.05, **P < 0.01 compared with control
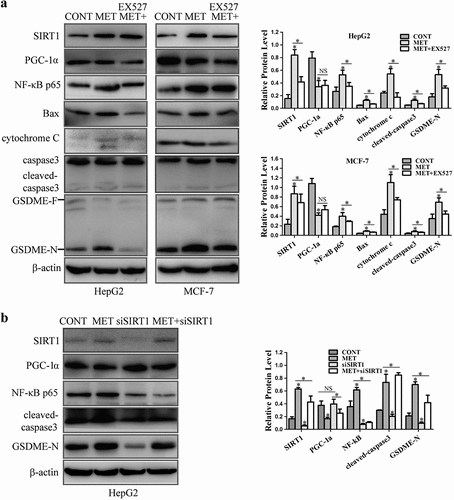
Figure 5. NF-κB activated pyroptosis-associated proteins treatment with metformin. (a) HepG2 and MCF-7 cells were incubated with 10 μM BAY 11–7082 for 1 h, and then treated with or without 20 mM MET for another 8 h. NF-κB p65, Bax, cleaved-caspase3 and GSDME-N expression were detected and their quantification. *P < 0.05, **P < 0.01 compared with control
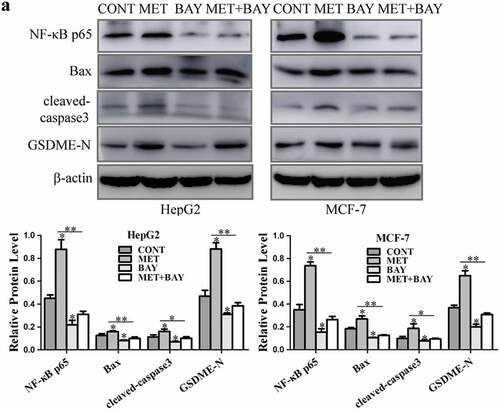
3.4. Metformin-induced mitochondrial dysfunction activated caspase3 and GSDME in cancer cells
Mitochondrial dysfunction may dissipate mitochondrial membrane potential (ΔΨm) [Citation32] and lead to increase of reactive oxygen species (ROS) [Citation33]. PGC-1α is considered as a central regulator of mitochondrial biogenesis in cancer cells [Citation34]. CCCP, a potential chemical uncoupler, induces mitochondrial depletion to trigger pyroptosis by cleavage of pyroptosis-associated proteins in melanoma A375 cells [Citation3]. In order to determine the association between mitochondrial function and pyroptosis induction after metformin treatment, the effects of metformin on mitochondrial function were evaluated. JC-1 assay was performed and results showed that metformin decreased mitochondrial membrane potential in HepG2 and MCF-7 cells (). The ROS level was elevated after metformin treatment by using SpectraMax M5 microplate reader (). To consolidate this result, DCFH.DA staining was performed and the analysis of fluorescent images revealed that metformin increased the level of ROS significantly (). Metformin inhibited expression of PGC-1α in HepG2 and MCF-7 cells, while CCCP was added to the cells, PGC-1α protein level was further repressed efficiently (), and cleaved-caspase3 and GSDME-N levels were enhanced markedly (). These results imply that mitochondrial dysfunction might involve in caspase3/GSDME-mediated cancer cell pyroptosis in the presence of metformin.
Figure 6. Metformin-induced mitochondrial dysfunction cleaved pyroptosis-associated proteins in cancer cells. (a) Mitochondrial membrane potential was measured by JC-1 assay kit when treatment with 20 mM MET for 8 h in cells and their quantification. Scale bar is 100 μm. (b) ROS was measured by reactive oxygen species assay kit after HepG2 cells treated by 20 mM MET for 4, 8 and 12 h. (c) Fluorescent microscopy images of DCFH.DA assay upon 20 mM MET treatment for 8 h in MCF-7 cells. Scale bar is 50 μm. (d) PGC-1α protein was detected by western blotting in cells treated with 20 mM MET for 8 h in the absence or presence of 40 μM CCCP for 24 h. (e) Cleaved-caspase3, GSDME-F and GSDME-N expression after cells treated with 40 μM CCCP for 24 h. *P < 0.05, **P < 0.01 compared with control
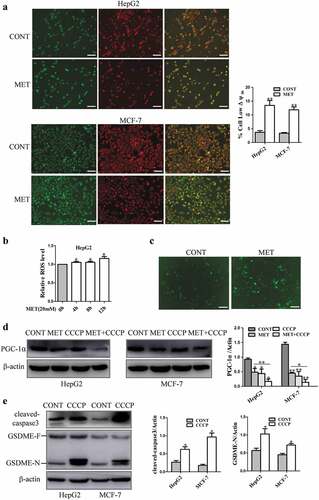
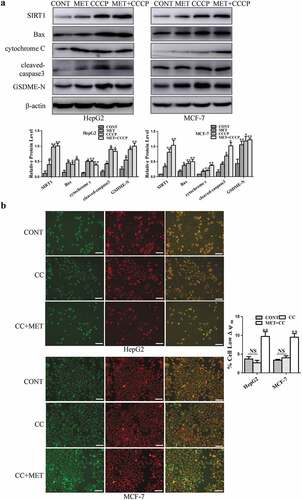
3.5. Mitochondrial dysfunction induced AMPK/SIRT1 pathway to drive pyroptotic death upon metformin treatment
It has been reported that mitochondrial uncoupler CCCP stimulates AMPK activation to inhibit artery constriction in vascular smooth muscle cells [Citation35]. The result in showed that metformin increased expression of p-AMPK and SIRT1. Thus, we tested whether mitochondrial dysfunction triggers cancer cell pyroptosis via AMPK/SIRT1 pathway induced by metformin. As illustrated in , expression of cytochrome c, Bax and pyroptosis-associated proteins were determined in HepG2 and MCF-7 cells. The results showed that metformin increased these protein expression. When CCCP was added to the cells, expression of SIRT1 and metformin-induced these proteins were further enhanced dramatically. In contrast, cancer cells were exposed to AMPK inhibitor Compound C for 24 h, there was no effect on mitochondrial membrane potential (), suggesting that AMPK activation could not induce mitochondrial dysfunction. Altogether these results indicate that mitochondrial dysfunction induced AMPK/SIRT1 signaling to cause cancer cell pyroptosis upon metformin treatment.
Figure 7. Mitochondrial dysfunction drived cancer cell pyroptosis via AMPK/SIRT1 pathway upon metformin treatment. (a) SIRT1, Bax, cytochrome c, cleaved-caspase3 and GSDME-N expression after cells treated with 20 mM MET in the absence or presence of 40 μM CCCP for 24 h in cells and their quantification. (b) Mitochondrial membrane potential was measured by JC-1 assay kit after 3 μM Compound C (CC) treatment for 24 h with or without 20 mM MET for 8 h in cells and their quantification. Scale bar is 100 μm; *P < 0.05, **P < 0.01 compared with control
4. Discussion
Cancer is a disease with high mortality and poor prognosis owing to its high incidence of relapse and metastasis in advanced stage [Citation36–Citation38]. In both sexes combined, lung, breast, liver, prostate and colon cancers lead to the most cancer deaths in 2018. Among females, breast cancer is the most commonly diagnosed cancer and the leading cause of cancer death, followed by colorectal and lung cancer [Citation1]. Thus, developing new strategies for cancer treatment is essential. Pharmacological intervention is an attractive approach for cancer prevention. Metformin, an antidiabetic drug, exerts the potential antitumorigenic effects in multiple in vitro and in vivo studies on several cancer models [Citation39]. On one hand, high concentrations of metformin suppress ovarian cancer growth and migration in vitro and in vivo [Citation40], and lead to cell cycle arrest and induce cell apoptosis in prostate cancer cells [Citation41]. On the other hand, low concentrations of metformin induce cellular senescence to suppress tumor stem cell transformation and inhibit HCC tumor growth [Citation42,Citation43]. Metformin prolongs lifespan and represses the mammary adenocarcinoma (MAC) of HER-2/neu transgenic mice in vivo [Citation44]. Metformin can activate caspase3 to trigger the apoptotic pathway in human adrenocortical carcinoma and pancreatic cancer cells [Citation45,Citation46]. Up to date, there are two important programmed cell death (PCD) pathways that include apoptotic (non-lytic) and pyroptotic (lytic) death [Citation47]. It is well known that apoptosis is a non-inflammatory form of PCD mediated by activation of the apoptotic caspases and can occur either via extrinsic pathway (also called death receptor) or intrinsic mitochondrial pathway. Both pathways converge on the activation of the executioner caspases, which target several hundred substrates to coordinate morphological changes related to apoptosis [Citation48,Citation49]. Many studies have shown that apoptosis plays a significant role in the eukaryotic development and maintenance of organismal homeostasis [Citation50], inflammatory and immunity response [Citation51–Citation53] and cancer progression [Citation54–Citation57]. Pyroptosis is a newly identified pathway of proinflammatory programmed cell death [Citation58]. The cells undergoing pyroptosis release intracellular contents, which additionally excites an inflammatory outcome [Citation59]. It occurs when GSDMD is cleaved by inflammatory caspases to generate an N-terminal [Citation12,Citation60,Citation61]. GSDME shares the same N-terminal domain of GSDMD and perforates the plasma membrane to drive pyroptosis. This pyroptotic death at least partly relies on caspase3-mediated cleavage of GSDME in GSDME-expressing cells [Citation7]. GSDME is silenced in multiple cancer cells but expressed in many kinds of normal tissues. The higher expression of exogenous GSDME in SH-SY5Y neuroblastoma cells drives more extensive pyroptosis than that is observed in wild-type SH-SY5Y cells induced by chemotherapy drugs [Citation7]. GSDME thus has been implicated as a tumor suppressor in cancer [Citation15,Citation62]. GSDME-positive normal human primary cells undergo pyroptosis upon activation of caspase3 by chemotherapy drugs. GSDME−/− mice are prevented from chemotherapy-induced tissue damage. Relatively high expression of GSDME can override the induction of typical apoptosis in caspase3-activated cells [Citation7]. However, it is unclear whether metformin induces pyroptotic death and the underlying mechanism in cancer cells. In this study, we found that metformin reduced cell viability for diverse periods of time (0–24 h), which is consistent with previous findings [Citation63,Citation64]. Metformin (10 mM treatment for 12 and 24 h) induces mitochondrial apoptosis by increasing protein levels of Bax and cleaved-caspase3 in ovarian cancer cells [Citation18]. Sun et al report that metformin (0.4 mM treatment for 48 h) significantly promotes expression of cleaved-caspase3 and caspase9 to drive HepG2 and Bel-7402 cell apoptotic death [Citation65]. Our results showed that pyroptosis (as judged by cell swelling with large bubbles blowing from the plasma membrane) other than apoptosis occurred when 20 mM metformin treatment for 4, 8 and 12 h in cancer cells. Both two types of cell death are dependent on Bax accumulation and caspase3 cleavage. And different concentrations and treatment times of metformin may determine the type of cancer cell death. These cancer cells that progress to pyroptotic death constitutively express GSDME. Metformin could cleave GSDME to generate more GSDME-N by activation of caspase3 in the GSDME-expressing cells ( and ), indicating that metformin activated caspase3/GSDME to induce cancer cell pyroptosis.
Caspase3 can be cleaved by NF-κB to involve in process of apoptosis in colorectal cancer cells [Citation31]. In human neuroblastoma cells, NF-κB also lead to activation of intrinsic apoptotic pathway through a mechanism involving Bax accumulation, cytochrome c release and caspase3 cleavage [Citation26]. SIRT1 is an essential regulator of inflammation by altering NF-κB [Citation30], and the modulatory effect of SIRT1 on NF-κB are controversial in the following articles. SIRT1 inhibits NF-κB activity to regulate colorectal cancer cell migration and invasion, inflammation-associated diseases and cancer [Citation29,Citation66–Citation68]. In contrast, SIRT1 activates NF-κB in the biological processes of oxidative stress and inflammation in vivo and in vitro [Citation69]. Metformin directly targets mitochondrial complex I to suppress mitochondrial oxidative phosphorylation, leading to decrease of ATP production and increase of AMP/ATP ratio to stimulate AMPK activation [Citation70,Citation71], which regulates the expression of genes related to mitochondrial and lipid metabolism through activation of SIRT1 in C2C12 myotubes and skeletal muscle. AMPK also increases NAD+ synthesis and the activity of SIRT1 in cells with oxidative stress (H2O2)-induced senescence [Citation20,Citation21]. Our present study revealed that metformin activated AMPK/SIRT1 pathway in a time-dependent manner, and then increased NF-κB p65 protein levels in cancer cells. In contrast, depletion of SIRT1 using siRNA and EX527 significantly attenuated metformin-induced expression of NF-κB p65 and its downstream targets, indicating that metformin promoted NF-κB p65 expression by activating SIRT1 ( and ). Furthermore, inhibition of NF-κB p65 reversed metformin-induced expression of Bax and pyroptosis-associated proteins (). These data suggest that metformin activated AMPK/SIRT1/NF-κB pathway to stimulate caspase3/GSDME-mediated pyroptotic death in cancer cells.
Mitochondrial uncouplers decrease cellular ATP levels and activate AMPK. CCCP considered as a classical mitochondrial uncoupler increases the cellular ADP/ATP ratio to activate AMPK in thoracic aorta vascular smooth muscle A10 cells [Citation72]. AMPK activates SIRT1 to result in lung cancer cell apoptosis [Citation73]. Stimulation of the mitochondrial apoptotic pathway by Bax leads to caspase3 activation to cleave GSDME in GSDME-expressing 293 T cells [Citation15]. CCCP is able to induce mitochondrial depletion via the mitophagy pathway. It initiates ROS signaling to facilitate Tom20-Bax-caspase3-GSDME pathway, eventually executing cell-lytic pyroptosis in melanoma cells [Citation3]. Recently, molecular targeted therapies with protein kinase inhibitors can drive caspase3/GSDME-dependent pyroptosis in lung cancer cells through the mitochondrial intrinsic apoptotic pathway [Citation74]. It is generally believed that metformin induces mitochondrial dysfunction to inhibit tumor growth in human diabetic patients [Citation17,Citation75]. In line with these studies, we demonstrated that metformin significantly induced mitochondrial dysfunction in HepG2 and MCF-7 cells. In response to CCCP, cleaved-caspase3 and GSDME-N expression were elevated clearly by induction of mitochondrial damage ( and ), suggesting that metformin-induced mitochondrial dysfunction might trigger caspase3/GSDME-mediated cancer cell pyroptosis. Furthermore, the results in have shown that metformin increased SIRT1 expression and stimulated cleavage of pyroptosis-associated proteins by inducing mitochondrial dysfunction. CCCP administration further elevated expression of SIRT1 significantly. Additionally, we observed that AMPK inhibition could not modulate mitochondrial dysfunction (). Thus, mitochondrial dysfunction induced AMPK/SIRT1 pathway to trigger caspase3/GSDME-mediated cancer cell pyroptosis upon metformin treatment.
In summary, our study demonstrated for the first time that metformin induced cell pyroptosis to exert anticancer effects. Metformin activated AMPK/SIRT1/NF-κB pathway to facilitate Bax accumulation and cytochrome c release, eventually triggering activation of caspase3 and cleavage of GSDME in cancer cells. Moreover, Metformin promoted mitochondrial dysfunction to induce AMPK/SIRT1 pathway, driving pyroptosis . Therefore, the present study identifies a previously unknown role of AMPK/SIRT1/NF-κB pathway in cancer cell pyroptotic death. Our data may also provide a theoretical basis for the application of metformin in the treatment of GSDME-expressing cancers.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data that support the findings of this study are available from the corresponding author, [author initials], upon reasonable request.
Additional information
Funding
References
- Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
- Bogdan AR, Miyazawa M, Hashimoto K, et al. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci. 2016;41:274–286.
- Zhou B, Zhang JY, Liu XS, et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28:1171–1185.
- Thakur B, Kumar Y, Bhatia A. Programmed necrosis and its role in management of breast cancer. Pathol Res Pract. 2019;215(11):152652.
- Gonzalez LC, Ghadaouia S, Martinez A, et al. Premature aging/senescence in cancer cells facing therapy: good or bad? Biogerontology. 2016;17:71–87.
- Wang Y, Yin B, Li D, et al. GSDME mediates caspase-3-dependent pyroptosis in gastric cancer. Biochem Biophys Res Commun. 2018;495:1418–1425.
- Wang Y, Gao W, Shi X, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103.
- Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42:245–254.
- Chen LM, Kaniga K, Galan JE. Salmonella spp. are cytotoxic for cultured macrophages. Mol Microbiol. 1996;21:1101–1115.
- Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265:130–142.
- Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121.
- Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158.
- Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116.
- Zhang CC, Li CG, Wang YF, et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis. 2019;24:312–325.
- Rogers C, Fernandes-Alnemri T, Mayes L, et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128.
- Mai FY, He P, Ye JZ, et al. Caspase-3-mediated GSDME activation contributes to cisplatin- and doxorubicin-induced secondary necrosis in mouse macrophages. Cell Prolif. 2019;52:e12663.
- Sanchez-Alvarez R, Martinez-Outschoorn UE, Lamb R, et al. Mitochondrial dysfunction in breast cancer cells prevents tumor growth: understanding chemoprevention with metformin. Cell Cycle. 2013;12:172–182.
- Wu Y, Gao WN, Xue YN, et al. SIRT3 aggravates metformin-induced energy stress and apoptosis in ovarian cancer cells. Exp Cell Res. 2018;367:137–149.
- Choi YK, Park KG. Metabolic roles of AMPK and metformin in cancer cells. Mol Cells. 2013;36:279–287.
- Han X, Tai H, Wang X, et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell. 2016;15:416–427.
- Canto C, Gerhart-Hines Z, Feige JN, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060.
- Ji S, Zheng Z, Liu S, et al. Resveratrol promotes oxidative stress to drive DLC1 mediated cellular senescence in cancer cells. Exp Cell Res. 2018;370:292–302.
- Nennig SE, Schank JR. The role of NFkB in drug addiction: beyond inflammation. Alcohol Alcohol. 2017;52:172–179.
- Liu T, Liu S, Ma L, et al. Oogenesis, vitellogenin-mediated ovarian degeneration and immune response in the annual fish Nothobranchius guentheri. Fish Shellfish Immunol. 2017;66:86–92.
- Shan S, Liu R, Feng H, et al. Identification and functional characterization of the transcription factor NF-kappaB subunit p65 in common carp (Cyprinus carpio L.). Fish Shellfish Immunol. 2019;95:25–34.
- Posadas I, Santos P, Cena V. Acetaminophen induces human neuroblastoma cell death through NFKB activation. PLoS One. 2012;7:e50160.
- Wang P, Lv C, Zhang T, et al. FOXQ1 regulates senescence-associated inflammation via activation of SIRT1 expression. Cell Death Dis. 2017;8:e2946.
- Wang H, Han L, Zhao G, et al. hnRNP A1 antagonizes cellular senescence and senescence-associated secretory phenotype via regulation of SIRT1 mRNA stability. Aging Cell. 2016;15:1063–1073.
- Liu S, Zheng Z, Ji S, et al. Resveratrol reduces senescence-associated secretory phenotype by SIRT1/NF-kappaB pathway in gut of the annual fish nothobranchius guentheri. Fish Shellfish Immunol. 2018;80:473–479.
- Vachharajani VT, Liu T, Wang X, et al. Sirtuins link inflammation and metabolism. J Immunol Res. 2016;2016:8167273.
- Buhrmann C, Shayan P, Goel A, et al. Resveratrol regulates colorectal cancer cell invasion by modulation of focal adhesion molecules. Nutrients. 2017;9:1073.
- Polo M, Alegre F, Moragrega AB, et al. Lon protease: a novel mitochondrial matrix protein in the interconnection between drug-induced mitochondrial dysfunction and endoplasmic reticulum stress. Br J Pharmacol. 2017;174:4409–4429.
- Correia-Melo C, Marques FD, Anderson R, et al. Mitochondria are required for pro-ageing features of the senescent phenotype. Embo J. 2016;35:724–742.
- Luo C, Widlund HR, Puigserver P. PGC-1 coactivators: shepherding the mitochondrial biogenesis of tumors. Trends Cancer. 2016;2:619–631.
- Tai Y, Li L, Peng X, et al. Mitochondrial uncoupler BAM15 inhibits artery constriction and potently activates AMPK in vascular smooth muscle cells. Acta Pharm Sin B. 2018;8:909–918.
- Lordick F, Lorenzen S, Yamada Y, et al. Optimal chemotherapy for advanced gastric cancer: is there a global consensus? Gastric Cancer. 2014;17:213–225.
- Hu Y, Ma Z, He Y, et al. LncRNA-SNHG1 contributes to gastric cancer cell proliferation by regulating DNMT1. Biochem Biophys Res Commun. 2017;491:926–931.
- Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90.
- Pierotti MA, Berrino F, Gariboldi M, et al. Targeting metabolism for cancer treatment and prevention: metformin, an old drug with multi-faceted effects. Oncogene. 2013;32:1475–1487.
- Zheng Y, Zhu J, Zhang H, et al. Metformin inhibits ovarian cancer growth and migration in vitro and in vivo by enhancing cisplatin cytotoxicity. Am J Transl Res. 2018;10:3086–3098.
- Tong D, Liu Q, Liu G, et al. Metformin inhibits castration-induced EMT in prostate cancer by repressing COX2/PGE2/STAT3 axis. Cancer Lett. 2017;389:23–32.
- Yi G, He Z, Zhou X, et al. Low concentration of metformin induces a p53-dependent senescence in hepatoma cells via activation of the AMPK pathway. Int J Oncol. 2013;43:1503–1510.
- Gou S, Cui P, Li X, et al. Low concentrations of metformin selectively inhibit CD133(+) cell proliferation in pancreatic cancer and have anticancer action. PLoS One. 2013;8:e63969.
- Anisimov VN, Egormin PA, Piskunova TS, et al. Metformin extends life span of HER-2/neu transgenic mice and in combination with melatonin inhibits growth of transplantable tumors in vivo. Cell Cycle. 2010;9:188–197.
- Poli G, Cantini G, Armignacco R, et al. Metformin as a new anti-cancer drug in adrenocortical carcinoma. Oncotarget. 2016;7:49636–49648.
- Shi Y, He Z, Jia Z, et al. Inhibitory effect of metformin combined with gemcitabine on pancreatic cancer cells in vitro and in vivo. Mol Med Rep. 2016;14:2921–2928.
- Wallach D, Kang TB, Dillon CP, et al. Programmed necrosis in inflammation: toward identification of the effector molecules. Science. 2016;352:aaf2154.
- Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241.
- Barber GN. Host defense, viruses and apoptosis. Cell Death Differ. 2001;8:113–126.
- Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. 2019;20:175–193.
- Yazdani R, Fatholahi M, Ganjalikhani-Hakemi M, et al. Role of apoptosis in common variable immunodeficiency and selective immunoglobulin A deficiency. Mol Immunol. 2016;71:1–9.
- Urbonaviciute V, Furnrohr BG, Meister S, et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205:3007–3018.
- Liang YY, Rainprecht D, Eichmair E, et al. Serum-dependent processing of late apoptotic cells and their immunogenicity. Apoptosis. 2015;20:1444–1456.
- Kunac N, Sundov Z, Vilovic K. Apoptosis as a prognostic factor in colorectal carcinoma: comparison of TUNEL method and immunohistochemical expression of Caspase-3. Appl Immunohistochem Mol Morphol. 2019;27:e22–e27.
- Koornstra JJ, Rijcken FE, De Jong S, et al. Assessment of apoptosis by M30 immunoreactivity and the correlation with morphological criteria in normal colorectal mucosa, adenomas and carcinomas. Histopathology. 2004;44:9–17.
- Roth C, Pantel K, Muller V, et al. Apoptosis-related deregulation of proteolytic activities and high serum levels of circulating nucleosomes and DNA in blood correlate with breast cancer progression. BMC Cancer. 2011;11(1):4.
- Khan S, Simpson J, Lynch JC, et al. Racial differences in the expression of inhibitors of apoptosis (IAP) proteins in extracellular vesicles (EV) from prostate cancer patients. PLoS One. 2017;12:e0183122.
- Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–114.
- Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8:1812–1825.
- Rathkey JK, Benson BL, Chirieleison SM, et al. Live-cell visualization of gasdermin D-driven pyroptotic cell death. J Biol Chem. 2017;292:14649–14658.
- Aglietti RA, Dueber EC. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol. 2017;38:261–271.
- Kim MS, Chang X, Yamashita K, et al. Aberrant promoter methylation and tumor suppressive activity of the DFNA5 gene in colorectal carcinoma. Oncogene. 2008;27:3624–3634.
- Hu ZZ, Xia Q, Liu Z, et al. Metformin attenuates hepatoma cell proliferation by decreasing glycolytic flux through the HIF-1alpha/PFKFB3/PFK1 pathway. Life Sci. 2019;116966. DOI:10.1016/j.lfs.2019.116966
- Esparza-Lopez J, Alvarado-Munoz JF, Escobar-Arriaga E, et al. Metformin reverses mesenchymal phenotype of primary breast cancer cells through STAT3/NF-kappaB pathways. BMC Cancer. 2019;19:728.
- Sun R, Zhai R, Ma C, et al. Combination of aloin and metformin enhances the antitumor effect by inhibiting the growth and invasion and inducing apoptosis and autophagy in hepatocellular carcinoma through PI3K/AKT/mTOR pathway. Cancer Med. 2019;9:1141–1151.
- Yu S, Zhou R, Yang T, et al. Hypoxia promotes colorectal cancer cell migration and invasion in a SIRT1-dependent manner. Cancer Cell Int. 2019;19:116.
- Kulkarni SS, Canto C. The molecular targets of resveratrol. Biochim Biophys Acta. 2015;1852:1114–1123.
- Ren Z, Wang L, Cui J, et al. Resveratrol inhibits NF-kB signaling through suppression of p65 and IkappaB kinase activities. Pharmazie. 2013;68:689–694.
- Zhang Y, Tao X, Yin L, et al. Protective effects of dioscin against cisplatin-induced nephrotoxicity via the microRNA-34a/sirtuin 1 signalling pathway. Br J Pharmacol. 2017;174:2512–2527.
- Thakur S, Daley B, Klubo-Gwiezdzinska J. The role of the antidiabetic drug metformin in the treatment of endocrine tumors. J Mol Endocrinol. 2019. DOI:10.1530/JME-19-0083
- Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60:1577–1585.
- Zhang YQ, Shen X, Xiao XL, et al. Mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone induces vasorelaxation without involving KATP channel activation in smooth muscle cells of arteries. Br J Pharmacol. 2016;173:3145–3158.
- Zhang Y, Sun C, Xiao G, et al. S-nitrosylation of the Peroxiredoxin-2 promotes S-nitrosoglutathione-mediated lung cancer cells apoptosis via AMPK-SIRT1 pathway. Cell Death Dis. 2019;10:329.
- Lu H, Zhang S, Wu J, et al. Molecular Targeted Therapies Elicit Concurrent Apoptotic and GSDME-Dependent Pyroptotic Tumor Cell Death. Clin Cancer Res. 2018;24:6066–6077.
- Muti P, Berrino F, Krogh V, et al. Metformin, diet and breast cancer: an avenue for chemoprevention. Cell Cycle. 2009;8:2661.