
ABSTRACT
Acute myeloid leukemia (AML) is a common hematopoietic malignancy with a generally poor prognosis. Long non-coding RNA (lncRNA) urothelial carcinoma-associated 1 (UCA1) has been identified as an oncogene in various malignancies including AML. However, the role and mechanisms of UCA1 in AML tumorigenesis were incompletely understood. Hence, this study aims to investigate whether UCA1 regulates AML progression by miR-296-3p/Myc axis. Cell proliferation and apoptosis were evaluated by MTT assay and flow cytometry, respectively. Luciferase reporter assay was performed to analyze the interaction between miR-296-3p and UCA1 or Myc. The results showed that UCA1 knockdown inhibited proliferation and induced apoptosis in AML cells (U937 and HL60). Mechanistically, UCA1 acted as a sponge of miR-296-3p by binding to miR-296-3p. Myc, a target of miR-296-3p, was positively regulated by UCA1. Functional assay showed that the anti-AML effect of UCA1 knockdown could be abrogated by miR-296-3p inhibition and Myc overexpression. Moreover, UCA1 knockdown inhibited AML cell tumorigenesis in vivo, which was associated with regulation of miR-296-3p and Myc expression. In conclusion, UCA1 modulates AML progression by regulating miR-296-3p/Myc axis.
Introduction
Acute myeloid leukemia (AML) is a highly heterogeneous hematopoietic malignant tumor characterized by uncontrolled proliferation of myeloid progenitor cells, resulting in abnormal accumulation of immature precursor cells and insufficient production of normal mature blood cells [Citation1]. Despite significant improvements in the treatment of AML, the prognosis of patients with AML remains poor with a 5-year overall survival of less than 40% [Citation2,Citation3]. Therefore, mechanisms underlying the pathogenesis of AML require illumination to improve the treatment of AML.
Increasing evidence shows that long non-coding RNAs (lncRNAs) play an important regulatory role in the development of AML [Citation4,Citation5]. LncRNAs are a type of RNA with lengths exceeding 200 nucleotides, which regulate gene expression and participate in various biological processes [Citation6]. Recently, Gan et al. [Citation7] reported that silencing of lncRNA zinc finger antisense 1 (ZFAS1) inhibited proliferation and facilitated apoptosis in AML cells and suppressed AML xenograft growth in vivo. Qi et al. [Citation8] demonstrated that a novel lncRNA H22954, lowly expressed in AML patients, inhibited AML cell growth in a Bcl-2-dependent mechanism.
LncRNA urothelial carcinoma associated 1 (UCA1) was initially identified in 2006 as a sensitive and specific unique marker for bladder carcinoma [Citation9]. Existing evidence has indicted that UCA1 was highly expressed and played an oncogenic role in multiple malignancies [Citation10–Citation12]. With regard to AML, Sun and his colleagues provided evidence that UCA1 knockdown inhibited cell proliferation, migration and invasion while induced cell apoptosis in human AML cell lines HL60 [Citation13]. Another study demonstrated that UCA1 knockdown played a positive role in overcoming the chemoresistance of pediatric AML by inhibiting glycolysis through the microRNA (miR)-125a/hexokinase 2 pathway [Citation14]. However, the mechanisms of UCA1 in AML tumorigenesis were incompletely understood.
In the current study, we further investigated the roles and molecular mechanisms of UCA1 in the development of AML. microRNA (miR)-296-3p has been shown to exert tumor-suppressing effects in various tumors by targeting oncogenic factors [Citation15,Citation16]. However, its role in AML remains unclear. Myc has been shown to exert a pro-AML action [Citation3]. Our findings confirmed Myc as a target of miR-296-3p. Furthermore, UCA1 knockdown suppressed AML progression by acting as a competitive endogenous RNA (ceRNA) of miR-296-3p to upregulate Myc expression. Our findings provide a novel view of the mechanisms involved in AML progression that may illuminate therapeutic strategies based on molecular crosstalk manipulation.
Materials and methods
Cell culture
Human AML cells (KG1, U937, THP-1, and HL60) and normal HS-5 bone marrow cells were obtained from BeNa Culture Collection (Beijing, China) and have been inspected for phenotypic variation and mycoplasma contamination. Cells were grown in RPMI-1640 medium (for U937, THP-1; HyClone, Logan, UT, USA), IMDM medium (for KG1 and HL60; Gibco, Carlsbad, CA, USA), or DMEM medium (for HS-5; Gibco), supplemented with 10% fetal bovine serum (Gibco) and L-glutamine (2 mM; Gibco).
Cell transfection
UCA1 overexpression vector (pcDNA3.1-UCA1), empty vector, UCA1-siRNA, negative control siRNA (NC-siRNA), miR-296-3p mimic, miR-296-3p inhibitor, mimic NC, and inhibitor NC were purchased from GenePharma (Shanghai, China). Cells were transfected with these plasmids or siRNA using Lipofectamine™ 2000 Transfection Reagent (Invitrogen, Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Cell proliferation assay
Cells in logarithmic growth phase were seeded in a 96-well plate at a density of 1 × 104 cells per well in a final volume of 100 μL. After transfection for 48 h, 20 μL of 5 g/L MTT solution (Sigma-Aldrich, St. Louis, MO, USA) was added to each well. After incubation for 4 h, the supernatant was discarded and 150 μL of DMSO was added to each well. After 15 min of shaking in the dark, the optical density (OD) at 490 nm was examined with a microplate reader (Multiskan MK3, Thermo Labsystems, Finland).
Cell apoptosis assay
Cells in logarithmic growth phase were seeded in a 24-well plate at a density of 1 × 105 cells per well in a final volume of 500 μL. After transfection for 48 h, cells were harvested, washed twice with cold PBS, and then resuspended in 100 μL of Binding Buffer provided in the Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) cell apoptosis kit (Sigma-Aldrich). Afterward, 5 μL Annexin V-FITC and 5 μL PI were added for 10 min of incubation in the dark. The mixtures were then analyzed using the FACScan flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA).
Bioinformatics analysis
To obtain the candidate miRNAs list which could be further investigated, TargetScan (http://www.targetscan.org/vert_72/) was used to predict the candidate miRNAs which may target on Myc mRNA. Moreover, starBase (http://starbase.sysu.edu.cn/) was used to identify miRNAs which may bind to UCA1.
Luciferase reporter assay
The fragments of UCA1 and 3ʹ-untranslated region (UTR) of Myc containing the predicted wild-type (WT) binding sites of miR-296-3p or mutated miR-296-3p binding sites (Mut) were amplified by PCR and inserted into a pMIR-REPORT luciferase reporter vector (Ambion, Austin, TX, USA), named as UCA1 WT, UCA1 Mut, Myc WT, and Myc Mut. For the luciferase reporter assay, 293 cells were co-transfected with 200 ng constructed luciferase reporter vectors, 25 ng pRL-TK (expressing renilla luciferase as the internal control) and 20 μM miR-296-3p mimic or mimic NC using Lipofectamine 2000. The luciferase activity was examined after 48 h of transfection using the luciferase assay kit (Promega, Madison, WI, USA) and the Promega GloMax 20/20 machine.
Animal experiments
Fifteen female nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice (age, 4-week-old; weight, 15–16 g) were obtained from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China). The animals were given free access to food and water at 22°C under a 12-h light/dark cycle. All experiments were performed in compliance with the guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health. This study was approved by the Ethics Committee of The First Affiliated Hospital of Bengbu Medical College. Every effort was made to minimize animal suffering and the number of animals used.
After acclimation for 1 week, mice were randomly divided into three groups (n = 5 per group), namely, control, LV-sh-NC (lentiviruses bearing scramble shRNA), and LV-sh-UCA1 (lentiviruses bearing UCA1 shRNA). HL60 cells were infected with LV-sh-UCA1 or LV-sh-NC. At 24 h after infection, infected cells (1 × 107) stably expressing LV-sh-UCA1 or LV-sh-NC (Hanbio, Shanghai, China) were subcutaneously injected into the underarm of NOD/SCID mice. The mice in the control group received a subcutaneous injection of untreated HL60 cells. Tumor volume (V, mm3) was monitored every 1 week after injection using a caliper and calculated following the formula: V = (ab2)/2, wherein a is the longest dimension (mm) and b is the perpendicular width (mm). Animals were sacrificed under anesthesia with pentobarbital (50 mg/kg body weight, intraperitoneally) at 5 weeks post-injection and tumor tissues were separated. Expression of UCA1 and miR-296-3p, as well as mRNA expression of Myc were examined by quantitative real-time PCR (qRT-PCR). Protein levels of Myc, proliferating cell nuclear antigen (PCNA), Bax, and Bcl-2 were measured by western blot.
qRT-PCR analysis
Total RNA was extracted from tumor tissues and cells by using Trizol reagent (Invitrogen) and then reverse transcribed into cDNA using a reverse transcription kit (Takara, Dalian, China). After that, the expression of UCA1 and Myc was detected using the SYBR premix (Takara) whereas the expression of miR-296-3p was using the miRNA qRT‐PCR Detection kit (GeneCopoeia, Rockville, MD, USA) using primers listed in . U6 and GAPDH were used as internal references for miRNA and mRNA/lncRNA, respectively.
Table 1. The sequence of primers used for qRT-PCR.
Western blot
Total proteins were extracted from tumor tissues and cells using the radioimmunoprecipitation assay lysis buffer (Beyotime, Shanghai, China). Then the protein samples were loaded and separated with 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and transferred to polyvinylidene difluoride membrane. After the blockade of nonspecific protein signals, the membranes were probed overnight at 4 °C with the following primary antibodies: Myc, PCNA, Bax, Bcl-2, and GAPDH (all from Abcam, Cambridge, UK; 1:1000). The membranes were then incubated with the horseradish peroxidase-conjugated secondary antibody (1:2000; Abcam) for 1 h at room temperature. Blots were examined by an Enhanced Chemiluminescence (ECL) Detection kit (Pierce Biotechnology, Rockford, IL, USA).
Statistical analysis
Student’s t-test and one-way ANOVA were used to analyze data with SPSS 22.0 software (IBM, Chicago, IL, USA). P < 0.05 was considered to indicate a statistically significant difference.
Results
Decreased miR-296-3p expression and upregualted UCA1 and Myc expression in AML cells
Expression of UCA1 was significantly higher in human AML cells (KG1, U937, THP-1, and HL60) than that in normal HS-5 bone marrow cells ()). Conversely, miR-196-3p expression was notably downregulated in these four AML cells ()). The mRNA and protein levels of Myc were significantly higher in these AML cells than that in HS-5 cells (,)).
Figure 1. Expression of UCA1, miR-296-3p, and Myc in human AML cells. qRT-PCR analysis of (a) UCA1 expression, (b) miR-296-3p expression, and (c) Myc mRNA expression, and (d) western blot analysis of Myc protein level in human AML cells (KG1, U937, THP-1, and HL60) and normal HS-5 bone marrow cells. *P < 0.05, **P < 0.01, vs. HS-5.
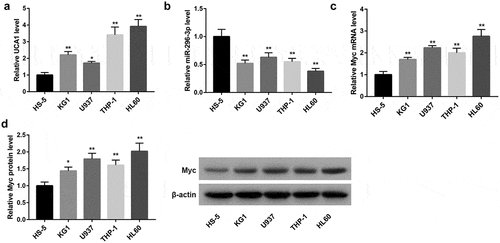
UCA1 knockdown inhibited AML cell proliferation and induced apoptosis
To assess the biological role of UCA1 in AML, we explored the effect of UCA1 knockdown on the proliferation and apoptosis of U937 and HL60 cells. Data from qRT-PCR confirmed that UCA1 expression was distinctly downregulated following transfection with UCA1-siRNA (,)). MTT assay demonstrated that the growth of cells transfected with UCA1-siRNA was notably impaired (,)). Furthermore, flow cytometry analysis indicated that the downregulation of UCA1 greatly facilitated apoptosis in these cells (,)). Moreover, protein levels of PCNA (a proliferative marker) and anti-apoptotic Bcl-2 were noticeably downregulated whereas expression of pro-apoptotic Bax was strikingly upregulated following UCA1 knockdown in both U937 and HL60 cells (,)).
Figure 2. Effect of UCA1 knockdown on AML cell proliferation and apoptosis. (a–b) UCA1 expression determined by qRT-PCR analysis, (c–d) cell viability determined by MTT assay, (e–f) cell apoptosis determined by flow cytometry, and (g–h) protein levels of PCNA, Bax, and Bcl-2 determined by western blot analysis in U937 and HL60 cells which were transfected with NC-siRNA and UCA1-siRNA. The data are presented as the mean ± SD (n = 3). *P < 0.05, **P < 0.01, vs. NC-siRNA.
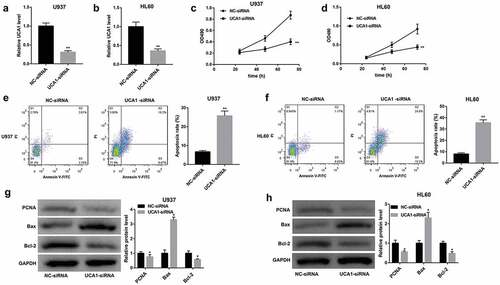
UCA1 acted as a ceRNA of miR-296-3p to elevate Myc expression
To unravel the mechanism of UCA1 behind biological behaviors of AML cells, we performed bioinformatics analysis and found that UCA1 harbors putative binding sites of miR-296-3p (Starbase). Furthermore, the proto-oncogene Myc (also called c-Myc) is identified as a putative target of miR-296-3p (Targetscan). We further determined the relationship among UCA1, miR-296-3p, and Myc. UCA1 knockdown remarkably upregulated miR-296-3p expression (,)) and downregulated Myc mRNA and protein levels (,,,)) in both U937 and HL60 cells. A dual‐luciferase reporter assay was performed to further analyze the interaction between UCA1 and miR-296-3p as well as miR-296-3p and Myc 3ʹUTR and the results showed that miR-296-3p mimic led to a significant decrease in luciferase activity in UCA1 WT group, whereas had no obvious effect on that in UCA1 Mut group, verifying the direct interaction between UCA1 and miR-296-3p ()). Furthermore, the luciferase activity in the Myc WT group was notably decreased following introduction of miR-296-3p mimic. However, the luciferase activity remained unchanged following miR-296-3p overexpression when the binding sites between Myc 3ʹUTR and miR-296-3p were mutated. These data suggested that Myc was a direct target of miR-296-3p ()).
Figure 3. Interaction between UCA1, miR-296-3p, and Myc. Effect of UCA1 knockdown on (a, d) miR-296-3p expression, (b, e) Myc mRNA expression, and (c, f) Myc protein level in U937 and HL60 cells was evaluated by qRT-PCR and western blot. (g) Predicted binding sites between UCA1 and miR-296-3p (Starbase) as well as mutant sites in UCA1 Mut reporter. Predicted binding sites between Myc 3ʹUTR and miR-296-3p (Targetscan) as well as mutant sites in Myc 3ʹUTR Mut reporter. Luciferase reporter assay was performed to evaluate the interaction between UCA1 and miR-296-3p as well as miR-296-3p and Myc 3ʹUTR. The data are presented as the mean ± SD (n = 3). **P < 0.01, vs. NC-siRNA or mimic NC.
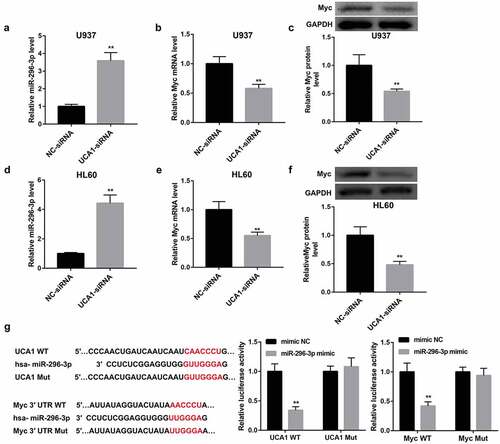
Also, introduction of miR-296-3p mimic significantly upregulated miR-296-3p expression (,)), downregulated UCA1 expression (,)) and Myc mRNA and protein levels (,,,)) in both U937 and HL60 cells. In contrast, inhibition of miR-296-3p by transfection with miR-296-3p inhibitor yielded the opposite effect on expression of miR-296-3p, UCA1 and Myc (–)). In contrast to miR-296-3p mimic, UCA1 overexpression remarkably upregulated mRNA and protein levels of Myc in both cells. More importantly, the inhibition of Myc by miR-296-3p mimic was attenuated by UCA1 overexpression (–)). Together, these findings indicated that UCA1 might act as a ceRNA for miR-296-3p and prevent it from binding to Myc mRNA, leading to upregulation of Myc.
Figure 4. miR-296-3p negatively regulated UCA1 and Myc expression. Effect of miR-296-3p overexpression and miR-296-3p inhibition on (a, e) miR-296-3p expression, (b, f) UCA1 expression, (c, g) Myc mRNA expression, and (d, h) Myc protein level in U937 and HL60 cells. The data are presented as the mean ± SD (n = 3). **P < 0.01, vs. mimic NC; &&P < 0.01, vs. inhibitor NC.
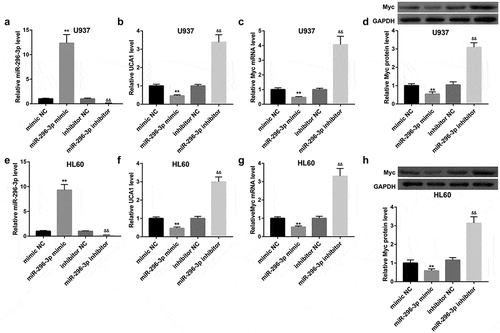
Figure 5. UCA1 overexpression attenuated the inhibition of Myc by miR-296-3p. (a, c) Myc mRNA expression and (b, d) Myc protein level in U937 and HL60 cells which were co-transfected with UCA1 overexpression vector/empty vector and miR-296-3p mimic/mimic NC. The data are presented as the mean ± SD (n = 3). *P < 0.05, **P < 0.01, vs. mimic NC+Vector; ##P < 0.01, vs. mimic NC+UCA1 or Vector+miR-296-3p mimic.
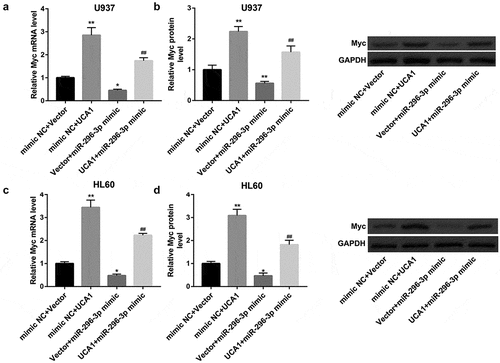
UCA1 knockdown inhibited AML cell proliferation and induced apoptosis by regulating miR-296-3p/Myc axis
To determine whether UCA1 knockdown inhibits AML cell proliferation and induces apoptosis by regulating miR-296-3p/Myc axis, we co-transfected U937 and HL60 cells with UCA1-siRNA and miR-296-3p inhibitor. MTT assay and flow cytometry assay revealed that the proliferation-inhibitory and pro-apoptotic effect of UCA1 knockdown was partially abrogated by inhibition of miR-296-3p (,)). Furthermore, the UCA1 knockdown-mediated downregulation of PCNA and Bcl-2 and upregulation of Bax protein levels were negated by miR-296-3p inhibition ()). Similarly, Myc overexpression also overturned the proliferation-inhibitory and pro-apoptotic effect of UCA1 knockdown in both U937 and HL60 cells (–)).
Figure 6. miR-296-3p inhibition attenuated the anti-AML action of UCA1 knockdown. (a) Cell viability determined by MTT assay, (b) cell apoptosis determined by flow cytometry, and (c) protein levels of PCNA, Bax, and Bcl-2 determined by western blot in U937 and HL60 cells which were co-transfected with NC-siRNA/UCA1-siRNA and inhibitor NC/miR-296-3p inhibitor. The data are presented as the mean ± SD (n = 3). *P < 0.05, **P < 0.01, vs. inhibitor NC+NC-siRNA; #P < 0.05, ##P < 0.01, vs. inhibitor NC+UCA1-siRNA.
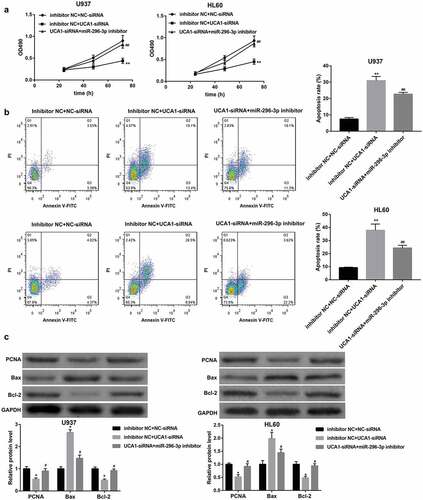
Figure 7. Myc overexpression attenuated the anti-AML action of UCA1 knockdown. (a) Cell viability determined by MTT assay, (b) cell apoptosis determined by flow cytometry, and (c) protein levels of PCNA, Bax, and Bcl-2 determined by western blot in U937 and HL60 cells which were co-transfected with NC-siRNA/UCA1-siRNA and empty vector/Myc overexpression vector. The data are presented as the mean ± SD (n = 3). *P < 0.05, **P < 0.01, vs. Vector+NC-siRNA; #P < 0.05, ##P < 0.01, vs. Vetcor+UCA1-siRNA.
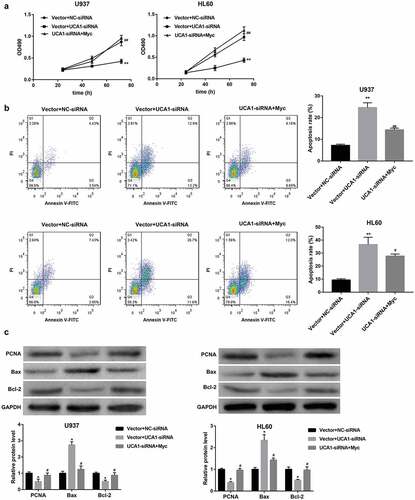
UCA1 knockdown inhibited AML cell tumorigenesis in vivo and regulated miR-296-3p and Myc
To investigate whether the expression level of UCA1 affect AML cell tumorigenesis, HL60 cells stably expressing LV-sh-UCA1 or control LV-sh-NC were inoculated into NOD/SCID mice. UCA1 knockdown led to a notable reduction of tumor volume in HL60 cells-induced mouse xenograft models of AML ()). Furthermore, UCA1 expression was confirmed to be significantly downregulated in tumors formed by LV-sh-UCA1-infected HL60 cells (). Consistent with the in vitro assay, miR-296-3p expression was greatly upregulated ()), whereas Myc mRNA and protein levels were notably downregulated (,)) following the knockdown of UCA1. Moreover, UCA1 knockdown noticeably decreased protein levels of PCNA and Bcl-2, whereas markedly increased that of Bax in tumors ()).
Figure 8. In vivo effect of UCA1 knockdown on xenograft tumor growth and miR-296-3p and Myc expression. (a) Images of representative tumors excised from mice and tumor growth curve reflected by tumor volume. qRT-PCR analysis of (b) UCA1 expression, (c) miR-296-3p expression, and (d) Myc mRNA expression. (e) Western blot analysis of Myc, PCNA, Bax, and Bcl-2 protein levels in tumor tissues. N = 5 in each group. *P < 0.05, **P < 0.01, vs. LV-sh-NC.
Discussion
LncRNAs and miRNAs play crucial roles in regulating multiple cellular processes and have been reported as potential therapeutic targets in AML [Citation17,Citation18]. miRNAs, a class of evolutionarily conserved, endogenous non-coding RNAs of about 22 nucleotides, can post-transcriptionally regulate gene expression of target mRNAs, resulting in translational repression or target degradation [Citation19]. It is well-established that lncRNAs can exert their functions by acting as a ceRNA to segregate miRNAs away from target mRNAs, leading to the downregulation of miRNAs, upregulation of miRNA target genes and suppression of miRNAs-mediated functional roles [Citation20,Citation21]. The ceRNA is an important mechanism of lncRNA‐regulated gene function. For instance, Tian et al. [Citation22] delineated that lncRNA SBF2-AS1 modulated AML cell proliferation by acting as a ceRNA of miR-188-5p to sequester it from its target zinc finger protein 91 (ZFP91).
LncRNA UCA1 is located on chromosome 19p13.12 with 1.4 kb in length and has been identified as an oncogene in multiple malignancies [Citation10–Citation12]. In leukemia, UCA1 has been reported to contribute to chemoresistance of leukemia including AML [Citation14,Citation23]. Furthermore, UCA1 knockdown suppressed cell proliferation, migration and invasion while facilitated cell apoptosis in human myelogenous leukemia cell lines K562 and HL60 [Citation13]. In the current study, we observed that UCA1 was highly expressed in human AML cells (KG1, U937, THP-1, and HL60) and as expected, UCA1 knockdown inhibited proliferation and induced apoptosis in both U937 and HL60 cells. To our knowledge, we demonstrated for the first time that UCA1 knockdown suppressed HL60 cell tumorigenesis in vivo. This study further supplements the cancer-promoting function of UCA1 in AML based on the existing literature, emphasizing UCA1 as a promising therapeutic target for AML.
The proposed mechanisms for the oncogenic functions of UCA1 in malignancies were complicated and one important one was its capacity of sponging certain miRNAs [Citation11,Citation13]. Using bioinformatics analysis and luciferase reporter assay, we found that UCA1 acted as a sponge of miR-296-3p by binding to miR-296-3p. Importantly, the proliferation-inhibitory and pro-apoptotic effect of UCA1 knockdown was overturned by miR-296-3p inhibition, indicating that UCA1 contributes to AML progression by sponging miR-296-3p. miR-296-3p has been shown to exert tumor-suppressing effects in various tumors by targeting oncogenic factors [Citation15,Citation16]. In the present study, we also identified Myc as a target of miR-296-3p. Myc is a proto-oncogene and acts as an important regulator of several cellular processes, including cell proliferation and apoptosis [Citation24,Citation25]. Myc has been identified as an AML-related oncogenic and anti-apoptosis gene and has been shown to exert a pro-AML action [Citation3]. It has been reported that lncRNA potassium voltage-gated channel subfamily Q member 1 overlapping transcript 1 (KCNQ1OT1) promoted AML progression by acting as a ceRNA of miR-326 to elevate Myc expression [Citation26]. Also, a recent study has shown that UCA1 can regulate expression of Myc by binding to certain miRNAs [Citation24]. Our present study adds to this research by demonstrating that UCA1 positively regulated Myc expression by sponging miR-296-3p to segregate it away from Myc mRNA. Moreover, in HL60 cells-induced mouse xenograft models of AML, the inhibition of tumor formation following UCA1 knockdown was accompanied by upregulation of miR-296-3p and downregulation of Myc. Together, UCA1/miR-296-3p/Myc axis was responsible for UCA1 promoting AML progression.
Conclusion
In conclusion, the results in the present study show that UCA1 modulates cell proliferation and apoptosis by regulating miR-296-3p/Myc axis in AML. Our findings may provide the feasibility for using lncRNA-based therapy in AML treatment.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Khasawneh MK, Abdel-Wahab O. Recent discoveries in molecular characterization of acute myeloid leukemia. Curr Hematol Malig Rep. 2014;9(2):93–99.
- Rubnitz JE. Current management of childhood acute myeloid leukemia. Paediatr Drugs. 2017;19(1):1–10.
- Yu Q, Xu Y, Zhuang H, et al. Aberrant activation of RPB1 is critical for cell overgrowth in acute myeloid leukemia. Exp Cell Res. 2019;384(2):111653.
- Lei L, Xia S, Liu D, et al. Genome-wide characterization of lncRNAs in acute myeloid leukemia. Brief Bioinform. 2018;19(4):627–635.
- Gourvest M, Brousset P, Bousquet M. Long noncoding RNAs in acute myeloid leukemia: functional characterization and clinical relevance. Cancers (Basel). 2019;11(11):1638.
- Kwok ZH, Tay Y. Long noncoding RNAs: lincs between human health and disease. Biochem Soc Trans. 2017;45(3):805.
- Gan S, Ma P, Ma J, et al. Knockdown of ZFAS1 suppresses the progression of acute myeloid leukemia by regulating microRNA-150/Sp1 and microRNA-150/Myb pathways. Eur J Pharmacol. 2019;844:38–48.
- Qi X, Jiao Y, Cheng C, et al. H22954, a novel long non-coding RNA down-regulated in AML, inhibits cancer growth in a BCL-2-dependent mechanism. Cancer Lett. 2019;454:26–36.
- Wang XS, Zhang Z, Wang HC, et al. Rapid identification of UCA1 as a very sensitive and specific unique marker for human bladder carcinoma. Clin Cancer Res. 2006;12(16):4851–4858.
- Xue M, Chen W, Li X. Urothelial cancer associated 1: a long noncoding RNA with a crucial role in cancer. J Cancer Res Clin Oncol. 2016;142(7):1407–1419.
- Yu H, Yang C, Jian L, et al. Sulfasalazine‑induced ferroptosis in breast cancer cells is reduced by the inhibitory effect of estrogen receptor on the transferrin receptor.. Oncol Rep. 2019;42(2):826–838.
- Li LB, Chai R, Zhang S, et al. Iron exposure and the cellular mechanisms linked to neuron degeneration in adult mice. Cells. 2019;8(2):undefined.
- Sun MD, Zheng Y-Q, Wang L-P, et al. Long noncoding RNA UCA1 promotes cell proliferation, migration and invasion of human leukemia cells via sponging miR-126. Eur Rev Med Pharmacol Sci. 2018;22(8):2233–2245.
- Zhang Y, Liu Y, Xu X. Knockdown of LncRNA-UCA1 suppresses chemoresistance of pediatric AML by inhibiting glycolysis through the microRNA-125a/hexokinase 2 pathway. J Cell Biochem. 2018;119(7):6296–6308.
- Wang X, Hu Y, Cui J, et al. Coordinated targeting of MMP-2/MMP-9 by miR-296-3p/FOXCUT exerts tumor-suppressing effects in choroidal malignant melanoma. Mol Cell Biochem. 2018;445(1–2):25–33.
- Wang L, Chen R, Zhang Y. miR-296-3p targets APEX1 to suppress cell migration and invasion of non-small-cell lung cancer. Oncol Lett. 2019;18(3):2612–2618.
- Peng L, Zhang Y, Xin H. lncRNA SNHG3 facilitates acute myeloid leukemia cell growth via the regulation of miR-758-3p/SRGN axis. J Cell Biochem. 2020;121(2):1023–1031.
- Niu M, Zhang N, Wang R, et al. MiR-340 is a biomarker for selecting treatment between chemotherapy and allogeneic transplantation in acute myeloid leukemia. Front Oncol. 2019;9:1058.
- Xu W, Hang M, Yuan C-Y, et al. MicroRNA-139-5p inhibits cell proliferation and invasion by targeting insulin-like growth factor 1 receptor in human non-small cell lung cancer. Int J Clin Exp Pathol. 2015;8(4):3864–3870.
- Lu M-H, Tang B, Zeng S, et al. Long noncoding RNA BC032469, a novel competing endogenous RNA, upregulates hTERT expression by sponging miR-1207-5p and promotes proliferation in gastric cancer. Oncogene. 2016;35(27):3524–3534.
- Cesana M, Cacchiarelli D, Legnini I, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147(2):358–369.
- Tian Y-J, Wang Y-H, Xiao A-J, et al. Long noncoding RNA SBF2-AS1 act as a ceRNA to modulate cell proliferation via binding with miR-188-5p in acute myeloid leukemia. Artif Cells Nanomed Biotechnol. 2019;47(1):1730–1737.
- Xiao Y, Jiao C, Lin Y, et al. lncRNA UCA1 contributes to imatinib resistance by acting as a ceRNA against miR-16 in chronic myeloid leukemia cells. DNA Cell Biol. 2017;36(1):18–25.
- Wang Y, Hou Z, Li D. Long noncoding RNA UCA1 promotes anaplastic thyroid cancer cell proliferation via miR135amediated cmyc activation. Mol Med Rep. 2018;18(3):3068–3076.
- Adams CM, Hiebert SW, Eischen CM. Myc induces miRNA-mediated apoptosis in response to HDAC inhibition in hematologic malignancies. Cancer Res. 2016;76(3):736–748.
- Huang P, Wang L, Li Q, et al. Atorvastatin enhances the therapeutic efficacy of mesenchymal stem cells derived exosomes in acute myocardial infarction via up-regulating long non-coding RNA H19. Cardiovasc Res. 2019;116(2):353–367.