
ABSTRACT
Tumor suppressor p53 is the most frequently mutated gene in human cancer. Mutant p53 (mutp53) not only loses the tumor suppressive activity of wild type p53, but often gains new oncogenic activities to promote tumorigenesis, defined as mutp53 gain of function (GOF). While the concept of mutp53 GOF is well-established, its underlying mechanism is not well-understood. AKT has been suggested to be activated by mutp53 and contribute to mutp53 GOF, but its underlying mechanism is unclear. In this study, we found that the activation of the Rac1 signaling by mutp53 mediates the promoting effect of mutp53 on AKT activation. Blocking Rac1 signaling by RNAi or a Rac1 inhibitor can inhibit AKT activation by mutp53. Importantly, targeting Rac1/AKT can greatly compromise mutp53 GOF in tumorigenesis. Results from this study uncover a new mechanism for AKT activation in tumors, and reveal that activation of AKT by mutp53 via the Rac1 signaling contributes to mutp53 GOF in tumorigenesis. More importantly, this study provides Rac1 and AKT as potential targets for therapy in tumors containing mutp53.
Introduction
p53 plays a key role in maintaining genomic stability and tumor suppression [Citation1]. p53 is the most frequently mutated gene in human cancers; more than half of all human cancers contain mutations in the p53 gene [Citation2,Citation3]. Majority of tumor associated-p53 mutations are missense mutations occurring in the DNA binding domain (DBD) of the p53 protein. These p53 missense mutations not only abrogate the tumor suppressive function of wild type (WT) p53, but also render mutant p53 (mutp53) proteins oncogenic activities, defined as the mutp53 gain of function (GOF) [Citation3,Citation4]. Studies have demonstrated an array of mutp53 GOFs, including promoting cell proliferation, migration, invasion and metastasis of tumor cells [Citation5–Citation7].
While it is well-established that many tumor-associated mutp53 proteins show GOF activities, the underlying mechanism of mutp53 GOFs is not well-understood. It has been shown that some GOF mutp53 proteins can interact with transcriptional factors and co-activators, including p63, p73, SREBP, ETS2, etc., and regulate their transcriptional activities, which contributes to mutp53 GOFs [Citation4,Citation8,Citation9]. In addition, mutp53 has been reported to exert its GOFs through activation of some oncogenic signaling pathways by interaction with some proteins in these oncogenic pathways. For instance, mutp53 (R248Q) can interact with STAT3 to activate STAT3, promoting tumorigenesis [Citation10]. Intriguingly, our recent study showed that mutp53 directly binds to small GTPase Rac1 and activates the Rac1 signaling in cancer cells [Citation11]. Aberrant activation of the Rac1 signaling in cancer cells has been reported to promote proliferation, migration, invasion and metastasis of cancer cells [Citation12,Citation13]. Previous reports have shown that some tumor-associated mutp53 proteins activate AKT in lung, breast and endometrial cancer cell lines [Citation6,Citation14,Citation15]. p53 mutations are positively correlated with elevated AKT phosphorylation levels in clinical colon and breast cancer specimens [Citation6,Citation15,Citation16]. However, the mechanism whereby mutp53 activates AKT is not well-understood.
In this study, we identified a new mechanism underlying AKT activation by mutp53. Rac1 is a positive regulator of AKT [Citation17]. Rac1 was reported to activate AKT through its critical downstream effectors, PAKs (p21 activated kinases) [Citation18,Citation19]. We found that the activation of the Rac1 signaling by mutp53 mediates the promoting effect of mutp53 on AKT activation. Blocking the Rac1 signaling by RNAi or a Rac1 inhibitor inhibits AKT activation by mutp53. Importantly, targeting the Rac1/AKT axis greatly compromises mutp53 GOFs. Results from this study demonstrate that Rac1 plays a critical role in mediating mutp53-induced AKT activation, which in turn promotes tumorigenesis. This study reveals a new mechanism whereby mutp53 exerts its GOF and provides Rac1 and AKT as potential targets for therapy in tumors containing mutp53.
Results
Mutp53 activates AKT
To validate the association of mutp53 with AKT activation, we analyzed the association of p53 mutation status and the phosphorylation levels of AKT at Ser473 (p-AKTS473), which reflects AKT activity [Citation20], in a breast invasive carcinoma cohort from TCGA (TCGA-BRCA). The mutation status of the p53 gene of these specimens was determined by sequencing and the levels of p-AKTS473 were determined by Reverse phase protein arrays (RPPA) [Citation21]. Tumors containing mutp53 (n = 689) had higher p-AKTS473 levels than those containing WT p53 (n = 251) ()). The average levels (log2) of p-AKTS473 in tumors containing mutp53 and WT p53 are 0.3 ± 0.06 and 0.2 ± 0.03, respectively (p = 0.0128, Student’s t-test) ()).
Figure 1. Mutp53 is associated with increased AKT phosphorylation levels in a breast cancer cohort and mutp53 activates AKT in cancer cells. (a) The mutation status of the p53 gene is associated with the phosphorylation levels of AKT at Ser473 (p-AKTS473) in human breast cancer specimens. An invasive breast cancer cohort from TCGA with known p53 mutation status (WT: n = 689; and mutp53: n = 251) and the levels of p-AKTS473 was used to analyze the association of p53 mutations with p-AKTS473 levels. Reverse phase protein arrays (RPPA) signals of p-AKTS473 were transformed to Log2 value. (b) Mutp53 increased AKT activity in human cancer cells as reflected by the levels of pAKTS473. Ectopic expression of mutp53 (R175 H, R248 W and R273 H) in p53 null H1299 and HCT116 p53−/- cells by transient transfection of expression vectors increased p-AKTS473 levels as determined by western-blot assays. (c) Higher levels of p-AKTS473 in cells with stable ectopic mutp53 expression. Isogenic cell lines, including HCT116 p53−/-, HCT116 p53R248 W/-, Saos2-Con and Saos2-mutp53 R175 H cells, were employed to analyze the levels of p-AKTS473 by western-blot assays. (d) Knockdown of endogenous mutp53 by siRNA in SK-BR-3, MDA-MB-468 and SW480 cells decreased p-AKTS473 levels.
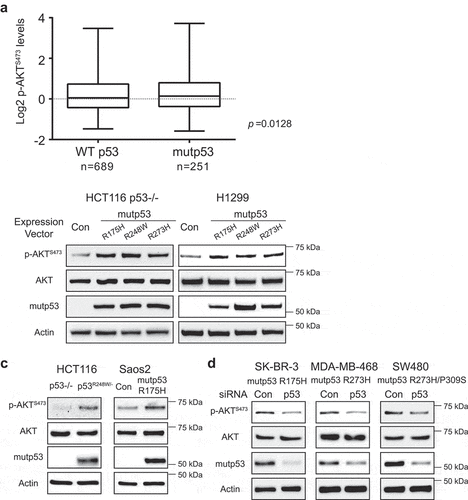
To confirm that mutp53 increases AKT activity, the levels of p-AKTS473 were determined in two p53 null human cancer cell lines, HCT116 p53-/- and H1299, with or without transient transfection of vectors expressing several tumor-associated hot spot mutp53 (R175 H, R248 W and R273 H). As shown in ), ectopic expression of mutp53 (R175 H, R248 W and R273 H) in both cell lines increased the levels of p-AKTS473 but did not affect the levels of total AKT as determined by western-blot assays, indicating that mutp53 enhances AKT activity but not total AKT levels in cells. The levels of p-AKTS473 were measured in HCT116 p53-/- cells and HCT116 p53R248 W/- cells, which contain a single copy of p53 gene with R248 W mutation, and p53 null osteosarcoma human Saos2 cells with or without stable expression of mutp53 (R175 H). The levels of p-AKTS473 were much higher in cells with mutp53 expression than their isogenic p53 null cells ()). Further, in a panel of human cancer cell lines containing a single allele of endogenous mutp53 gene, including breast SK-BR-3 (R175 H mutp53), MDA-MB-468 (R273 H mutp53) and colorectal SW480 (R273 H/P309 S mutp53) cells, knockdown of the endogenous mutp53 by siRNA in all three cell lines decreased the levels of p-AKTS473 ()). Together, these results demonstrate that mutp53 activates AKT in tumor cells.
Mutp53 activates AKT through the Rac1 signaling in vitro and in vivo
Our recent study showed that tumor-associated GOF mutp53 proteins activate small GTPase Rac1 through promoting Rac1 SUMOylation [Citation11]. The Rac1 signaling is involved in a diverse array of cellular processes. The Rac1 signaling is frequently activated in human cancers, which plays a crucial role in promoting tumor development and progression [Citation22]. Rac1 has been shown to be an upstream regulator of the PI3 K signaling [Citation17]. Rac1 cycles between an inactive Rac1-GDP form and an active Rac1-GTP form in cells [Citation12,Citation13]. Activation of Rac1 was reported to promote the phosphorylation PAKs, the critical downstream effectors of Rac1, which in turn activate AKT [Citation18,Citation19]. Here, we investigated whether the activation of Rac1 by mutp53 mediates AKT activation by GOF mutp53. Ectopic expression of Rac1 in H1299 cells by Rac1 vectors increased the levels of Rac1-GTP and the phosphorylation levels of PAK1 at Ser144 and PAK2 at Ser141 (p-PAK1/2), respectively, which reflect the Rac1 activity in cells [Citation23,Citation24], and the levels of p-AKTS473, but did not affect the levels of total PAK1/2 or AKT ()). Knockdown of endogenous Rac1 by shRNA vectors decreased the levels of Rac1-GTP, p-PAK1/2 and p-AKTS473, but did not have a clear effect on the levels of total PAK1/2 and AKT ()). Further, compared with cells with ectopic expression of Rac1 alone, simultaneous expression of Rac1 and mutp53 (R175 H) increased the levels of p-PAK1/2 and p-AKTS473 to a much greater extent, which supports the hypothesis that mutp53 activates the Rac1 signaling to increase AKT activity ()). Importantly, knockdown of endogenous Rac1 by shRNA vectors largely abolished the promoting effect of mutp53 (R175 H) on AKT activation as reflected by the levels of p-AKTS473 in H1299 cells ()). Consistently, blocking Rac1 signaling by ectopic expression of the dominant-negative Rac1 (DN-Rac1; Rac1-T17 N) decreased the levels of p-PAK1/2 and p-AKTS473, and notably, largely abolished the promoting effect of mutp53 (R175 H) on AKT activation in H1299 cells ()).
Figure 2. Mutp53 activates AKT through the Rac1 signaling in vitro and in vivo. (a) Ectopic Rac1 expression activated PAKs and AKT reflected by the increased phosphorylation levels of PAK1 at Ser144 and PAK2 at Ser141 (p-PAK1/2) and increased p-AKT S473 levels. Ectopic expression of Rac1 in p53 null H1299 cells by transient transfection of vectors increased Rac1-GTP, p-PAK1/2 and p-AKT S473 levels. (b) Knockdown of endogenous Rac1 by shRNA in H1299 cells decreased Rac1-GTP, p-PAK1/2 and p-AKT S473 levels. (c) The levels of p-PAK1/2 and p-AKT S473 were determined in H1299 cells with ectopic expression of Rac1 and mutp53 (R175 H) individually or simultaneously. (d) Knockdown of endogenous Rac1 abolished the increase of p-PAK1/2 and p-AKT S473 levels induced by mutp53 (R175 H) in H1299 cells. (e) Ectopic expression of the dominant-negative Rac1 (DN-Rac1) largely abolished the increase of p-PAK1/2 and p-AKT S473 levels induced by mutp53 (R175 H) in H1299 cells. (f) & (g) Mutp53 (R715 H) increased p-PAK1/2 and p-AKT S473 levels in H1299 xenograft tumors. Knockdown of Rac1 by shRNA (F) or expression of DN-Rac1 (G) largely abolished the increase of p-PAK1/2 and p-AKT S473 levels induced by mutp53 (R175 H) in H1299 xenograft tumors.
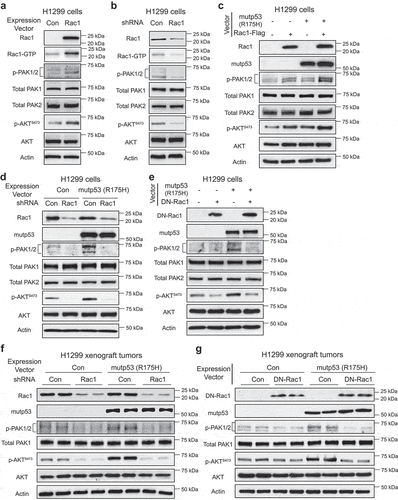
We further investigated the role of Rac1 in mediating the promoting effect of mutp53 on AKT activation in vivo. H1299 cells with or without ectopic expression of mutp53 (R175 H) (H1299-Con and H1299-R175 H cells) were infected with control shRNA vectors or shRNA targeting Rac1 and employed for xenograft tumor formation in nude mice. Tumors were harvested when the tumor volume reached ~200 mm3. H1299-R175 tumors displayed a much faster proliferation rate with higher IHC staining of Ki-67, a marker for cell proliferation, than H1299-Con tumors (Supplementary Fig 1). Knocking down endogenous Rac1 by shRNA vectors largely abolished the promoting effect of mutp53 on cell proliferation in H1299 xenograft tumors (Supplementary Fig 1). The activities of the Rac1 signaling and AKT were much higher in xenograft tumors formed by H1299-R175 H cells compared with tumors formed by H1299-Con cells, indicating that mutp53 increases the activities of Rac1 and AKT in vivo ()). Knockdown of Rac1 by shRNA vectors clearly decreased AKT activity ()). Notably, knockdown of Rac1 largely abolished the promoting effect of mutp53 on AKT activation ()). Similar results were obtained when Rac1 activity was blocked by ectopic expression of the DN-Rac1 in H1299-Con and H1299-R175 H tumors. DN-Rac1 expression largely abolished the promoting effect of mutp53 on cell proliferation in H1299 xenograft tumors (Supplementary Fig 1). Tumors with DN-Rac1 expression showed decreased levels of p-PAK1/2 and p-AKTS473 ()). Importantly, DN-Rac1 expression largely abolished the promoting effect of mutp53 on AKT activation in tumors ()). Together, these results strongly suggest that the activation of Rac1 by mutp53 mediates the promoting effect of mutp53 on AKT activation in tumor cells.
Rac1 inhibitor inhibits AKT activation by mutp53
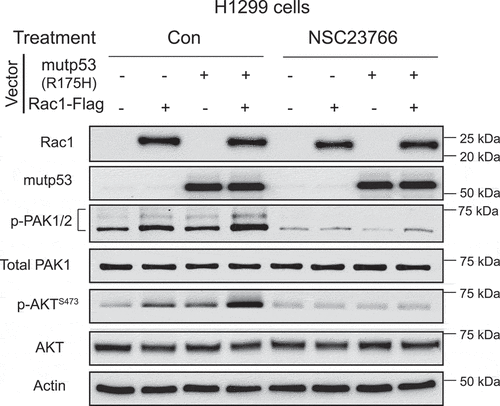
We further investigated whether targeting Rac1 by employing a small molecule inhibitor targeting Rac1 can block AKT activation promoted by mutp53. Small molecule compound NSC23766 is a widely-used potent Rac1 inhibitor, which can competitively block the binding loop of Rac1-specific guanine nucleotide exchange factors Rac1 activators to prevent the conversion of the inactive Rac1-GDP form to the active Rac1-GTP form [Citation25,Citation26]. H1299 cells with ectopic expression of Rac1 and mutp53 (R175 H) individually or simultaneously were treated with NSC23766 or vehicle. NSC23766 treatment decreased the levels of p-PAK1/2, indicating the inhibition of the Rac1 signaling (). Notably, NSC23766 clearly decreased the levels of p-AKTS473 and largely abolished the promoting effect of Rac1 and mutp53 on AKT activation (). These results support that mutp53 activates AKT through the Rac1 signaling, and targeting Rac1 can inhibit AKT activation by mutp53.
Figure 3. The Rac1 inhibitor abolishes AKT activation by mutp53 (R175 H). H1299 cells with or without ectopic expression of Rac1 and mutp53 were treated with the Rac1 inhibitor (NSC23766; 1 µM).
Rac1 SUMOylation promoted by mutp53 is crucial for mutp53-induced AKT activation
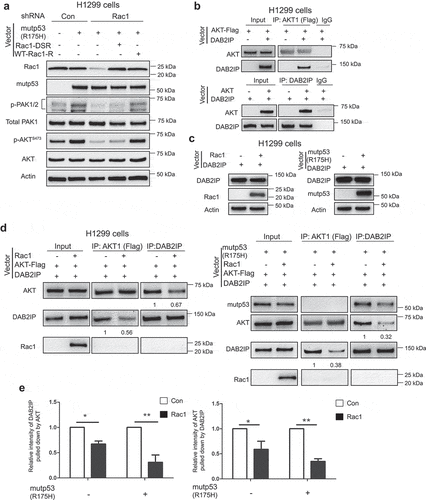
Our recent report revealed that mutp53 activates Rac1 through increasing Rac1 SUMOylation levels [Citation11]. Rac1 SUMOylation is critical in maintaining the active Rac1-GTP form to enhance Rac1 activity in cells [Citation27]. Here, we investigated whether Rac1 SUMOylation promoted by mutp53 contributes to mutp53-induced AKT activation. Vectors expressing wild type Rac1 (WT-Rac1-R) or SUMOylation site mutated Rac1 (Rac1 -DSR) that are resistant to Rac1 shRNA were transfected into H1299 cells with knockdown of endogenous Rac1. The expression levels of WT-Rac1-R and Rac1-DSR were comparable to endogenous Rac1 levels in cells ()). In H1299-R175 H cells, knockdown of endogenous Rac1 abolished AKT activation induced by mutp53, which can be restored by expression of WT-Rac1-R but not Rac1-DSR ()). These results suggest that promoting Rac1 SUMOylation by mutp53 plays an important role in mutp53-induced AKT activation.
Figure 4. Mutp53 activates AKT through Rac1 activation and Rac1 activation by mutp53 inhibits DAB2IP-AKT interaction. (a) H1299 cells with or without knockdown of endogenous Rac1 were transfected with vectors as indicated. Ectopic expression of mutp53 increased the levels of p-PAK1/2 and p-AKT S473 in cells which was abolished by knockdown of endogenous Rac1. The effect of mutp53 on increasing of p-PAK1/2 and p-AKT S473 levels was restored in cells with knockdown of endogenous Rac1 by expression of wild type Rac1 resistant to Rac1 shRNA (WT-Rac1-R) but not SUMOylation site mutant Rac1 resistant to Rac1 shRNA (Rac1-DSR). (b) AKT interacted with DAB2IP in H1299 cells transfected with vectors expressing AKT and DAB2IP, respectively. The DAB2IP-AKT interaction was determined by co-IP assays using anti-Flag and anti-DAB2IP antibodies, respectively, followed by western-blot assays. (c) H1299 cells were transfected with vectors expressing DAB2IP together with vectors expressing Rac1 or mutp53 (R175 H) as indicated. The protein levels of DAB2IP were determined by western-blot assays. (d) Rac1 reduced the DAB2IP-AKT interaction, especially in the presence of mutp53. H1299 cells were transfected with vectors as indicated. The DAB2IP-AKT interaction was determined by co-IP assays followed by western-blot assays. (e) Quantification of the DAB2IP-AKT interaction by measuring the relative intensity of DAB2IP pulled down by AKT-Flag (left panel) and the relative intensity of AKT-Flag pulled down by DAB2IP (right panel) in co-IP assays. Data are presented as mean ± SD, n = 3. * p < 0.05, ** p < 0.01, Student’s t-test.
Rac1 activation by mutp53 inhibits DAB2IP-AKT interaction
DAB2IP (DODC-2/DAB2-interacting protein) is a Ras-GAP protein that negatively regulates multiple oncogenic pathways and functions as a tumor suppressor [Citation28]. DAB2IP was reported to bind to AKT and inhibit its activation [Citation16,Citation29]. DAB2IP is frequently down-regulated or inactivated in cancers and its underlying mechanism remains unclear. A previous study showed that DAB2IP can be functionally inactivated through physical interaction with mutp53 proteins [Citation30]. A recent study reported that mutp53 can increase insulin-induced AKT activation by binding to and inhibiting the function of DAB2IP [Citation16]. We confirmed the interaction of AKT with DAB2IP in H1299 cells transfected with expression vectors of AKT1 and DAB2IP by co-Immunoprecipitation (co-IP) assays ()). Neither Rac1 nor mutp53 appears to change DAB2IP levels; expression of Rac1 or mutp53 R175 H did not affect DAB2IP protein levels in H1299 cells ()). Interestingly, we found that Rac1 inhibits the interaction of DAB2IP and AKT in H1299 cells. H1299 cells were co-transfected with vectors expressing AKT, DAB2IP, along with vectors expressing Rac1 or control vectors. The interaction of DAB2IP and AKT was determined by co-IP assays. Rac1 clearly decreased the interaction between DAB2IP and AKT (), left panels). Results from co-IP assays also confirmed the interaction between mutp53 and DAB2IP (), right panels), and showed no obvious interaction between mutp53 and AKT1 (), right panels). Notably, the inhibitory effect of Rac1 on the interaction between DAB2IP and AKT was much stronger in H1299 cells with mutp53 expression compared with cells without mutp53 expression. These results suggest that Rac1 activation by mutp53 contributes to the inhibition of DAB2IP-AKT interaction, which in turn activates AKT () and )).
Targeting the Rac1/AKT axis by inhibitors largely blocks mutp53 GOFs
Tumor-associated mutp53 proteins often gain new oncogenic activities to promote tumor development and progression, such as promoting proliferation, migration and invasion of cancer cells [Citation31–Citation34]. The AKT signaling is often activated in many types of cancers, and plays an important role in promoting survival, proliferation and migration of cancer cells [Citation35–Citation37]. Given that mutp53 activates AKT, we tested whether AKT activation by mutp53 contributes to mutp53 GOF in tumorigenesis. To this end, inhibitors targeting the Rac1/AKT axis, including NSC23766 and Wortmannin, which are specific inhibitors for Rac1 and AKT, respectively, were employed to treat H1299 cells with or without ectopic expression of mutp53 (R715 H) (H1299-Con and H1299-R175 H cells). Expression of mutp53 (R175 H) in H1299 cells significantly promoted cell proliferation, colony formation and anchorage-independent cell growth (–c)). NSC23766 and Wortmannin significantly inhibited cell proliferation, colony formation and anchorage-independent growth in both H1299-Con and H1299-R175 H cells. Most importantly, these inhibitors largely abolished mutp53 GOF in promoting cell proliferation, colony formation and anchorage-independent growth (–c)).
Figure 5. Targeting the Rac1/AKT axis by inhibitors greatly compromises mutp53 GOF in promoting cell proliferation, anchorage-independent cell growth and migration in H1299 cells. (a) Mutp53 (R175 H) promoted proliferation of H1299 cells, which was greatly compromised by Rac1 inhibitor NSC23766 and AKT inhibitor Wortmannin. H1299 cells with or without ectopic mutp53 (R175 H) expression were treated with NSC23766 (1 µM) or Wortmannin (5 µM). (b) Mutp53 (R175 H) promoted the colony formation of H1299 cells, which was greatly compromised by NSC23766 (100 nM) and Wortmannin (500 nM). Left panel: representative images. Right panel: quantification of the area of colonies/field. (c) Mutp53 (R175 H) promoted the anchorage-independent growth of H1299 cells, which was greatly compromised by NSC23766 (100 nM) and Wortmannin (500 nM). Left panel: representative images. Right panel: quantification of colony numbers per field. (d) Mutp53 (R175 H) promoted the migration ability of H1299 cells, which was greatly compromised by NSC23766 and Wortmannin. Left panel: representative images. Right panel: quantification of average number of migrated cells/field. H1299 cells with or without ectopic mutp53 (R175 H) were treated with NSC23766 (2 µM) or Wortmannin (1 µM) for 24 h. For B-D, data were present as mean ± SD. n ≥ 3. *** p < 0.001, Student’s t-test.
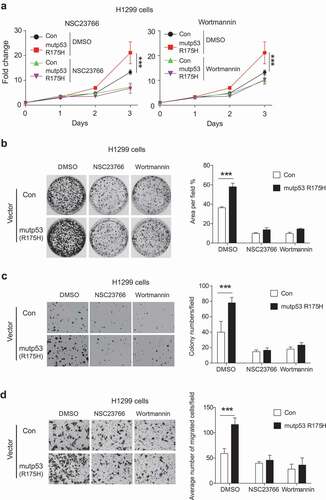
Furthermore, mutp53 (R175 H) significantly promoted migration of H1299 cells as determined by trans-well assays ()). Treatment of NSC23766 or Wortmannin significantly inhibited the migration of both H1299-Con and H1299-R175 H cells, and notably, largely abolished mutp53 GOF in promoting migration of cells ()).
We further investigated whether AKT functions as a down-stream effector of the Rac1 signaling to promote mutp53 GOF. In H1299-Con and H1299-R175 cells, blocking Rac1 function by knockdown of Rac1 inhibited the proliferation, colony formation, and anchorage-independent cell growth, and importantly, largely abolished mutp53 GOF in promoting proliferation and anchorage-independent cell growth (–c)). Notably, Wortmannin treatment exhibited limited inhibitory effects on cell proliferation, colony formation and anchorage-independent growth in H1299-Con and H1299-R175 H cells with knockdown of endogenous Rac1 (–c)). Similarly, knockdown of Rac1 inhibited migration of H1299-Con and H1299-R175 H cells, and importantly, largely abolished mutp53 GOF in promoting migration of cells ()). While Wortmannin treatment greatly inhibited the migration of H1299-Con and H1299-R175 H cells and largely abolished mutp53 GOF in promoting migration, it exhibited a limited inhibitory effect on migration of both cell lines with knockdown of endogenous Rac1 ()). These results indicate that AKT functions as an important down-stream effector of the Rac1 signaling to promote mutp53 GOF.
Figure 6. AKT activation is a downstream effector of the Rac1 signaling to promote mutp53 GOF in cell proliferation (a), colony formation (b), anchorage-independent cell growth (c) and migration (d). H1299 cells with or without ectopic expression of mutp53 (R175 H) were subjected to knockdown of endogenous Rac1 by shRNA vectors and/or treatment of Wortmannin. For B-D, data were present as mean ± SD. n ≥ 3. *** p < 0.001, Student’s t-test.
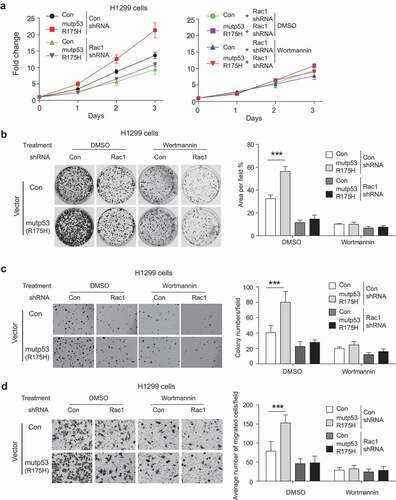
Together, these results strongly suggest that AKT activation by mutp53 via the Rac1 signaling contributes to mutp53 GOF in tumorigenesis, and targeting the Rac1/AKT axis suppresses mutp53 GOF in tumorigenesis.
Discussion
AKT, a serine/threonine kinase, is a major player in the PI3 K/AKT signaling that regulates many important biological functions, including cell proliferation, survival and metabolism, etc [Citation20,Citation38]. AKT is frequently activated in many human cancers, which contributes to tumor initiation and progression [Citation20,Citation38]. Activation of certain oncogenes, such as ErbB2 and EGFR, and inactivation of some tumor suppressor genes, such as PTEN that regulate PI3 K/AKT signaling, are important mechanisms underlying AKT activation in human cancers [Citation20,Citation39,Citation40]. The phosphorylation of AKT at Ser473 and Thr308 leads to its activation and regulation of its downstream substrates, such as FOXOs, GSK3, p27, to exert its biological functions [Citation41,Citation42].
Previous studies on the cross-talk between AKT and p53 and its underlying mechanisms were mainly focused on WT p53 [Citation43–Citation45]. It was reported that the activation of AKT can phosphorylate MDM2, a key negative regulator of p53, at Ser166 and Ser186, which results in the translocation of MDM2 to the nucleus to promote ubiquitination and degradation of WT p53 [Citation43–Citation45]. At the same time, WT p53 negatively regulates AKT via its target gene PTEN [Citation46,Citation47]. The coexistence of p53 mutations and AKT activation in cancers has been reported previously [Citation6,Citation14,Citation15]. Previous reports have also shown that some tumor-associated mutp53 proteins activate AKT in cancer cells [Citation6,Citation14,Citation15]. Results from this study demonstrated that GOF mutp53 activates AKT in cancer cells, and analysis of a breast cancer cohort in TCGA database shows a significant correlation between p53 mutation and AKT activation, which is consistent with previous reports [Citation6,Citation14,Citation15]. However, the mechanism whereby mutp53 activates AKT is not well-understood and cannot be explained by the cross-talk between AKT and WT p53.
Many tumor-associated mutp53 proteins exhibit GOFs, including promoting cell proliferation, survival, migration, invasion, metabolic reprogramming, and chemoresistance [Citation5–Citation7,Citation48]. Mutp53 proteins often accumulate to high levels in tumors, which is crucial for its GOF activities [Citation3,Citation49–Citation51]. Mutp53 can accumulate in both nuclei and cytoplasm and interact with different proteins to regulate their functions, which is an important mechanism by which mutp53 exerts its GOFs [Citation4,Citation8,Citation9]. For example, in the nucleus, mutp53 interacts with a group of transcriptional factors and/or cofactors and regulate their transcription activities to promote tumor progression [Citation4,Citation9,Citation52–Citation55]. Our recent study showed that mutp53 binds to small GTPase Rac1 in the cytoplasm [Citation11]. The mutp53-Rac1 interaction activates Rac1 through inhibiting de-SUMOylation of Rac1. SUMOylation of Rac1 is critical to maintain Rac1 activity in cells [Citation27]. The inhibition of Rac1 de-SUMOylation by mutp53 leads to increased Rac1 activity in cells [Citation11]. Rac1 is an upstream regulator of the PI3 K/AKT signaling and can activate AKT via its downstream effectors, PAKs [Citation17]. These findings suggest that the activation of AKT through the Rac1 signaling could be an important mechanism whereby mutp53 activates AKT to exert its GOF.
Results from this study showed that mutp53 does not interact with AKT in cells as examined by co-IP assays ()). Interestingly, our results show that blocking Rac1 by RNAi or inhibitors largely inhibited AKT activation by mutp53. We found that Rac1 SUMOylation promoted by mutp53 is crucial for mutp53-induced AKT activation; in H1299 cells with knockdown of endogenous Rac1 and ectopic expression of the Rac1 with a mutation at the SUMOylation site, mutp53 showed a minimal effect on AKT activation. We further found that Rac1 activation by mutp53 inhibits the interaction of DAB2IP, a negative regulator of AKT, with AKT, which contributes to AKT activation by mutp53. These results suggested that the activation of Rac1 by mutp53 mediates mutp53-induced AKT activation. Results from this study further demonstrated that the activation of AKT by mutp53 contributes to mutp53 GOF. Wortmannin, the inhibitor targeting AKT, largely abolished mutp53 GOF in promoting cell proliferation, colony formation, anchorage-independent growth and migration ability in H1299 cells (). Combined treatment using inhibitors targeting Rac1 and AKT did not lead to an additive or synergistic effect on inhibiting mutp53 GOF (). These results suggest that the Rac1/AKT axis contributes to mutp53 GOF.
Together, this study reveals that the activation of AKT through the Rac1 signaling is an important mechanism whereby mutp53 exerts its GOF. AKT is an important signaling hub with over 100 downstream target substrates [Citation56,Citation57]. Further studies to characterize the levels and functions of these substrates in tumors containing mutp53 will help further understand the mechanism of mutp53 GOF and the contribution of AKT activation to mutp53 GOF. It is worth noting that p53 mutations are scattered throughout the DNA binding domain, and it is still unclear whether p53 mutations at different residues have a similar effect on AKT activation. Further studies on mutp53 at different residues will help address this question. Results from this study also suggest that Rac1 and AKT are potential therapeutic targets in tumors containing GOF mutp53.
Materials and methods
Cell cultures, vectors and reagents
Human cancer cell lines H1299, SK-BR-3, SW480, Saos2 and MDA-MB-468 were purchased from ATCC. HCT116 p53−/- and HCT116 p53R248W/- were gifts from Dr. Bert Vogelstein at Johns Hopkins University. H1299-Con and H1299-R175 H cells were established as previously described [Citation11]. Vectors (mutp53, Rac1, DN-Rac1, WT-Rac1-R and Rac1-DSR) and shRNAs (p53 and Rac1) were constructed previously [Citation11]. Flag-tagged AKT1 plasmid was a gift from Dr. William Sellers (Addgene plasmid #9021). pLPC-mDAB2IP plasmid was a gift from Dr. Collavin’s lab (National Laboratory CIB, Italy). siRNA oligos targeting p53 were purchased from Dharmacon. The Rac1 inhibitor (NSC23766) and the AKT inhibitor (Wortmannin) were purchased from Sigma.
Western-blot assays
Protein lysates from cells and xenograft tumors were used for western-blot assays. The following antibodies were used in this study: anti-p53 (Santa Cruz Biotechnology), anti-Rac1 (05–389, Millipore), anti HA (Roche), anti-PAK1 (Cell Signaling Technology), anti-PAK2 (Cell signaling technology), anti-p-PAK1/2 (Cell Signaling Technology), anti-AKT (Santa Cruz Biotechnology), anti-p-AKT (s473) (Cell Signaling Technology) and anti-DAB2IP (Abcam) antibodies.
Rac1 activity analysis
Rac1 activity was analyzed by using a Rac1 activation assay kit (Millipore) to perform the GST-p21-binding domain of PAK1 pull-down assays which specifically pull down Rac1-GTP in cells as previously described [Citation24]. The p21-binding domain of the Rac1 effector protein PAK1 specifically binds to Rac1-GTP [Citation58]. The levels of precipitated Rac1-GTP were measured by western-blot assays.
Co-IP assays
Co-IP assays were performed as previously described [Citation48]. Anti-Flag and anti-DAB2IP antibodies were used to pull down Flag-AKT and DAB2IP, respectively. The protein complex pulled-down by above-mentioned antibodies was used for western-blot assays.
Cell proliferation assays
Cells (1 × 104) were seeded in 24-well plates, and treated with DMSO, Rac1 inhibitor NSC23766 (1 µM) or AKT inhibitor Wortmannin (5 µM). Cell proliferation was monitored by IncuCyte ZOOM live-cell analysis system (Essen Bioscience).
Colony formation
Exponentially growing cells (1 × 103) were seeded in 6-well plates, and treated with DMSO, Rac1 inhibitor NSC23766 (100 nM) or AKT inhibitor Wortmannin (500 nM) for 14 days. Colonies were then stained by crystal violet. Colony area was analyzed by using Image J (vendor) software.
Anchorage-independent cell growth
Anchorage-independent cell growth assays were performed as previously described [Citation11]. Briefly, 6-well plates were coated with 0.6% agarose, and cells (1 × 103) were cultured in it with medium containing 0.3% agarose. Cells were treated with DMSO, Rac1 inhibitor NSC23766 (100 nM) or AKT inhibitor Wortmannin (500 nM) for 14 days. The colonies were stained using crystal violet, and quantified by Image J (vendor) software.
Migration assays
Migration assays were performed by using the trans-well system (BD Biosciences) as previously described [Citation11]. Briefly, cells were seeded on the top chamber with FBS-free medium and the bottom chamber was filled with medium containing 10% FBS. Rac1 inhibitor NSC23766 (2 µM) or AKT inhibitor Wortmannin (1 µM) were added into the medium in both chambers. Cells on the lower surface of the top chambers were fixed by methanol, stained by violet crystal for counting after culturing for 24 h.
Xenograft tumor formation
Cells were inoculated subcutaneously (s.c.) into BALB/c athymic male nude mice (Taconic) to form xenograft tumors as previously described [Citation59]. Tumors were harvested when the tumor volume reached ~200 mm3 for western-blot assays. The mouse experiments were performed with the approval of the Institutional Animal Care and Use Committee of University.
Statistical analysis
Quantitative data were presented as mean ± SD. The Student’s t-test was used to analyze the difference between two groups. Values of p < 0.05 were considered to be significant.
Author Contributions
X.Y. carried out the experiments, analyzed data and wrote manuscript; F.W., Y.L., J.L. C.Z., M.B., K.M. carried out experiments; Z.F. and J.G. analyzed data and wrote manuscript; W.H. designed experiments, analyzed data and wrote manuscript.
Supplemental Material
Download PDF (76.8 KB)Acknowledgments
We thank Dr. Bert Vogelstein for HCT116 p53−/- and HCT116 p53R248W/- cell lines, Dr. William Sellers for Flag-tagged AKT1 plasmid; and Dr. Collavin for pLPC-mDAB2IP plasmid.
Disclosure statement
Authors declare no competing interests.
Supplementary materials
Supplemental data for this article can be accessed here.
Related Research Data
References
- Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–758.
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431.
- Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15:2–8.
- Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268–1286.
- Freed-Pastor WA, Mizuno H, Zhao X, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244–258.
- Muller PA, Caswell PT, Doyle B, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139:1327–1341.
- Zhang C, Liu J, Liang Y, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935.
- Yue X, Zhao Y, Xu Y, et al. Mutant p53 in cancer: accumulation, gain-of-function, and therapy. J Mol Biol. 2017;429:1595–1606.
- Di Agostino S, Strano S, Emiliozzi V, et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell. 2006;10:191–202.
- Schulz-Heddergott R, Stark N, Edmunds SJ, et al. Therapeutic ablation of gain-of-function mutant p53 in colorectal cancer inhibits stat3-mediated tumor growth and invasion. Cancer Cell. 2018;34:298–314 e7.
- Yue X, Zhang C, Zhao Y, et al. Gain-of-function mutant p53 activates small GTPase Rac1 through SUMOylation to promote tumor progression. Genes Dev. 2017;31:1641–1654.
- Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701.
- Bid HK, Roberts RD, Manchanda PK, et al. RAC1: an emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol Cancer Ther. 2013;12:1925–1934.
- Dong P, Xu Z, Jia N, et al. Elevated expression of p53 gain-of-function mutation R175H in endometrial cancer cells can increase the invasive phenotypes by activation of the EGFR/PI3K/AKT pathway. Mol Cancer. 2009;8:103.
- Tan BS, Tiong KH, Choo HL, et al. Mutant p53-R273H mediates cancer cell survival and anoikis resistance through AKT-dependent suppression of BCL2-modifying factor (BMF). Cell Death Dis. 2015;6:e1826.
- Valentino E, Bellazzo A, Di Minin G, et al. Mutant p53 potentiates the oncogenic effects of insulin by inhibiting the tumor suppressor DAB2IP. Proc Natl Acad Sci U S A. 2017;114:7623–7628.
- Keely PJ, Westwick JK, Whitehead IP, et al. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature. 1997;390:632–636.
- Murga C, Zohar M, Teramoto H, et al. Rac1 and RhoG promote cell survival by the activation of PI3K and Akt, independently of their ability to stimulate JNK and NF-kappaB. Oncogene. 2002;21:207–216.
- Higuchi M, Onishi K, Kikuchi C, et al. Scaffolding function of PAK in the PDK1-Akt pathway. Nat Cell Biol. 2008;10:1356–1364.
- Manning BD, Toker A. AKT/PKB Signaling: navigating the Network. Cell. 2017;169:381–405.
- Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
- Kazanietz MG, Caloca MJ. The Rac GTPase in cancer: from old concepts to new paradigms. Cancer Res. 2017;77:5445–5451.
- Kumar R, Gururaj AE, Barnes CJ. p21-activated kinases in cancer. Nat Rev Cancer. 2006;6:459–471.
- Zhang C, Liu J, Zhao Y, et al. Glutaminase 2 is a novel negative regulator of small GTPase Rac1 and mediates p53 function in suppressing metastasis. Elife. 2016;5:e10727.
- Gao Y, Dickerson JB, Guo F, et al. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004;101:7618–7623.
- Cancelas JA, Lee AW, Prabhakar R, et al. Rac GTPases differentially integrate signals regulating hematopoietic stem cell localization. Nat Med. 2005;11:886–891.
- Castillo-Lluva S, Tatham MH, Jones RC, et al. SUMOylation of the GTPase Rac1 is required for optimal cell migration. Nat Cell Biol. 2010;12:1078–1085.
- Bellazzo A, Di Minin G, Collavin L. Block one, unleash a hundred. Mechanisms of DAB2IP inactivation in cancer. Cell Death Differ. 2017;24:15–25.
- Xie D, Gore C, Zhou J, et al. DAB2IP coordinates both PI3K-Akt and ASK1 pathways for cell survival and apoptosis. Proc Natl Acad Sci U S A. 2009;106:19878–19883.
- Di Minin G, Bellazzo A, Dal Ferro M, et al. Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Mol Cell. 2014;56:617–629.
- Duan W, Gao L, Jin D, et al. Lung specific expression of a human mutant p53 affects cell proliferation in transgenic mice. Transgenic Res. 2008;17:355–366.
- Scian MJ, Stagliano KE, Ellis MA, et al. Modulation of gene expression by tumor-derived p53 mutants. Cancer Res. 2004;64:7447–7454.
- Adorno M, Cordenonsi M, Montagner M, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137:87–98.
- Wang SP, Wang WL, Chang YL, et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat Cell Biol. 2009;11:694–704.
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501.
- Wendel HG, De Stanchina E, Fridman JS, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–337.
- Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004.
- Scheid MP, Woodgett JR. PKB/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol. 2001;2:760–768.
- Tomas A, Futter CE, Eden ER. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 2014;24:26–34.
- Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007;26:6469–6487.
- Li Y, Wang Z, Kong D, et al. Regulation of Akt/FOXO3a/GSK-3beta/AR signaling network by isoflavone in prostate cancer cells. J Biol Chem. 2008;283:27707–27716.
- Liang J, Zubovitz J, Petrocelli T, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–1160.
- Gottlieb TM, Leal JF, Seger R, et al. Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene. 2002;21:1299–1303.
- Zhou BP, Liao Y, Xia W, et al. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001;3:973–982.
- Abraham AG, O’Neill E. PI3K/Akt-mediated regulation of p53 in cancer. Biochem Soc Trans. 2014;42:798–803.
- Mayo LD, Donner DB. The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem Sci. 2002;27:462–467.
- Feng Z. p53 regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb Perspect Biol. 2010;2:a001057.
- Yue X, Zhao Y, Liu J, et al. BAG2 promotes tumorigenesis through enhancing mutant p53 protein levels and function. Elife. 2015;4. doi:10.7554/eLife.08401.
- Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010;2:a001107.
- Liu J, Zhang C, Hu W, et al. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015;356:197–203.
- Terzian T, Suh YA, Iwakuma T, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337–1344.
- Dupont S, Mamidi A, Cordenonsi M, et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell. 2009;136:123–135.
- Sampath J, Sun D, Kidd VJ, et al. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem. 2001;276:39359–39367.
- Walerych D, Lisek K, Sommaggio R, et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat Cell Biol. 2016;18:897–909.
- Stambolsky P, Tabach Y, Fontemaggi G, et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell. 2010;17:273–285.
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274.
- Mundi PS, Sachdev J, McCourt C, et al. AKT in cancer: new molecular insights and advances in drug development. Br J Clin Pharmacol. 2016;82:943–956.
- Hayashi-Takagi A, Takaki M, Graziane N, et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci. 2010;13:327–332.
- Zheng T, Wang J, Zhao Y, et al. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nat Commun. 2013;4:2996.