
ABSTRACT
Nasal epithelial cells are the first barrier against allergen infiltration in allergic rhinitis (AR), and the relationship between nasal epithelial cells and mast cell-mediated hypersensitivity remains unclear. This study aimed to investigate the possible association between allergen-challenged nasal epithelial cells (AR-HNEpC) and mast cell degranulation in AR. Our data revealed that calcium influx and degranulation were increased in AR-HNEpC-co-cultured mast cells. Expression of IL-33, a factor that binds to ST2 receptors on mast cells and regulates their degranulation, was elevated in AR-HNEpC. Blocking IL-33/ST2 pathway activated autophagy and inhibited degranulation and inflammatory factor release in mast cells. Furthermore, PI3K/mTOR was increased in IL-33-treated mast cells. Inhibition on PI3K/mTOR pathway enhanced autophagy and inhibited degranulation. Analysis using an in vivo AR model supported the above findings. In conclusion, IL-33 from epithelial cells promotes degranulation of mast cells in AR through inhibition on ST2/PI3K/mTOR-mediated autophagy, which provides a potential therapeutic target for the disease.
Abbreviations: AR: allergic rhinitis; IL: interleukin; TNF-α: tumor necrosis factor-alpha; INF-γ: interferon-gamma; HNEpC: human nasal epithelial cell line; ATCC: American Type Culture Collection; C48/80: compound 48/80; 3-MA: 3-methyladenine; qPCR: quantitative PCR; AR-HNEpC: dust mite allergen-treated nasal epithelial cells; IgE: immunoglobulin E; Atg7: autophagy-related gene 7
KEYWORDS:
Introduction
Allergic rhinitis (AR) is a type of nasal mucosal inflammatory disease caused by allergen exposure [Citation1]. Commonly seen in otolaryngological clinics, AR is prevalent around the world and is closely associated with asthma, nasosinusitis, and nasal polyp [Citation2,Citation3]. Previous studies on the pathogenesis of AR focused much on the inflammatory cells and responses in the nasal mucosa tissue [Citation3–Citation5], while nasal epithelial cells, the first defending line against allergen infiltration, were seldom studied, especially the interactions between epithelial cells and inflammatory cells in AR.
Mast cells play key roles in the allergic inflammation in AR through release of histamine and inflammation factors, a process which is called degranulation [Citation6]. In this process, within seconds after stimulation of allergens or environmental factors, mast cells release a diverse array of factors, including histamine, heparin, serotonin, and serine proteases, which are responsible for the acute symptoms in allergic conditions [Citation7,Citation8]. Autophagy, which is another important cellular process, recycles intracellular components and helps cell survive [Citation9]. Dysfunction of autophagy is involved in many diseases. For example, autophagy exerts tumor-suppressive or tumor-promoting effects in different circumstances in cancer [Citation9]. Deficiency in autophagy promotes tumorigenesis in genetically engineered mouse model, e.g. liver tumors [Citation10], while in advanced cancer, autophagy suppresses inflammation in tumor regions and promotes survival of cancer cells [Citation11]. Defects of autophagy were also observed in metabolic disorders and inflammatory bowel disease [Citation12]. For example, mutation in ATG16L1 gene was linked to increased risk of Crohn’s disease in genome-wide association studies [Citation13]. Upregulation of autophagy acted as protective roles in neurodegenerative diseases [Citation14]. In Huntington’s disease, chaperone-mediated autophagy was up-regulated to compensate the impaired macroautophagy in early stage of the disease [Citation15]. Previous studies found that autophagy was crucial in the degranulation of mast cells. Bone marrow-derived mast cells from mice lacking autophagy-related gene 7 (Atg7) showed severely impaired degranulation, and light chain 3 type 2 (LC3-II), an important marker for autophagy, was missing in the secretory granules of those mast cells [Citation16,Citation17]. However, it is still unknown if epithelial cells play any role in the autophagy and degranulation of mast cells.
Previous study revealed that the expression levels of interleukin (IL)-33 and its receptor ST2 were significantly elevated in epithelial cells in AR [Citation18]. IL-33 is a recently discovered cytokine that is produced by specialized epithelial cells and plays important roles in the pathogenesis of various allergic diseases [Citation19–Citation21]. In addition, IL-33 is able to suppress autophagy and exerts neuroprotection functions [Citation22,Citation23] and ameliorates experimental colitis by promoting autophagy of macrophages [Citation24]. It is, therefore, possible that epithelial cells regulate autophagy and degranulation of mast cells through IL-33, which could be a potential mechanism of the pathogenesis of AR.
In our study, we investigated the relationship between epithelial cell and mast cell degranulation in AR. Using shRNAs targeting IL-33, we further demonstrated the role of epithelial-cell-released IL-33 in the degranulation of mast cells and also revealed the underlying pathway, which serves as a novel view angle for the investigations on pathogenesis and treatment of AR.
Methods
Cell culture
Human nasal epithelial cell line (HNEpC) and mast cell line (LAD2) were supplied by American Type Culture Collection (ATCC, USA). HNEpC cells were cultured in RPMI2650 (Invitrogen, USA) and LAD2 cells were cultured in minimum essential medium (GIBCO, USA), with both culture media supplemented with 10% fetal calf serum (GIBCO, USA). Cells were cultured in a humid atmosphere at 37°C with 5% carbon dioxide. Transwell cell culture inserts (Corning, USA) were used to co-culture different types of cells. Compound 48/80 (C48/80, Sigma-Aldrich) was used at 0.1 mg/ml to stimulate the degranulation of mast cells [Citation25]. In order to suppress autophagy, 3-methyladenine (3-MA, Sigma-Aldrich) was used at 5 mmol/l. IL-33 (Sigma-Aldrich) treatment was performed at 10 ng/ml. Human recombinant ST2 protein (sST2) (ab219662, Abcam, USA) was used at 0.7 μg/ml [Citation26]. Dust mite allergen (Derp1, Greer Laboratories) was administered at 1 μg/ml or 10 μg/ml.
Cell transfection and vector construction
ShRNA targeting IL-33 (sh-IL-33) was obtained from Genechem (Shanghai, China). HNEpC cells were transfected with sh-IL-33 using Lipofectamine 2000 (Invitrogen, USA) as per the manufacturer’s protocol. Briefly, cells were seeded in 24-well plate and cultured until ~80% confluence. One microliter lipofectamine reagent was mixed with 25 μl Opti-MEM Medium, and 240 ng DNA were diluted in 25 μl Opti-MEM Medium. The two mixtures were combined within 30 min after preparation and incubated at room temperature for 5 min. Fifty microliters of the final working solution were then added into each well, mixed gently, and incubated for 2 days at 37°C.
Extraction of total RNA and quantitative PCR
For extraction of total RNA, cells were homogenized in 0.3 ml TRIzol Reagent, and isopropanol was added into the homogenates. The mixture was incubated for 10 mins and centrifuged at 12,000 x g for 10 min at 4°C. After re-suspending the pellet in 75% ethanol, the mixture was centrifuged at 7,500 x g for 5 min at 4°C. The pellet was air-dried for 5 mins and re-suspended in 20 μl RNase-free water. Absorbance at 260 nm and 280 nm was measured to calculate RNA concentration, and 1 μg RNA from each sample was used for reverse transcription at 60°C for 10 min. The cDNA template was mixed with master mix, assay buffer (ABI, USA), and specific primers for IL-33: forward 5ʹGCCTGTCAACAGCAGTCTACTG-3ʹ, reverse 5ʹ-TGTGCTTAGAGAAGCAAGATACTC-3ʹ. The reaction mixture was then transferred into a 96-well reaction plate and sealed with appropriate cover. Quantitative PCR was performed using Applied Biosystems 7500 System (ABI, USA).
Protein extraction and Western blot
After being homogenized in HLB buffer with protease inhibitors, cell lysates were mixed with SDS lysis buffer and boiled. Protein contents in cell lysates were measured using BCA Protein Assay Reagent (Pierce, USA), and equal amount of protein was loaded onto SDS gels, separated by electrophoresis, and then transferred to nitrocellulose membranes. After blocking in 5% skimmed milk, nitrocellulose membranes were incubated overnight with primary antibodies against light chain 3 (LC3, ab128025, Abcam), p62 (ab56416, Abcam), Beclin-1 (ab207612, Abcam), PI3 K (ab32089, Abcam), Akt (ab179463, Abcam), mTOR (ab134903, Abcam), phospho-PI3 K (p-PI3 K, ab182651, Abcam), phospho-Akt (p-Akt, ab38449, Abcam), phospho-mTOR (p-mTOR, ab109268, Abcam), or β-actin (ab8227, Abcam). Membranes were then incubated with goat anti-rabbit IgG (ab6721, Abcam) or goat anti-mouse IgG (ab6789, Abcam) secondary antibody for 2 h. Proteins were then visualized using a gel-imaging system.
ELISA measurement
ELISA kits were used to measure protein levels of β-hexosaminidase (orb440028, biorbyt, UK), histamine (ab213975, Abcam), tryptase (orb348936, biorbyt, UK), IL-33 (ab223865, Abcam), IL-4 (ab215089, Abcam), IL-6 (ab46027, Abcam), INF-γ (ab100538, Abcam), and TNF-α (ab181421, Abcam) as per the manufacturer’s instructions. Briefly, 50 μl of samples or standards were added to appropriate wells. Antibody cocktail was added into each well, and plate was sealed and incubated at room temperature for 1 h with gentle shaking at 400 rpm. Each well was washed 3 times with wash buffer. TMB substrate was added to each well and plates were incubated for 10 min in the dark with shaking at 400 rpm. After adding stop solution into each well, optical density values were measured at 450 nm using a microplate reader.
Measurement of calcium concentration using fluo-3AM
Intracellular calcium levels were measured using fluo-3 AM (F1242, Invitrogen). Briefly, fluo-3 AM stock solution was diluted into 1 μM working solution using PBS. Fluo-3 AM working solution was then incubated with cells for 60 min at room temperature. After the incubation, wells were washed with PBS for 3 times and incubated for another 30 min. Results were obtained using a microplate reader (485/20 nm excitation and 528/20 nm emission).
Toluidine blue staining
Tissue samples were fixed in 4% formaldehyde, dehydrated and embedded in paraffin. Slides were sectioned at 5 μm and then de-paraffinized and hydrated before the staining. Slides were stained in Toluidine blue for 1 min and then washed under de-ionized water. After dehydration, slides were mounted and covered with coverslip before examination under microscope.
Establishment of in vivo AR model
A total of 20 SD rats were obtained and housed in our institute animal-holding unit with 12 h:12 h light/dark cycle. After accommodation, animals were randomly allocated into four groups (control group, AR model group, AR model group with sST2 treatment, and AR model group with rapamycin treatment). In vivo AR model was generated by treating nasal mucosa of rats using 1 μg/ml Derp1 (10 ml) on day 0 and 10 μg/ml Derp1 (10 ml) from day 7 to day 11, while normal saline was used for control group. For AR model group with sST2 treatment and AR model group with rapamycin treatment, at 40 min before the second sensitization, 10 ml solution containing 0.7 μg/ml sST2, or 10 μg/ml Derp1 and 0.2 nM rapamycin was administered to the nasal mucosa of rats. All the procedures were reviewed and approved by the Institutional Animal Care and Use Committee.
Statistical analysis
For the statistical analysis performed in this article, GraphPad Prism 5 (GraphPad Software, USA) was used, and differences between different groups were calculated using Student t-test or one-way ANOVA if there are more than two groups. Difference was considered statistically significant if P < 0.05.
Results
Dust mite allergen-treated nasal epithelial cells (AR-HNEpC) promoted degranulation of mast cells
To generate AR-HNEpC cells, HNEpC cells were treated with 5 μg/ml dust mite allergen (Derp1) for 24 h. AR-HNEpC cells or untreated HNEpC cells were co-cultured overnight with LAD2 cells in Transwell inserts. LAD2 cells were then treated with 0.1 mg/ml C48/80, a mast cell degranulator [Citation25], for 10 min. ELISA results showed that expression levels of mast cell degranulation markers (β-hexosaminidase, histamine, and tryptase) were increased in LAD2 cells which were co-cultured with AR-HNEpC cells, compared to control (), indicating that AR-HNEpC cells could promote mast cell degranulation. Furthermore, calcium levels were augmented in AR-HNEpC-co-cultured LAD2 cells ()). Toluidine blue staining further convinced the findings that degranulation activities were stimulated in AR-HNEpC-co-cultured LAD2 cells ()). In all, those results demonstrated that allergen-treated epithelial cells enhanced the degranulation of mast cells.
Figure 1. AR-HNEpC cells promoted mast cell degranulation. In the mast cells (LAD2) co-cultured with AR-HNEpC cells (Derp1-treated HNEpC) or untreated HNEpC (HNEpC), levels of granule components (a) β-hexosaminidase, (b) histamine, and (c) tryptase were upregulated in Derp1+ HNEpC group. (d) Intracellular calcium levels were stimulated in Derp1-HNEpC group. (e) Toluidine blue showed stronger staining in mast cell cytoplasm in Derp1-treated HNEpC group. **P < 0.01
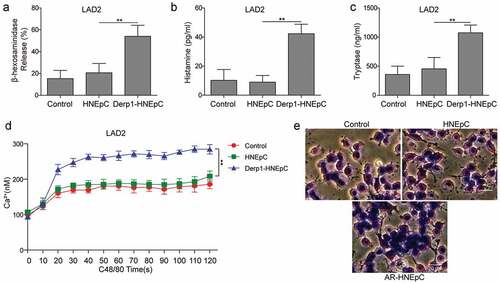
AR-HNEpC cells inhibited autophagy and promoted the degranulation of mast cells through IL-33/ST2
IL-33 expression was upregulated in AR-HNEpC cells ()), indicating that IL-33 expression was elevated in epithelial cells when challenged with allergen (Derp1). In order to investigate if the elevation in IL-33 expression in HNEpC could influence the autophagy and degranulation of mast cells, AR-HNEpC cells were transfected with sh-IL-33. ELISA results demonstrate that AR-HNEpC increased protein levels of IL-33, which was inhibited by transfection of sh-IL-33 in AR-HNEpC ()). Moreover, LAD2 cells were treated with sST2 which blocked IL-33/ST2 signaling pathway [Citation27] and co-cultured with AR-HNEpC cells (AR+sST2 group). Subsequently, expression levels of autophagy-related proteins (LC3-I/II, p62 and Beclin-1) and degranulation in LAD2 were evaluated. Western blot assay illustrated that autophagy was activated in LAD2 cells which were co-cultured with AR-HNEpC cells and depressed after silencing IL-33 or blocking IL-33/ST2 pathway ()). These results suggested that allergen treatment stimulated secretion of IL-33 from HNEpC, which then inhibited autophagy of mast cells through IL-33/ST2 signaling pathway.
Figure 2. AR-HNEpC cells regulated autophagy and degranulation of mast cells through IL-33/ST2 pathway. (a) Expression of IL-33 mRNA was upregulated in AR-HNEpC (Derp1-induced HNEpC cells). (b) Protein expression of IL-33 was upregulated in AR-HNEpC, which was inhibited when AR-HNEpC cells were transfected with sh-IL-33. (c) LAD2 cells that were co-cultured with AR-HNEpC cells showed lower LC3 I/II and Beclin-1 expression, while levels of those factors were higher after transfection of sh-IL-33 in AR-HNEpC cells (AR+sh-IL-33), or treatment of sST2 in LAD2 cells. (d) After inhibiting IL-33 in AR-HNEpC cells or inhibiting ST2 in mast cells (LAD2), mast cell degranulation was decreased, and (e) β-hexosaminidase, (f) IL-4, and (g) IL-6 expression levels were also lower compared to control groups (AR and AR+sh-NC). *P < 0.05; **P < 0.01
Furthermore, Toluidine blue staining manifested that mast cell degranulation was suppressed after silencing IL-33 or blocking IL-33/ST2 pathway ()). Further results from ELISA measurement testified that secretion of β-hexosaminidase, IL-4, and IL-6 in LAD2 cells were aggravated by AR-HNEpC, which were counteracted by knockdown of IL-33 in AR-HNEpC or treatment of sST2 on LAD2 cells (). All the above data indicated that AR-HNEpC cell suppressed autophagy and promoted degranulation of mast cells through IL-33/ST2 pathway.
IL-33/ST2 regulated autophagy of mast cells via PI3K/Akt/mTOR pathway
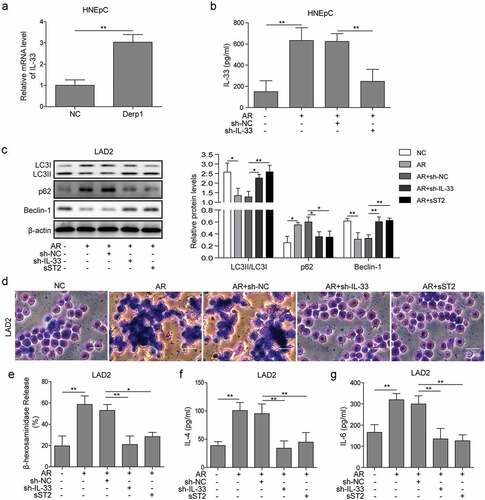
In order to investigate the role of IL-33/ST2 and PI3 K/mTOR pathway in autophagy of mast cell, LAD2 cells were treated with 10 ng/ml IL-33 for 24 h, together with treatment of sST2 or selective inhibitor on PI3 K (LY294002), Akt (AZD5363), or mTOR (Rapamycin). The results showed that levels of p-PI3 K, p-Akt, and p-mTOR were elevated in IL-33-treated LAD2 cells, while treatment of sST2 or selective inhibitors on PI3 K, Akt, or mTOR resulted in blocking of the PI3K/Akt/mTOR pathway ()). Furthermore, IL-33 decreased LC3-I/II and Beclin-1 levels and increased p62 levels, indicating inhibited autophagy in LAD2 cells, while sST2 or selective inhibitors counteracted the effects of IL-33 on autophagy ()). Taken together, those results indicated that IL-33/ST2 pathway inhibited autophagy of mast cells through regulation on PI3K/Akt/mTOR pathway.
Figure 3. IL-33/ST2 pathway regulated autophagy through PI3K/mTOR pathway in mast cells. (a) Levels of p-PI3K, p-Akt, and p-mTOR were increased in IL-33-treated LAD2 cells, which was selectively inhibited by sST2 (IL-33/sST2 blocker), LY294002 (PI3K inhibitor), AZD5363 (Akt inhibitor), or rapamycin (mTOR inhibitor). (b) IL-33 treatment inhibited expression of LC3-I/II and Beclin-1 and promoted expression of p62, which were reversed by treatment of selective inhibitors on ST2, PI3 K, Akt, or mTOR.*P < 0.05; ** P < 0.01; ***P < 0.001
AR-HNEpC cells regulated degranulation of mast cells via PI3K/mTOR pathway and autophagy
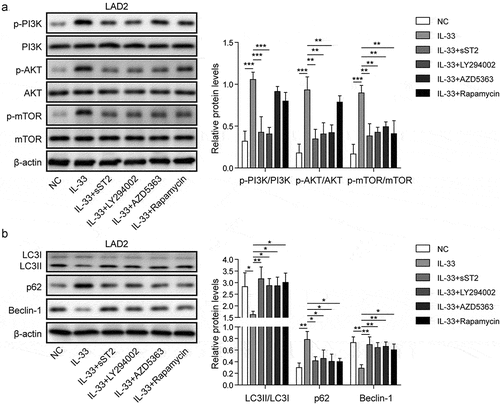
Firstly, LAD2 cells were treated with 3-MA, an autophagy inhibitor [Citation28], and then stimulated by C48/80. Results showed that release of β-hexosaminidase, histamine, IL-4, and IL-6 was remarkably accelerated, and intracellular calcium level and degranulation were increased (), which was similar as the increase of degranulation in AR-HNEpC-co-cultured LAD2 cells (). Moreover, investigation showed that treatment of rapamycin in co-cultured AR-HNEpC and LAD2 cells (AR+rapamycin) decreased intracellular calcium level and degranulation of mast cells, together with lower release of β-hexosaminidase, histamine, IL-4, and IL-6 (). Those findings demonstrated that AR-HNEpC cells regulated degranulation of mast cells through autophagy and PI3K/Akt/mTOR pathway.
Figure 4. AR-HNEpC cells regulated degranulation of mast cells via PI3K/mTOR pathway and autophagy. Mast cells (LAD2) were treated with 3-MA (NC+3-MA) or co-cultured with AR-HNEpC cells and treated with rapamycin (AR+rapamycin). After stimulation using C48/80, release of (a) β-hexosaminidase, (b) histamine, (c) IL-4, and (d) IL-6 was promoted in NC+3-MA group but not in AR+rapamycin group; (e) levels of intracellular calcium, (f) and degranulation of mast cells were elevated in NC+3-MA group, which was not observed in AR+rapamycin group. **P < 0.01
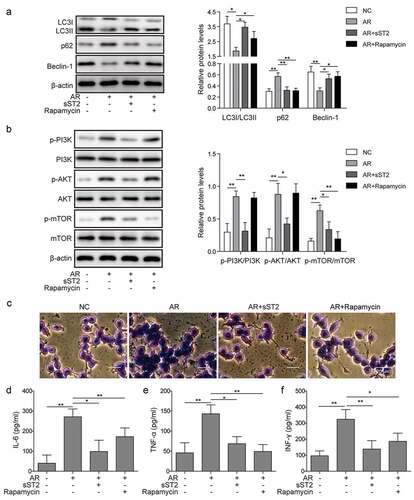
Allergen exposure regulated autophagy and degranulation of mast cells through IL-33/ST2 and PI3K/Akt/mTOR pathways in an AR model
To further explore the roles of IL-33/ST2 and PI3K/Akt/mTOR pathways in the regulation of epithelial cells on the autophagy and degranulation of mast cells, SD rats were used to establish AR model. Levels of autophagy were decreased in AR model (AR), while recovered by co-treatment of Derp1 with sST2 or rapamycin ()). Additionally, PI3K/Akt/mTOR pathway was activated in AR group, while it was blocked by sST2 or rapamycin. Furthermore, mast cell degranulation and release of inflammatory factors: interleukin-6(IL-6), interferon-gamma (INF-γ), and tumor necrosis factor-alpha (TNF-α) were stimulated in AR group, which were inhibited by sST2 or rapamycin (). Those data supported our in vitro findings that allergen exposure regulated autophagy and degranulation of mast cell through IL-33/ST2 and PI3K/Akt/mTOR pathways.
Figure 5. Allergen exposure regulated autophagy and degranulation of mast cells through IL-33/ST2 and PI3K/mTOR pathways in an AR model. An AR model was generated using SD rats and treated with sST2 (AR+sST2) or rapamycin (AR+rapamycin). (a) AR group showed that expression levels of LC3 I/II and Beclin-1 were decreased, and p62 was increased, which were reversed by sST2 or rapamycin treatment. (b) AR treatment enhanced the activation of PI3K/Akt/mTOR pathway proteins, which were blocked by sST2 or rapamycin. (c) Mast cell degranulation and (d–f) levels of IL-6, TNF-α and INF-γ were promoted by AR treatment, which was neutralized by sST2 or rapamycin treatment. *P < 0.05; **P < 0.01
Discussion
AR affects a great number of population worldwide, and as a chronic disease, symptoms of AR can severely influence patients’ quality of life, sleep, and work productivity [Citation29–Citation31]. Symptoms of AR result from a complex allergen-driven inflammatory response in nasal mucosa, which involves immunoglobulin E (IgE) and different types of inflammatory cells and pro-inflammatory factors, e.g. cytokines [Citation3,Citation31]. Among those inflammatory cells, mast cells and IgE were found to mediate immediate hypersensitivity response through their interactions, which triggers mast cells to release biologically active products from both pre-formed products in granules, a process called degranulation, and newly formed lipid-derived mediators and certain cytokines [Citation32,Citation33]. On the other hand, as the first barrier against allergen infiltration, epithelial cells respond to allergen exposure through releasing a wide array of factors [Citation34,Citation35]. Therefore, investigation on their interaction with mast cells is an interesting new angle in the AR research.
Consistent with previous findings [Citation18], our preliminary data demonstrated that IL-33 expression was elevated in AR-HNEpC cells (). Functionally, IL-33 has been found to play important roles in the pathogenesis of allergic disorders [Citation19]. On allergen exposure or parasite infection, IL-33 was produced by specialized epithelial cells, which initiated the pathogenic immune response as observed in those allergic disorders [Citation19]. Previous studies found that IL-33/ST2 signaling pathway regulated the functions of different immune cells (T cell, B cell, macrophage, mast cell, etc.) [Citation36] and mediated the production of several pro-inflammatory factors (TNF-α, IL-6, etc.) [Citation37]. In addition, IL-33, which was found to ameliorate experimental colitis in mice by inhibiting autophagy of macrophages [Citation24], have a neuroprotective effect by inhibition on autophagy [Citation22,Citation23]. Similarly, our results also showed that IL-33/ST2 signaling pathway regulated the degranulation of mast cells through autophagy ( and ). Therefore, IL-33/ST2 acts as the linkage between epithelial cells and mast cells in AR. Upon allergen exposure, epithelial cells release IL-33 which binds to ST2 receptor on mast cells and regulate their degranulation and hypersensitivity.
Previous investigations showed that autophagy played essential roles in the pathogenesis of chronic respiratory disease [Citation38] and allergic inflammation [Citation39]. Indeed, autophagy was found to be involved in many aspects of innate and adaptive immunity and inflammatory responses, such as cytokine production, phagocytosis, antigen presentation, and survival and development of lymphocytes [Citation40,Citation41]. Abnormalities in autophagy are associated with abnormal immune functions, which are found in several types of diseases, e.g. cancer, asthma, allergic airway inflammation, chronic respiratory disease, etc. [Citation38,Citation39,Citation42,Citation43]. Autophagy also plays a crucial role in degranulation of mast cells. In the previous study by Ushio et al. [Citation16], deletion of Atg7 severely impaired degranulation of mast cells through impaired co-localization and release of LC3-II and CD63. Interestingly, our results showed that allergen-treated epithelial cells elevated mast cell degranulation through impairment of autophagy, e.g. decrease in LC-3-I/II and Beclin-1 and increase in p62 levels ()). In addition, inhibition on autophagy using 3-MA also increased calcium influx and degranulation of mast cells (). Those conflicting results of the relationship between autophagy impairment and degranulation of mast cells could be due to the different inhibition methods used (Atg7 deletion, or 3-MA treatment/allergen treatment) which regulate autophagy and degranulation through different pathways. Further investigations are required to clarify those mechanisms.
Well known for its roles in the development and treatment of cancer [Citation44], PI3K/Akt/mTOR pathway was also involved in the regulation of cellular metabolism and immune system [Citation45]. Our results showed that IL-33 could increase the expression and activation of PI3K/Akt/mTOR pathway in mast cells ( and ), while inhibition on PI3K pathway elevated autophagy of mast cells ()) and attenuated AR-HNEpC-promoted calcium influx and degranulation of mast cells ( and ). Those findings were consistent with previous studies that PI3K pathway mediated the degranulation of mast cells [Citation46], and its inhibition attenuated mast cell degranulation [Citation47,Citation48]. In addition, it was reported that inhibition on PI3K pathway reduced the secretion of IL-6 in mast cells [Citation49]. Consistently, our results also revealed that inhibition on PI3K pathway reduced the release of several inflammatory factors in mast cells, including IL-4, IL-6, TNF-α, and INF-γ ( and ). Those findings support our hypothesis that PI3K/Akt/mTOR pathway regulates the AR-mediated autophagy and degranulation of mast cells.
In summary, our study results suggested that upon allergen exposure, nasal epithelial cells released IL-33 which then regulated degranulation and excretion of inflammatory factors of mast cells through activation of PI3K/Akt/mTOR pathway and inhibition of autophagy. Further clinical investigations are on-going. Currently, very few studies focused on the interactions between different types of cells in AR, and our study suggested a new angle to understand this disease. Through this new angle, our study suggested a novel hypothesis on the pathogenesis of AR. Future studies following this angle may unfold more mechanisms of AR and possibly provide new therapeutic targets for the disease.
Disclosure statement
The authors declare no conflict of interest.
Data availability statement
All data generated or analyzed during this study are included in this published article.
Additional information
Funding
References
- Khan DA. Allergic rhinitis and asthma: epidemiology and common pathophysiology. Allergy Asthma Proc. 2014;35(5):357–361.
- Greiner AN, Hellings PW, Rotiroti G, et al. Allergic rhinitis. Lancet. 2011;378(9809):2112–2122.
- Eifan AO, Durham SR. Pathogenesis of rhinitis. Clin Exp Allergy. 2016;46(9):1139–1151.
- Melum GR, Farkas L, Scheel C, et al. A thymic stromal lymphopoietin-responsive dendritic cell subset mediates allergic responses in the upper airway mucosa. J Allergy Clin Immunol. 2014;134(3):613–621 e7.
- Peng H, Wang J, Ye XY, et al. Histamine H4 receptor regulates IL-6 and INF-gamma secretion in native monocytes from healthy subjects and patients with allergic rhinitis. Clin Transl Allergy. 2019;9:49.
- Amin K. The role of mast cells in allergic inflammation. Respir Med. 2012;106(1):9–14.
- Modena BD, Dazy K, White AA. Emerging concepts: mast cell involvement in allergic diseases. Transl Res. 2016;174:98–121.
- da Silva EZ, Jamur MC, Oliver C. Mast cell function: a new vision of an old cell. J Histochem Cytochem. 2014;62(10):698–738.
- Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30(17):1913–1930.
- Takamura A, Komatsu M, Hara T, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795–800.
- Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64.
- Lapaquette P, Guzzo J, Bretillon L, et al. Cellular and Molecular Connections between Autophagy and Inflammation. Mediators Inflamm. 2015;2015:398483.
- Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39(5):596–604.
- Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16(6):345–357.
- Koga H, Martinez-Vicente M, Arias E, et al. Constitutive Upregulation of Chaperone-Mediated Autophagy in Huntington’s Disease. J Neurosci. 2011;31(50):18492–18505.
- Ushio H, Ueno T, Kojima Y, et al. Crucial role for autophagy in degranulation of mast cells. J Allergy Clin Immunol. 2011;127(5):1267–76 e6.
- Nakano H, Ushio H. An unexpected role for autophagy in degranulation of mast cells. Autophagy. 2011;7(6):657–659.
- Kamekura R, Kojima T, Takano K, et al. The role of IL-33 and its receptor ST2 in human nasal epithelium with allergic rhinitis. Clin Exp Allergy. 2012;42(2):218–228.
- Gupta RK, Gupta K, Dwivedi PD. Pathophysiology of IL-33 and IL-17 in allergic disorders. Cytokine Growth Factor Rev. 2017;38:22–36.
- Takatori H, Makita S, Ito T, et al. Regulatory Mechanisms of IL-33-ST2-Mediated Allergic Inflammation. Front Immunol. 2018;9:2004.
- Cayrol C, Girard JP. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol Rev. 2018;281(1):154–168.
- Gao Y, Ma L, Luo C-L, et al. IL-33 Exerts Neuroprotective Effect in Mice Intracerebral Hemorrhage Model Through Suppressing Inflammation/Apoptotic/Autophagic Pathway. Mol Neurobiol. 2017;54(5):3879–3892.
- Gao Y, Zhang M-Y, Wang T, et al. IL-33/ST2L Signaling Provides Neuroprotection Through Inhibiting Autophagy, Endoplasmic Reticulum Stress, and Apoptosis in a Mouse Model of Traumatic Brain Injury. Front Cell Neurosci. 2018;12:95.
- Wang Z, Shi L, Hua S, et al. IL-33 ameliorates experimental colitis involving regulation of autophagy of macrophages in mice. Cell Biosci. 2019;9:10.
- Schemann M, Kugler EM, Buhner S, et al. The mast cell degranulator compound 48/80 directly activates neurons. PLoS One. 2012;7(12):e52104.
- Akimoto M, Maruyama R, Takamaru H, et al. Soluble IL-33 receptor sST2 inhibits colorectal cancer malignant growth by modifying the tumour microenvironment. Nat Commun. 2016;7:13589.
- Oshio T, Komine M, Tsuda H, et al. Nuclear expression of IL-33 in epidermal keratinocytes promotes wound healing in mice. J Dermatol Sci. 2017;85(2):106–114.
- Jin Y, Lei J, Lin Y, et al. Autophagy Inhibitor 3-MA Weakens Neuroprotective Effects of Posttraumatic Brain Injury Moderate Hypothermia. World Neurosurg. 2016;88:433–446.
- Mims JW. Epidemiology of allergic rhinitis. Int Forum Allergy Rhinol. 2014;4(Suppl 2):S18–20.
- Kakli HA, Riley TD. Allergic Rhinitis. Prim Care. 2016;43(3):465–475.
- Settipane RA, Schwindt C. Chapter 15: allergic rhinitis. Am J Rhinol Allergy. 2013;27(Suppl 1):S52–5.
- Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med. 2012;18(5):693–704.
- Gilfillan AM, Austin SJ, Metcalfe DD. Mast cell biology: introduction and overview. Adv Exp Med Biol. 2011;716:2–12.
- Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. 2015;16(1):27–35.
- Moon HG, Kim S-J, Jeong JJ, et al. Airway Epithelial Cell-Derived Colony Stimulating Factor-1 Promotes Allergen Sensitization. Immunity. 2018;49(2):275–287 e5.
- Lu J, Kang J, Zhang C, et al. The role of IL-33/ST2L signals in the immune cells. Immunol Lett. 2015;164(1):11–17.
- Fattori V, Hohmann MSN, Rossaneis AC, et al. Targeting IL-33/ST2 signaling: regulation of immune function and analgesia. Expert Opin Ther Targets. 2017;21(12):1141–1152.
- Racanelli AC, Kikkers SA, Choi AMK, et al. Autophagy and inflammation in chronic respiratory disease. Autophagy. 2018;14(2):221–232.
- Liu JN, Suh DH, Trinh HK, et al. The role of autophagy in allergic inflammation: a new target for severe asthma. Exp Mol Med. 2016;48(7):e243.
- Puleston DJ, Simon AK. Autophagy in the immune system. Immunology. 2014;141(1):1–8.
- Shibutani ST, Saitoh T, Nowag H, et al. Autophagy and autophagy-related proteins in the immune system. Nat Immunol. 2015;16(10):1014–1024.
- Jin Y, Hong Y, Park C, et al. Molecular Interactions of Autophagy with the Immune System and Cancer. Int J Mol Sci. 2017;18(8):1694.
- Jiang X, Fang L, Wu H, et al. TLR2 Regulates Allergic Airway Inflammation and Autophagy Through PI3K/Akt Signaling Pathway. Inflammation. 2017;40(4):1382–1392.
- Noorolyai S, Shajari N, Baghbani E, et al. The relation between PI3K/AKT signalling pathway and cancer. Gene. 2019;698:120–128.
- Fruman DA, Chiu H, Hopkins BD, et al. The PI3K Pathway in Human Disease. Cell. 2017;170(4):605–635.
- Alsaleh NB, Persaud I, Brown JM. Silver Nanoparticle-Directed Mast Cell Degranulation Is Mediated through Calcium and PI3K Signaling Independent of the High Affinity IgE Receptor. PLoS One. 2016;11(12):e0167366.
- Xu H, Gu L-N, Yang Q-Y, et al. MiR-221 promotes IgE-mediated activation of mast cells degranulation by PI3K/Akt/PLCgamma/Ca(2+) pathway. J Bioenerg Biomembr. 2016;48(3):293–299.
- Yi Z, Yi Z, Huang K, et al. Propofol attenuates mast cell degranulation via inhibiting the miR-221/PI3K/Akt/Ca(2+) pathway. Exp Ther Med. 2018;16(2):1426–1432.
- Yang Q, Xu H, Yang J, et al. MicroRNA-223 affects IL-6 secretion in mast cells via the IGF1R/PI3K signaling pathway. Int J Mol Med. 2016;38(2):507–512.