
ABSTRACT
Long non-coding RNA metastasis associated with lung adenocarcinoma transcript 1 (MALAT1) contributes to chemotherapy resistance in some cancers, but the role of MALAT1 in sunitinib (SU) chemoresistance of carcinoma (RCC) is still unknown. In this study, MALAT1 expression in SU-resistance tumor tissues and cells was tested by qRT-PCR. Then, CCK-8, Annexin V-FITC/PI, transwell, and Western blotting assays were used to evaluate cell viability and IC50, apoptosis, cell invasion, and resistance of SU-resistance RCC cells after transfected with small interfering RNA against MALAT1. Further, RNA pull-down and luciferase reporter assay were applied to investigate the underlying mechanism of MALAT1 in SU resistance. The results showed that MALAT1 expression was dramatically upregulated in SU-resistance RCC tissues and cell lines. Knockdown of MALAT1 inhibited proliferation, invasion, and SU chemoresistance, but induced apoptosis in RCC cells. The results of RNA pull-down and luciferase reporter assay indicated that MALAT1 could interact with miR-362-3p and miR-362-3p interact with RasGAP SH3-domain-Binding Protein 1 (G3BP1). Moreover, G3BP1 also played a role in SU chemoresistance of RCC cells, and MALAT1 could perform as a miR-362-3p sponge to modulate G3BP1 expression. Rescue experiments suggested that downregulation of miR-362-3p and overexpression of G3BP1 can reverse the SU chemosensitivity of MALAT1 knockdown in RCC cells. In conclusion, depletion of LncRNA MALAT1 inhibited SU chemoresistance through modulating G3BP1 via sponging miR-362-3p in RCC cells, suggesting that targeting MALAT1 may be a potential therapeutic strategy for SU-resistance RCC.
Introduction
Renal cell carcinoma (RCC) is one of the most common malignancies in the urinary system and accounts for 2%~3% of cancers in adults with the incidence rates increasing 2% per year [Citation1,Citation2]. Clear cell Renal cell carcinoma (ccRCC) is the majority subtype and is responsible for approximately 70% of RCC [Citation3,Citation4]. Although sunitinib (SU)-based chemotherapy is an effective method used in the treatment of RCC, due to an inherent tumor resistance of RCC, the 5-year overall survival rate is still only 8% for metastatic and advanced disease [Citation5,Citation6]. Therefore, understanding of the SU-resistance mechanism and the specific targets contributing to SU-resistance RCC is paramount importance for improving the overall prognosis of RCC patients.
Long non-coding RNAs (lncRNAs) are important class of non-coding RNAs that are characterized by their length longer than 200 nt [Citation7,Citation8]. It has been proved that LncRNAs play a vital role in diverse biological processes, including cell differentiation, metabolism, and cancer progression [Citation9,Citation10]. In addition, LncRNAs can regulate molecules by multiple mechanisms, such as epigenetic regulation, genomic imprinting, RNA stability, RNA alternative splice, and microRNA regulation [Citation11,Citation12]. Emerging evidence suggests that the dysregulation of lncRNAs is widely involved in the drug-resistance development. For example, lncRNA GAS5 enhances the sensitivity of SU in RCC cells via interacting with miR-21-mediated SOX5 [Citation13]; lncRNA HOXD-AS1 promotes castration resistance and chemoresistance in prostate cancer by recruiting WDR5 [Citation14]. Therefore, lncRNAs can be an effective molecular therapeutic target of drug-resistance cancers.
LncRNA metastasis-associated in lung adenocarcinoma transcript 1 (MALAT1) encode gene is located on chromosome 11q13 and contains more than 8000 nucleotides. Numerous studies disclosed MALAT1 exerts oncogenic functions in massive cancers via interacting with its target miRNA [Citation15,Citation16]. Furthermore, recent researches also reported that MALAT1 enhances chemo-induced autophagy and chemoresistance of gastric cancer. MALAT1 also can promote docetaxel resistance via modulating miR-145-5p-mediated AKAP12 expression in prostate cancer cells [Citation17,Citation18]. Although the involvement of MALAT1 in the chemoresistance is frequently observed in various cancers, the role of MALAT1 in the SU-resistance remains to be well elucidated.
In the current study, we explored the expression of lncRNA MALAT1 in SU-resistance or nonresistance RCC patients and cell lines. Furthermore, the roles of MALAT1 in the progression and SU chemoresistance were investigated in vitro to better understanding its function and mechanism in SU-resistance RCC. Our data revealed that MALAT1 is a target therapeutic molecule for SU-resistance RCC.
Materials and methods
Patients and clinical samples
All 96 pairs of human renal cell carcinoma (RCC) tissues were obtained from Yulin No.2 Hospital. None of the patients received any chemotherapy and radiotherapy before. The RCC patients received sunitinib (SU) chemotherapy every 3 weeks according to NCCN guidelines, totaling four cycles. After chemotherapy for 3 months, 26 patients with tumors shrink by more than 30% or disappear was defined as non-drug resistance; 35 patients with tumor enlargement by more than 30% or emergence of new metastases were defined as drug resistance. The fresh nonresistance tumor tissues (n = 26), SU-resistance tumor tissues (n = 35) and adjacent non-tumor tissues (n = 35) of RCC patients were rapidly frozen in −80°C. All the cancer tissue samples were collected with written informed consent in accordance with the Declaration of Helsinki and with the approval of the Ethical Committee of Yulin No.2 Hospital.
Cell lines and cell culture
HEK-293T, HK-2, CAKI-2, ACHN, 769P, 786O cell lines were purchased from Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China), the cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, USA) adding 10% Fetal Bovine Serum (FBS, Thermo Fisher Scientific, USA) at 37°C with 5% CO2 in a humidified incubator. SU-resistance 769P (769P-SU) and SU-resistance 786O (786O-SU) cells were derived from 769P-SU and 786O-SU tumor xenografts. The half-maximal inhibitory concentration (IC50) for SU was significantly increased in 786O-SU and 769P-SU cells, compared with 786O and 769P cells, as indicated in Supplement of Additional File 1. 769P-SU and 786O-SU cells were maintained in the medium containing 2 μM SU (Sigma Chemical Co, USA).
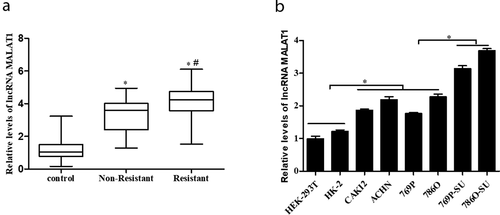
Figure 1. MALAT1 expression was upregulated in RCC tissue specimens, cell lines, and SU-resistance RCC cells. (a) qRT-PCR analysis of MALAT1 expression level in nonresistance tumor tissues (n = 26), SU-resistance tumor tissues (n = 35) and adjacent non-tumor tissues (n = 35) of RCC patients (the average expression level of the control group was defined as 1). *p < 0.05 vs control, #p < 0.05 vs nonresistance group. (b) Comparison of MALAT1 level among human normal renal epithelial cell lines (HEK-293 T and HK-2), RCC cell lines (CAKI-2, ACHN, 769P and 786O) and SU-resistance RCC strains (769P-SU and 786O-SU). * p < 0.05 vs control
Cell proliferation
Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, Inc.) was performed to detect the cell viability in this study. Briefly, cells were seeded at a density of 2 × 103 cells per well in 96-well plates. After cultured for 48 h, per well cells were incubated with 10 μl CCK-8 solution at room temperature for 2 h, and OD values were measured at 450 nm.
Apoptosis assessment
The RCC cells were harvested and resuspended in binding buffer, and then incubated with 0.25 mg/ml Annexin V-FITC (Dojindo, Japan) and 10 mg/ml propidium iodide (PI) (Dojindo, Japan) at 25°C in the dark for 30 min. Finally, apoptosis evaluated by a flow cytometry (BD Biosciences, USA).
Transwell assay
The invasion of RCC cells was detected by transwell assay. In brief, 5 × 104 RCC cells were seeded in the upper Chambers (8 mm, BD Biosciences), and cultured in 200 ml serum-free DMEM for 48 h at 37°C. After incubation, the upper surface of the membrane was wiped with a cotton tip and cells attached to the lower surface were stained with crystal violet for 5 min, and then cells were counted.
Quantitative reverse-transcriptase PCR (qRT-PCR)
The total RNA of treated or non-treated cells were extracted by TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). The TaqMan MicroRNA Reverse Transcription Kit and TaqMan miRNA assay (Qiagen) were used to the quantification of miRNA expression. U6 was performed as the internal control. Prime ScriptTM RT reagent Kit (Takara) and DyNamo SYBR1 Green qPCR Kit (Takara) was used to measure the mRNAs expression, according to the manufacturer’s protocol. GAPDH served as an internal control. The results were evaluated using the 2−ΔΔ CT. The primers are following: MALAT1 (Forward 5ʹ- GCGACGAGTTGTGCTGCTATCT −3ʹ; Reverse 5ʹ- ACACTGCTCTGGGTCTGCTTTT −3ʹ), miR-362-3p (Forward 5ʹ-ATATACATGAGAGTGAGACTTG −3ʹ; Reverse 5ʹ- AACACACCTATTCAAGGATTCA −3ʹ), G3BP1 (Forward 5ʹ- CATTAACAGTGGTGGGAAA −3ʹ; Reverse 5ʹ-AGGCTTTGAGGAGATTCAT −3ʹ), U6 (Forward 5ʹ-CGCTTCGGCAGCACATATAC-3ʹ; Reverse 5ʹ-AAATATGGAACGCTTCACGA-3ʹ), GAPDH (Forward 5ʹ- CACCATCTTCCAGGAGCGAG-3ʹ; Reverse 5ʹ- TCACGCCACAGTTTCCCGA −3ʹ).
Transfection assay
The G3BP1 overexpression vector (pcDNA-G3BP1), the small interfering RNAs (siRNAs) against MALAT1, and miR-362-3p inhibitor and mimics were all constructed and purchased from GenePharma (China). The transfection assay was performed using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s protocol. Primer sequences used in transfection are following: pCDH_G3BP1 (5ʹ-CUCACGCUCUUGUUGCUUC-3ʹ), siMALAT1 (5ʹ- GGGCUUCTCUUAACAUUUA −3ʹ), siRNA-NC (5ʹ-UUCUCCGAACGUGUCACGU −3ʹ), miR-362-3p inhibitor (5ʹ-GCCUAACAAGGUUCAAA-3ʹ), inhibitor-NC (5ʹ- GGCUCAUCGAUAGUCAUUA −3ʹ), miR-362-3p mimics (5ʹ-GUGUGGAUUGUUCCAAGUUU −3ʹ).
Pull-down assay with biotinylated RNA
Cells were transfected with biotinylated RNA (50 nm) and harvested after transfection for 48 h. The cells were washed with PBS and then cultured in a solution of ice for 10 min. The lysates were cleared by centrifugation and then 50 ml sample was aliquoted for input. The remaining lysates were cultured with M-280 streptavidin magnetic beads (Sigma). The bound RNAs were purified by Trizol for the analysis. Probe sequence used in RNA pull-down assay is following: MALAT1 (5ʹ- GATCTGCAGAGGATCCTAGACCAGCATGC −3ʹ), control probe (5ʹ- TGATGTCTAGCGCTTGGGCTTTG −3ʹ).
Western blotting
769P-SU cells were harvested and lysed Radio Immunoprecipitation Assay (RIPA) buffer (Thermo Scientific) with a protease inhibitor cocktail (Millipore, MA, USA). The protein extraction resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and subsequently transferred onto a PVDF membrane (Millipore). After blocked by 5% nonfat milk for 1 h at room temperature, the membrane was incubated with primary antibody at 4°C overnight. The primary antibodies including anti-Bax antibody (1:500, Abcam, ab32503), anti-Bcl-2 antibody (1:500, Abcam, ab32124), anti-P-gp antibody (1:500, Abcam, ab103477), anti-ERCC1 antibody (1:500, Abcam, ab129267), anti-GAPDH antibody (1:500, Abcam, ab181150). Then, the membrane was treated with secondary antibody conjugated to horseradish peroxidase for 2 h at room temperature. Subsequently, protein bands were visualized using SuperSignal West Femto Maximum Sensitivity Substrate solution (Thermo).
Luciferase activity assay
The pmirGLO-G3BP1-WT and pmirGLO-G3BP1-MUT, which were synthesized by GenePharma Co. (Shanghai, China), were transfected into cells (150 ng/well) using Lipofectamine 3000 (Invitrogen). Then, those cells were infected with indicated adenovirus. At 48 h after infection, luciferase activity was measured.
Statistical analysis
All experiments were repeated at least three independent times. Statistical analysis was performed using SPSS 22.0 software (SPSS Inc., USA). The data are expressed as the mean ± SEM. The results value of *p < 0.05, ***p < 0.01 and #p < 0.05 were considered statistically significant.
Results
1 MALAT1 expression was upregulated in RCC tissue specimens, cell lines, and SU-resistance RCC cells
To determine whether MALAT1 participates in the development of human RCC, we first detected MALAT1 expression level in SU-resistance and nonresistance RCC tissues by qRT-PCR. As shown in ), the expression level of MALAT1 was significantly upregulated in SU-resistance RCC tissues compared with nonresistance RCC tissues (p < 0.05). Then, expression of MALAT1 was also examined by qRT-PCR in various renal cells, including nonmalignant renal cell lines HEK-293 T/HK-2, RCC cell lines CAKI-2/ACHN/769P/786O, and the SU-resistance RCC strains 769P-SU/786O-SU. As shown in ), MALAT1 expression was significantly increased in SU-resistance RCC strains (p < 0.05). Thus, our data suggest that MALAT1 might play a role in SU-resistance RCC.
2 si-MALAT1 inhibited cell proliferation, invasion, and resistance in SU-resistance RCC
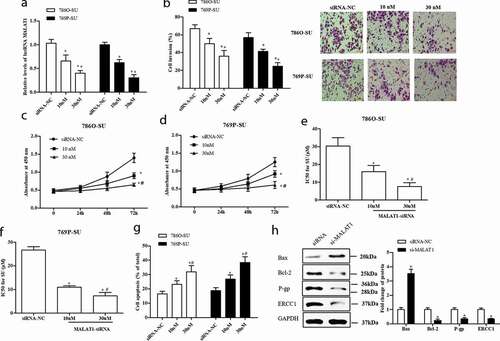
To further investigate the function of MALAT1 in SU-resistance RCC, small interfering RNAs (siRNAs) against MALAT1 were transfected into the 786O-SU and 769P-SU cells. As shown in ), the expression level of MALAT1 of SU-resistance RCC cells was decreased by MALAT1 siRNAs (si-MALAT1) in a dose-dependent manner (p < 0.05). In addition, incubation of SU-resistance RCC cells with si-MALAT1 induced dose-dependent reduction of the cell invasion and viability, the differences were significant (p < 0.05, )). The results of IC50 value, apoptosis assessment, and Western blotting assay showed that si-MALAT1 enhanced apoptosis and inhibited drug-resistance in SU-resistance RCC cells (p < 0.05, )). Hence, these results indicate that MALAT1 could act as an oncogene and enhanced the SU-resistance in SU-resistance RCC.
Figure 2. Si-MALAT1 inhibited cell proliferation, invasion, and resistant of SU-resistance RCC. The 786O-SU and 769P-SU cells were harvested after siRNA-NC, 10 nM, 30 nM si-MALAT1 transfection for 48 h. (a) qRT-PCR analysis of MALAT1 expression level in treated cells. (b) Transwell assay was performed to detect the cell invasion of treated cells, scale bar: 100 µm. (c, d) CCK-8 assay detected the cell proliferation of treated 786O-SU (c) and 769P-SU (d) cells. (e, f) The half-maximal inhibitory concentration (IC50) for SU in treated 786O-SU and 769P-SU cells (μg/mL). (g) The apoptosis rate of 786O-SU and 769P-SU cells. (h) The 786O-SU cells were harvested after 30 nM si-MALAT1 transfection for 48 h. Western blotting was used to measure the protein levels of apoptosis-related genes (Bax, Bcl-2) and resistant-related genes (P-gp, ERCC1), with GAPDH was performed as internal control. *p < 0.05 vs siRNA-NC, #p < 0.05 vs 10 nM si-MALAT1
3 G3BP1 enhanced resistant in SU-resistance RCC
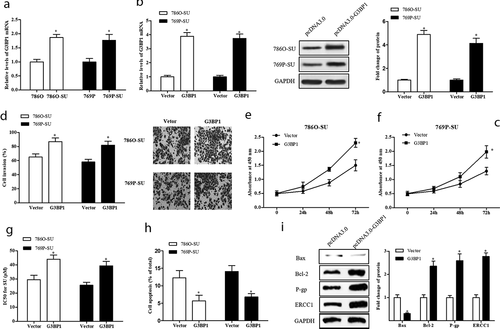
RasGAP SH3-domain-Binding Protein 1 (G3BP1) has been implicated in cell growth, migration, and metastasis of various cancers, and it also can promote the cell proliferation and metastasis of RCC in vivo [Citation17–21]. To explore the role of G3BP1 in SU-resistance RCC, the constructed overexpression vector (pcDNA-G3BP1) was transfected into SU-resistance and nonresistance RCC cells. As shown in ), G3BP1 expression was upregulated in SU-resistance RCC cells compared with nonresistance cells, and pcDNA-G3BP1 significantly increased G3BP1 expression level in SU-resistance RCC cells (p < 0.05). In addition, pcDNA-G3BP1 significantly promoted the cell invasion and viability in SU-resistance RCC cells (p < 0.05, )). Further, the results of IC50 value, apoptosis rate, and Western blotting showed that pcDNA-G3BP1 suppressed cell apoptosis and promoted drug-resistance in SU-resistance RCC cells (p < 0.05, )). In a word, our data suggest that G3BP1 contributed to SU resistance in RCC cells.
Figure 3. G3BP1 enhanced cell proliferation, invasion, resistant of SU-resistance RCC. (a) qRT-PCR analysis of G3BP1 expression level in nonresistance RCC cells and SU-resistance RCC cells. GAPDH as control. The 786O-SU and 769P-SU cells were harvested after 1 μg/mL vector (pcDNA3.0) or 1 μg/mL G3BP1 overexpression vector (pcDNA-G3BP1) transfection for 48 h. (b) qRT-PCR analysis of G3BP1 expression level in treated cells. (c) Western blotting analysis of the protein levels of G3BP1 in treated cells, with GAPDH was used as internal control. (d) The cell invasion of treated cells, scale bar: 100 µm. (e, f) CCK-8 assay detected the cell proliferation of treated 786O-SU and 769P-SU cells. (g) The IC50 for SU in treated 786O-SU and 769P-SU cells (μg/mL). (h) The apoptosis rate of 786O-SU and 769P-SU cells. (i) The 786O-SU cells were transfected with 30 nM si-MALAT1 for 48 h. Western blotting analysis of the protein levels of Bax/Bcl-2 and P-gp/ERCC1. GAPDH as control. *p < 0.05 vs vector
4 MALAT1 sponged miR-362-3p to modulate G3BP1 expression
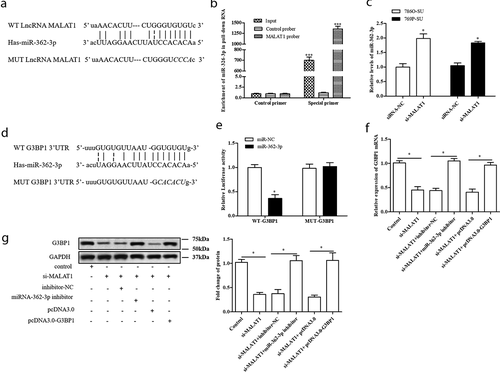
Accumulating evidence indicates that MALAT1 can exert its functions by regarding as a competing endogenous RNA (ceRNA) to regulate miRNAs. In this study, the bioinformatics prediction analysis was performed to identify microRNA that harbors complementary sequences to MALAT1. The result of ) displayed that miR-362-3p has complementary binding sequences of MALAT1. Then, we applied a biotin-avidin pull-down system to test whether miR-362-3p could directly bind to MALAT1. The result showed that miR-362-3p could be pull-downed by MALAT1 probe, but the mutations of control were not able to pull-down miR-362-3p (p < 0.01, )). In addition, si-MALAT1 significantly increased miR-362-3p expression level in SU-resistance RCC (p < 0.05, )). What is more, the result of biological databases software starBase determined that putative 3ʹ UTR sequence of RasGAP SH3-domain-Binding Protein 1 (G3BP1) was complementary to miR-362-3p ()). To validate the predicted binding sequence between G3BP1 and miR-362-3p, 786O cells were transfected with the constructs of wild-type promoter (WT-G3BP1) or the promoter with mutations in the binding site (MUT-G3BP1) for 48 h. The luciferase activity result showed that miR-362-3p significantly decreased luciferase activity in WT-G3BP1 transfected 786O cells, rather than MUT-G3BP1 transfected 786O cells (p < 0.05, )). According to these data, we speculated that MALAT1 could act as a ceRNA to regulate G3BP1 expression by interacting with miR-362-3p. The further result indicated that si-MALAT1 conspicuously downregulated the expression of G3BP1; however, miR-362-3p inhibitor and pcDNA-G3BP1 counteracted this effect (p < 0.05, )). Therefore, our findings demonstrate that MALAT1 could modulate G3BP1 expression by sponging miR-362-3p in SU-resistance RCC.
Figure 4. MALAT1 sponged miR-362-3p to modulate G3BP1 expression. (a) The binding sits between miRNA-362-3p and MALAT1 was predicted by biological databases software starBase. The RNA pull-down showed MALAT1 can bind to miRNA-362-3p in vitro. (b) 786O cells were incubated with a MALAT1 probe or a random probe-coated magnetic bead. After washing the enrichment of beads/RNA complex, miRNA-362-3p was eluted from the streptavidin beads and was analyzed by qRT-PCR. ***p < 0.001 vs control primer. (c) 786O-SU and 769P-SU cells were harvested after 30 nM si-MALAT1 transfection for 48 h, and qRT-PCR was used to detect the expression level of miRNA-362-3p. *p < 0.05 vs siRNA. (d) Schematic diagram showed the sequence alignment between 3ʹUTR of G3BP1 and miR-362-3p, with italics indicating the mutated seed region. (e) In 786O cells, the luciferase assay showed that miR-362-3p mimics significantly reduced the luciferase activity of WT-G3BP1 but not MUT-G3BP1, indicating G3BP1 as a direct target of miR-362-3p. *p < 0.05 vs mimics control. (f) The relative expression of G3BP1 mRNA in siMALAT1/miR-362-3p inhibitor/pcDNA-G3BP1 transfected 786O-SU cells. *p < 0.05 vs control. (g) The relative expression of G3BP1 protein in siMALAT1/miR-362-3p inhibitor/pcDNA-G3BP1 transfected 786O-SU cells. *p < 0.05 vs control
5 Depletion of MALAT1 inhibited SU resistance via miR-362-3p/G3BP1 axis
On the basis of the previous data, si-MALAT1 enhanced the expression of miR-362-3p and miR-362-3p inhibitor upregulated the expression of its target gene G3BP1. Therefore, we speculated that MALAT1 might play a role in SU-resistance RCC by regulating G3BP1 expression. As shown in ), si-MALAT1 conspicuously downregulated cell invasion, and miR-362-3p inhibitor and pcDNA-G3BP1 counteracted this effect (p < 0.05). Moreover, pcDNA-G3BP1 and miR-362-3p inhibitor ameliorated the inhibition of si-MALAT1 on IC50 value and the expression of resistant-related genes, abated the promotion of si-MALAT1 on apoptosis (p < 0.05, )). Therefore, as shown in the graphical abstract of , our study suggests that downregulation of MALAT1 inhibited cell invasion, cell apoptosis, and SU resistance by modulating miR-362-3p/G3BP1 axis.
Figure 5. Depletion of MALAT1 inhibited SU resistance via miR-362-3p/G3BP1 axis. The 786O-SU cells were harvested after transfected with si-MALAT1, inhibitor-NC, miR-362-3p inhibitor, pcDNA3.0 and pcDNA-G3BP1. (a) The invasion of each group cells, scale bar: 100 µm. (b) The IC50 for SU in each group cells (μg/mL). (c)The protein level of P-gp and ERCC1 in each group cells. GAPDH as control. (d) The apoptosis of each group cells. *p < 0.05 vs control
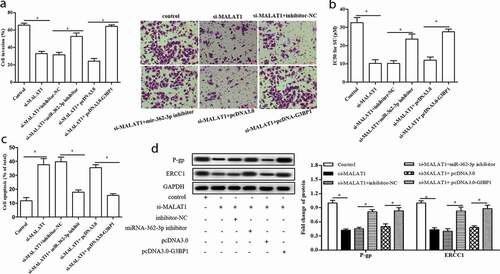
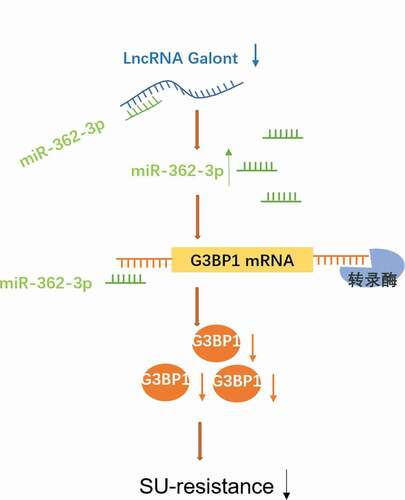
Figure 6. The graphical abstract of MALAT1/miR-362-3p/G3BP1 axis function in SU-resistance RCC
Discussion
Chemoresistance is still a headache for RCC chemotherapy and tremendously hinder therapeutic effect. Numerous lncRNAs have been reported involve in the drug resistance of cancers, which also can serve as biomarkers/targets of drug resistance in diverse cancer progression [Citation14,Citation19,Citation20]. In this study, we found that MALAT1 was overexpressed in SU-resistance RCC tissues and cells. In addition, knocking down MALAT1 inhibited chemoresistance, cell proliferation, and invasiveness, as well as enhanced cell apoptosis in SU-resistance RCC. The results of functional and mechanistic experiments revealed that MALAT1 acted as a ceRNA of miR-362-3p, which controlled its down-stream target G3BP1. Our findings demonstrate that MALAT1 was responsible for SU resistance in RCC cells via miR-362-3p/G3BP1 axis.
LncRNA MALAT1 was first recognized as a marker for metastasis development in the early stages of lung adenocarcinoma [Citation15,Citation21,Citation22]. Growing evidence shows that high MALAT1 expression in RCC tissues is inversely correlated with overall survival, and downregulation of MALAT1 inhibits ccRCC tumor growth by miR-182-5p regulation [Citation23,Citation24]. Furthermore, MALAT1 has been proved to play a positive role in drugs chemoresistance of various human cancers, and recent studies have reported that the dysregulation of MALAT1 also discovered in RCC [Citation17,Citation18,Citation25]. Our data indicated that MALAT1 expression was significantly increased in SU-resistance RCC tissues and cells, and inhibition of MALAT1 significantly suppressed SU-resistance in RCC cells.
CeRNA has become an accepted hypothesis, in which lncRNAs indirectly affect gene expression by sponging their target miRNAs [Citation11,Citation26,Citation27]. In this study, we determined that MALAT1 can perform as a sponge to up-regulate G3BP1 by sequestering miR-362-3p. It has been reported that miR-362-3p plays a tumor-suppressive role in a variety of carcinoma biological processes, including cell proliferation, invasion, resistance, and apoptosis [Citation28,Citation29]. A previous study shows that the upregulation of miR-362-3p suppresses the proliferation, migration, and invasion of the renal cancer cells [Citation30]. G3BP1 is an mRNA-binding protein that regulates mRNA stability and stress granule formation [Citation31]. What is more, it has been found that G3BP1 contributes to cell growth, migration, and metastasis of hepatocellular carcinoma [Citation32]. Further, Wang et al. indicated that G3BP1 promotes tumor progression and metastasis of RCC via IL-6/G3BP1/STAT3 signaling axis [Citation33]. Based on its crucial involvement in multiple biological processes of tumorigenesis, G3BP1 has been regarded as a molecular target for various oncologic investigations [Citation34,Citation35]. In the present study, we demonstrated that G3BP1 led to a promotion of the SU-resistance mechanism in HCC, and G3BP1 partly reversed the decreased SU chemosensitivity of MALAT1 knockdown in SU-resistance RCC cells.
Our study indicates that lncRNA MALAT1 is highly expressed in SU-resistance RCC cells compared with nonresistance RCC cells in vivo and in vitro, and knockdown of MALAT1 suppressed the SU resistance of RCC cells by regulating miR-362-3p/G3BP1 axis. Hence, our findings contribute to the comprehension of SU resistance of RCC, and MALAT1 can be a potential therapeutic target for SU-resistance RCC.
Supplemental Material
Download MS Word (224.7 KB)Acknowledgments
None.
Disclosure statement
The authors declare that they have no competing interests.
Supplemental data
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Zhang Y, Fang L, Zang Y, et al. CIP2A promotes proliferation, invasion and chemoresistance to cisplatin in renal cell carcinoma. J Cancer. 2018;9(21):4029.
- Zhao S, Wang Y, Luo M, et al. Long noncoding RNA small nucleolar RNA host gene 1 (SNHG1) promotes renal cell carcinoma progression and metastasis by negatively regulating miR-137. Med Sci Monit. 2018;24:3824.
- Yuan L, Zeng G, Chen L, et al. Identification of key genes and pathways in human clear cell renal cell carcinoma (ccRCC) by co-expression analysis. Int J Biol Sci. 2018;14(3):266. .
- Wang G, Zhang ZJ, Jian WG, et al. Novel long noncoding RNA OTUD6B-AS1 indicates poor prognosis and inhibits clear cell renal cell carcinoma proliferation via the Wnt/β-catenin signaling pathway. Mol Cancer. 2019;18(1):15. .
- Rini BI, Hutson TE, Figlin RA, et al. Sunitinib in patients with metastatic renal cell carcinoma: clinical outcome according to international metastatic renal cell carcinoma database consortium risk group. Clin Genitourin Cancer. 2018;16(4):298–304. .
- Guo J, Jin J, Oya M, et al. Safety of pazopanib and sunitinib in treatment-naive patients with metastatic renal cell carcinoma: asian versus non-Asian subgroup analysis of the COMPARZ trial. J Hematol Oncol. 2018;11(1):69. .
- Wang X, Sun W, Shen W, et al. Long non-coding RNA DILC regulates liver cancer stem cells via IL-6/STAT3 axis. J Hepatol. 2016;64(6):1283–1294. .
- Wang K, Liu F, Zhou LY, et al. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res. 2014;114(9):1377–1388. .
- Guo X, Yang Z, Zhi Q, et al. Long noncoding RNA OR3A4 promotes metastasis and tumorigenicity in gastric cancer. Oncotarget. 2016;7(21):30276–30294. .
- Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152(6):1298–1307.
- Paraskevopoulou MD, Hatzigeorgiou AG. Analyzing miRNA-lncRNA interactions. Methods Mol Biol. 2016;1402:271–286.
- Engreitz JM, Haines JE, Perez EM, et al. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature. 2016;539(7629):452. .
- Liu L, Pang X, Shang W, et al. Long non-coding RNA GAS5 sensitizes renal cell carcinoma to sorafenib via miR-21/SOX5 pathway. Cell Cycle. 2018;18(3):257–263.
- Gu P, Chen X, Xie R, et al. lncRNA HOXD-AS1 regulates proliferation and chemo-resistance of castration-resistant prostate cancer via recruiting WDR5. Mol Ther. 2017;25(8):1959–1973. .
- Li J, Wang J, Chen Y, et al. LncRNA MALAT1 exerts oncogenic functions in lung adenocarcinoma by targeting miR-204. Am J Cancer Res. 2016;6(5):1099.
- Hirata H, Hinoda Y, Shahryari V, et al. Long noncoding RNA MALAT1 promotes aggressive renal cell carcinoma through Ezh2 and interacts with miR-205. Cancer Res. 2015;75(7):1322. .
- Yiren H, Yingcong Y, Sunwu Y, et al. Long noncoding RNA MALAT1 regulates autophagy associated chemoresistance via miR-23b-3p sequestration in gastric cancer. Mol Cancer. 2017;16(1):174. .
- Xue D, Lu H, Xu HY, et al. Long noncoding RNA MALAT 1 enhances the docetaxel resistance of prostate cancer cells via miR‐145‐5p‐mediated regulation of AKAP 12. J Cell Mol Med. 2018;22(6):3223–3237.
- Fang Z, Zhao J, Xie W, et al. LncRNA UCA1 promotes proliferation and cisplatin resistance of oral squamous cell carcinoma by suppressing miR-184 expression. Cancer Med. 2017;6(12):2897–2908.
- Cao L, Chen J, Ou B, et al. GAS5 knockdown reduces the chemo-sensitivity of non-small cell lung cancer (NSCLC) cell to cisplatin (DDP) through regulating miR-21/PTEN axis. Biomed Pharmacother. 2017;93:570–579.
- Li S, Mei Z, Hu HB, et al. The lncRNA MALAT1 contributes to non‐small cell lung cancer development via modulating miR‐124/STAT3 axis. J Cell Physiol. 2018;233(9):6679–6688.
- Chen S, Ma P, Zhao Y, et al. Biological function and mechanism of MALAT-1 in renal cell carcinoma proliferation and apoptosis: role of the MALAT-1–Livin protein interaction. J Physiol Sci. 2017;67(5):577–585. .
- Zhang HM, Yang FQ, Chen SJ, et al. Upregulation of long non-coding RNA MALAT1 correlates with tumor progression and poor prognosis in clear cell renal cell carcinoma. Tumor Biol. 2015;36(4):2947–2955.
- Kulkarni P, Dasgupta P, Majid S, et al. miR-182-5p suppresses progression of renal cancer through cell cycle arrest by targeting lncRNA MALAT-1. Cancer Res. 2018. DOI:https://doi.org/10.1158/1538-7445.
- Li P, Zhang X, Wang H, et al. MALAT1 is associated with poor response to oxaliplatin-based chemotherapy in colorectal cancer patients and promotes chemoresistance through EZH2. Mol Cancer Ther. 2017;16(4):739–751. .
- Xie S, Qin C, Jin L, et al. Long noncoding RNA SNHG14 promotes breast cancer cell proliferation and invasion via sponging miR-193a-3p. Eur Rev Med Pharmacol Sci. 2019;23(6):2461–2468. .
- Ji N, Wang Y, Bao G, et al. LncRNA SNHG14 promotes the progression of cervical cancer by regulating miR-206/YWHAZ. Pathol Res Pract. 2019;215(4):668–675.
- Wang N, Feng Y, Xu J, et al. miR-362-3p regulates cell proliferation, migration and invasion of trophoblastic cells under hypoxia through targeting Pax3. Biomed Pharmacothe. 2018;99:462.
- Li M, Liu Q, Lei J, et al. MiR-362-3p inhibits the proliferation and migration of vascular smooth muscle cells in atherosclerosis by targeting ADAMTS1. Biochem Biophys Res Commun. 2017;493(1):270–276.
- Zou X, Zhong J, Li J, et al. miR-362-3p targets nemo-like kinase and functions as a tumor suppressor in renal cancer cells. Mol Med Rep. 2016;13(1):994–1002. .
- Liu Z-S, Cai H, Xue W, et al. G3BP1 promotes DNA binding and activation of cGAS. Nat Immunol. 2019;20(1):18. .
- Dou N, Chen J, Yu S, et al. G3BP1 contributes to tumor metastasis via upregulation of Slug expression in hepatocellular carcinoma. Am J Cancer Res. 2016;6(5):2641.
- Wang Y, Fu D, Chen Y, et al. G3BP1 promotes tumor progression and metastasis through IL-6/G3BP1/STAT3 signaling axis in renal cell carcinomas. Cell Death Dis. 2018;9(5):501. .
- Somasekharan SP, Elnaggar A, Leprivier G, et al. YB-1 regulates stress granule formation and tumor progression by translationally activating G3BP1. J Cell Biol. 2015;208(7):913–929. .
- Min L, Shen YR, Jia Z, et al. Overexpression of G3BP1 correlates with poor prognosis in gastric cancer patients. Histopathology. 2015;67(5):677–688. .