
ABSTRACT
Lysosomes are acidic, dynamic organelles that supervise catabolism, integrate signaling cascades, and tune cellular trafficking. Moreover, the loss of their integrity may jeopardize cell viability. In cancer cells, lysosomes are qualitatively and quantitatively modified for the tumor’s own benefit. For all these reasons, these organelles emerge as appealing intracellular targets to manipulate non-oncogene addiction. This is of particular interest for brain diseases, including neurodegenerative disorders and cancer, in which stem cells are exhausted and transformed, respectively. Recent publications had demonstrated that stem cells displayed disarmed lysosomes in terms of number and functions during aging and oncogenic progression. Likewise, our laboratory identified that the arginine protease MALT1, normally dedicated to the assembly of proper NF-kB activation and processing a number of substrates, arbitrates lysosome biogenesis and mTOR signaling in glioblastoma stem-like cells. Indeed, blocking either the expression or the activity of this enzyme leads to an aberrant increase of lysosomes, alongside of the down-regulation of the mTOR signaling. This surge of lysosomes eradicates glioblastoma stem-like cells. Targeting lysosomes might thus inspire the design of new strategies to face this devastating human cancer. Here, we provide an overview of the functions of the lysosome as well as its role as a cell death initiator, to highlight the potential of lysosomal drugs for glioblastoma therapy.
Glioblastoma Multiforme (GBM) is a deadly brain tumor in adults, for which treatments remain unsatisfactory. Not only do therapies need to reach the brain and selectively destroy the tumor within this privileged, protected tissue, but also the anticancer arsenal has to deal with high molecular and high cellular heterogeneity and plasticity. Thus, median survival plateaus at 15 months, and 5-years survival does not exceed 5% [Citation1–Citation3]. It is now well admitted that GBM arises from a pool of transformed initiating cells, termed as glioblastoma stem-like cells (GSCs) [Citation1,Citation4–Citation6]. This sub-population of tumor cells is capable of tumor initiation, expansion and relapse. Targeting GSCs is thus of paramount potential. Boosted by the advances in targeting intracellular homeostasis, such as metabolism and proteostasis, organelles and their functions had received increasing attention in the field of cancer research. In this perspective, we presented recent findings highlighting how lysosomes are paralyzed in glioblastoma stem-like cells [Citation7–Citation9].
1. Lysosomes: an overview
Lysosomes are acidic organelles, with an intraluminal pH ranging between 4.5 and 5.5, that were discovered by Christian de Duve’s group in 1955 [Citation10]. These intracellular entities pilot degradation and metabolic signaling. Lysosomes are composed of two main classes of proteins: hydrolases and membrane proteins. Hydrolases reside in the acidic lysosomal lumen, where each enzyme acts on a particular subset of cargos in order to fulfill their varied functions, including catabolism, antigen and pro-protein processing, extracellular matrix degradation, or apoptosis initiation [Citation11]. Lysosomal membrane proteins, by contrast, are heavily glycosylated and occupy the limiting membrane of the lysosome; their roles echelon to acidification, membrane fusion, and protein import and export [Citation12–Citation14].
1.1. Biogenesis and assembly
Seminal work from Andrea Ballabio’s group identified that many lysosomal genes contain in their promoter a consensus sequence, GTCACGTGAC, termed the CLEAR element (coordinated lysosomal expression and regulation). The basic helix-loop-helix leucine zipper transcription factors from the MITF (melanocyte inducing transcription factor) family, among which is TFEB (the transcription factor EB), bind to this site and regulate lysosomal biogenesis. In fact, TFEB overexpression promotes lysosomal gene transcription in HeLa cells [Citation15]. Subsequently, the same group uncovered that the CLEAR element is also located in the promoter of many autophagy-related genes, underlining an intermingled regulation between lysosomal biogenesis and autophagosome formation [Citation16]. TFEB is regulated via inactivating phosphorylation at multiple sites, which promotes its cytosolic retention. Upon dismantlement of lysosome functions or cellular starvation, TFEB is dephosphorylated and subsequently shuttled to the nucleus, promoting the transcription of its target genes [Citation17]. Both ERK2 and the mechanistic target of rapamycin complex (mTORC1) have been implicated in TFEB phosphorylation under nutrient-rich conditions [Citation18,Citation19]. As mTORC1 counteracts autophagy induction, this emphasizes the interplay between lysosomal biogenesis, autophagy, and mTORC1 signaling. Furthermore, recent work by Kevin Ryan’s laboratory demonstrated that bromodomain-containing protein 4 (BRD4) functions as a transcriptional repressor of the CLEAR network, independent of TFEB. Indeed, the knockdown or inhibition of BRD4 induced CLEAR network gene transcription even upon co-silencing of TFEB [Citation20]. Therefore, there may be alternate, yet unknown, mechanisms regulating lysosomal gene transcription.
Newly synthesized hydrolases traffic to the lysosome, from the trans-golgi network (TGN), via the mannose-6-phosphate receptor (M6PR) transport system. Clathrin-coated vesicles, containing M6PR and hydrolases, leave the TGN and travel to early endosomes. Upon endosome maturation, pH changes cause hydrolases to dissociate from M6PR, which can in turn recycle back to the TGN. Of note, M6PR also localizes at the cell surface to shuttle extracellular content to the lysosome via the endocytosis pathway [Citation21–Citation24]. Hence, lysosomes can acquire integral components via two routes, i.e. de novo synthesis and endocytic uptake.
1.2. Fusion of lysosomes to cellular membranes
Functionally, lysosomes act as a delivery receptacle for multiple degradative processes, including autophagy and endocytosis. Moreover, lysosomes can fuse with the plasma membrane and undergo exocytosis to expel certain contents. These varied functions converge on the fusion of lysosomes with different cellular membranes including autophagosomes, endosomes, and the plasma membrane ().
Figure 1. Overview of lysosomal functions.
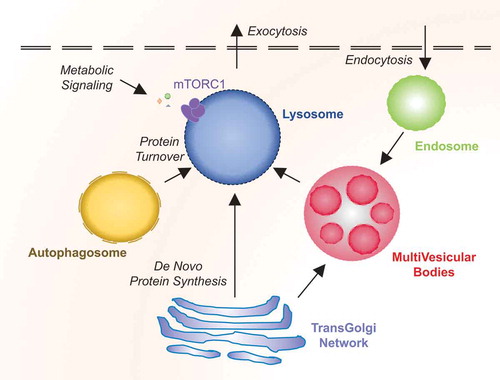
In addition to degrading internalized cargos, a key role of the lysosome in endocytosis resides in ligand/receptor recycling. Upon endocytosis, the acidic conditions drive low-density lipoprotein ligands to dissociate from receptors and to be consequently chopped by hydrolases. Transmembrane receptors can then travel back to the cell surface for further signaling [Citation25]. In this way, the lysosome can alter the duration of signaling cascades in the cell. Additionally, endocytosis serves to remodel the plasma membrane by removing transporters and adhesion molecules [Citation26].
Autophagy is a cellular process for bulk degradation in reaction to certain stimuli including stress, starvation, and hypoxia. Upon initiation, autophagy-related proteins (ATG) converge at punctate structures, termed as the phagophore assembly site (PAS). The phagophore isolates designated cargos or organelles, sealing off into double-membrane vesicles (autophagosomes). Autophagosomes then fuse with the lysosome to degrade their freight [Citation27]. Autophagy is tightly regulated by a variety of cues including at the transcriptional level (e.g. TFEB). Activated TFEB promotes the transcription of genes involved in the initiation of autophagy [Citation28]. Additionally, mTOR hinders autophagy via inhibitory phosphorylation of multiple pathway components, such as the Unc-51 like autophagy activating kinase ULK1 and ATG13 [Citation29–Citation32].
Finally, lysosomal exocytosis is a calcium-dependent process exploited by cells for plasma membrane repair, as well as pathogen removal [Citation33–Citation35]. In cancer cells, increasing evidence suggests that this pathway is also co-opted to digest the extracellular matrix of surrounding cells during invasion [Citation36].
Thus, lysosomes sit at the crossroad of multiple organizational and functional intracellular commands.
1.3. mTORC1 metabolic signaling
Lysosomes play a key role in metabolic signaling, since they act as docking sites for active mTORC1. Upon growth factor or amino acid stimulation, cytosolic mTORC1 migrates to the surface of the lysosome, where its kinase activity operates to stimulate mRNA translation and metabolism (via nucleotide, lipid, and glucose synthesis), while inhibiting protein turnover (via inhibition of autophagy and lysosomal biogenesis) [Citation37]. Besides controlling cell size, mTOR signaling orchestrates metabolism, proliferation and survival, as such, this pathway can be pirated to the cancer cell’s benefits. In fact, approximately 30% of human tumors experience hyperactivation of the cascade [Citation37]. Therefore, targeting mTOR signaling may be important for effective anti-cancer therapy.
2. Lysosome-induced cell death
The idea of lysosomes as initiators of cell death was suggested early on following their discovery. However, this capacity was not fully explored until recently, likely owing to the fact that their ultrastructure appeared unaltered during cellular demise [Citation38,Citation39]. Damage to lysosomes comprises alteration in hydrolase expression, as well as changes in their size, number, pH and cellular positioning [Citation40]. They also show deficiencies in autophagic flux.
Lysosomes work at acidic pH. Inhibition of the proton pump vacuolar-ATPase, e.g. with bafilomycin A1 increases intraluminal pH and thus reduces their catabolic capacity. This results in lysosomal dysfunction and defect in fusion with the autophagosome [Citation41,Citation42]. Similarly, the autophagic inhibitor chloroquine also suppresses the lysosomal mission. Rather than blocking acidification, this weak base accumulates in lysosomes, increasing de facto the pH. This also limits autophagosome-lysosome fusion and thus, clearance of cargos [Citation43] (). Inhibitors of mTOR can too alter lysosomal function in a cell. As already mentioned, there is a reciprocal interplay between mTOR inhibition and autophagy induction. Therefore, mTOR inhibitors can increase autophagic degradation, and thus switch on the lysosomal activity [Citation44].
Figure 2. Therapeutic targeting of the lysosome.
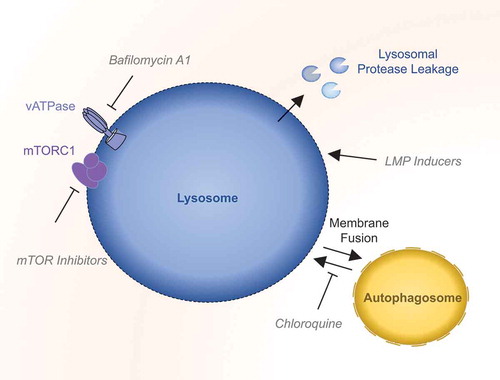
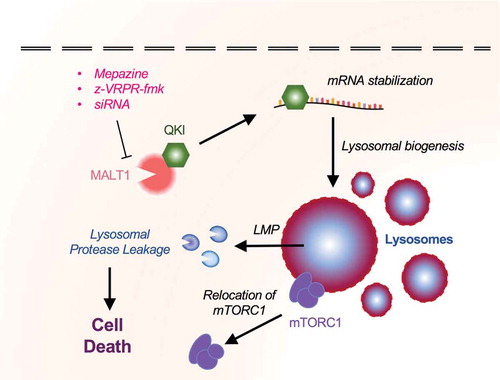
Figure 3. MALT1 interacts with QKI and regulates lysosomal homeostasis in Glioblastoma Stem-like Cells.
Furthermore, lysosomal membrane integrity is crucial to circumscribe degradative enzymes and to maintain confined acidic pH. Conversely, lysosomal membrane permeabilization (LMP) results in an exodus of the lysosome cargos toward the cytosol. This LMP term encompasses varying degrees of membrane alterations, from selective cathepsin release, instigating death via tunable signaling cascades, to total lysosomal lysis, which lowers cellular pH and prompts necrosis [Citation40,Citation45,Citation46]. LMP can be induced by a variety of methods. For instance, lysosomotropic agents are weak bases, which passively cross the lysosomal membrane and accumulate into the lumen. Amines with hydrophobic side-chains such as morphomine, cisprofloxacin, sphingosine, imidazole, and siramesine are all classified as lysosomotropic detergents [Citation39,Citation47–Citation49]. Furthermore, microtubule poisons, such as vincristine and paclitaxel also modify the stability of lysosomes [Citation50,Citation51].
One major method of altering the stability of lysosomal membranes, and thus inciting LMP, is arbitrated by the modification of sphingolipids. Acid sphingomyelinase (ASM) binds to both HSP70 and its docking lipid BMP (bis monoacylglycerophosphate), to foster membrane stability [Citation52,Citation53]. Drugs that target ASM displace it from BMP to promote its degradation, resulting in sphingomyelin accumulation. These drugs, ranging from antihistamines to antidepressants, are collectively known as cationic amphiphilic drugs (CAD), as they have hydrophilic amine groups and hydrophobic ring structures [Citation54,Citation55]. Marja Jäättelä’s group has done extensive research on CADs, including siramesine. They demonstrated that siramesine is a lysosomal detergent, which disrupts autophagy [Citation49]. Moreover, this compound shows selectivity toward cancer cells, and can help resensitizing them to chemotherapy [Citation51,Citation55]. Additionally, inhibitors of HSP70 also halt ASM stability and trigger LMP [Citation53,Citation56,Citation57]. Hence, drugs that revise lysosomal sphingolipids have an exciting anti-cancer potential.
When LMP is induced, cells engage several damage response mechanisms to avoid death. For instance, inactivation of mTORC1 prompts TFEB-dependent lysosomal biogenesis [Citation58,Citation59]. In this vein, TFEB nuclearization enhances with the addition of lysosomotropic agents [Citation60]. Moreover, minor perturbation in lysosomal membrane integrity stimulates the recruitment of the ESCRT (endosomal sorting complexes required for transport) machinery to heal membrane flaws. Indeed, knockdown of ESCRT components increased lysosomal damage-induced cell death [Citation61]. Concurrently, Phyllis Hanson’s laboratory demonstrated that ESCRT knockdown reduced lysosomal repair, using the cathepsin probe Magic Red [Citation62]. In contrast, numerous studies have demonstrated that more severe LMP leads to the recruitment of galectins 3, 8, and 9, as sensors that bind β-galactosides on the injured organelles to recruit the autophagic machinery for selective clearance (termed as lysophagy) [Citation63–Citation67]. Additionally, lysosomes can undergo exocytosis in response to anti-cancer agents [Citation68]. For instance, recent work demonstrated a calcium-dependent fusion of lysosomes with the plasma membrane following ionizing radiation [Citation69].
Lysosomal disorder efficiently induces cell death, providing an opportunity to exploit this process for novel anti-cancer therapies.
3. Targeting lysosomes in glioblastoma stem-like cells
Lysosomes not only engage in recycling and signaling but also supervise stem cell fate alongside development and aging. Recent work by Villegas et al. identified TFE3, a MITF family transcription factor, known to promote the transcription of lysosomal genes, as a major regulator of Embryonic Stem Cell (ESC) differentiation. Genome-wide CRISPR/Cas9 screen, performed in mouse ESCs, indeed illustrated that the self-renewal state is controlled by the activity of TFE3, thus linking stem cell fate to the lysosomes [Citation70]. Anne Brunet’s group provided additional evidence for a role of lysosomes in the maintenance of stem cell properties in brain. It is noteworthy that quiescent Neural Stem Cells (qNSCs) have more numerous and larger lysosomes than their activated counterparts (aNSCs). qNSCs are further involved in maintaining brain function and repair after injury [Citation71]. However, the lysosomal compartment is exhausted in older NSCs (19–22 months), as compared to young NSCs (3–4 months), together with the accumulation of protein aggregates [Citation71]. The weakening of differentiation potential in older qNSCs accompanied the decline of the lysosomal compartment, suggesting that the impact of aging on lysosomes alters stemness [Citation71]. Therefore, lysosomal homeostasis may be of great importance in the process of aging, culminating with neurodegenerative pathologies, such as Alzheimer's and Parkinson's diseases. This might further be the case in GBM development, as this cancer occurs more frequently in older patients [Citation72].
As briefly mentioned, GBM contains a subset of cells with stem properties, namely Glioblastoma Stem-like Cells (GSCs), which may arise from mutations in NSCs [Citation6,Citation73,Citation74], and consequently may show similar lysosomal vulnerability. As these cells may govern initiation, resistance to treatments, and relapse, how the lysosomal compartment contributes to their fate is of high interest. Pivotal findings by Shingu et al. identified QKI, an RNA binding protein highly expressed in the brain, as a regulator of lysosomes in NSCs and GSCs [Citation9]. Gene deletion of Qki in transformed NSCs leads to the maintenance of stemness properties, even outside the favorable environment of the NSC niche, while recapitulating GBM development in mice brains. QKI can bind specifically to and stabilize lysosomal RNAs in these cells [Citation9]. Correspondingly, QKI silencing restricts the endo-lysosomal compartment, resulting in reduced receptor recycling. This prolongs, in turn, self-renewal signaling emanating from receptors, and therefore culminates in maintaining their survival even in harsh environments [Citation9]. Conversely, one can envision that reversing lysosomal shutdown might be deleterious for GSCs. In keeping with this idea, several groups tried to take advantage of this putative frailty in GSCs to improve therapeutic targets in GBM ().
Table 1. Compounds reported to induce lysosome membrane permeabilization in glioblastoma.
From this view, lysosomotropic drugs penetrate cells, accumulate in the lysosomal lumen, and therefore may be interesting compounds to disrupt lysosomes in GBM [Citation68,Citation75]. For instance, betulinic acid derivative B10 kills glioblastoma cell lines in vitro, as well as patient-derived cell cultures, when combined with the PI3 K inhibitor GDC-0941 [Citation76]. From a molecular standpoint, PI3K inhibition activates TFEB-dependent accumulation of lysosomes, destabilized in turn by B10. This was hindered by Ca-074Me, a known cathepsin B inhibitor, arguing in favor of a lysosomal-mediated death [Citation76]. Lys05, another lysosomotropic molecule, also showed promising results for the treatment of GBM [Citation77]. This drug was reported to impair lysosomal function in glioma cell lines, as assessed by Lysotracker and acridine orange staining, two acid-activated fluorophores. Lys05 accumulation in lysosomes executes LMP, as evaluated by Galectin3 puncta formation [Citation77]. Viability was further rescued upon Ca-074Me administration. Lys05 radio-sensitizes glioma cell lines in vitro, suggesting the potential of combining lysosomal destabilization with standard-of-care therapies. Similar experiments were conducted with the previously described CAD, siramesine, in glioma cell lines and patient-derived cells in vitro [Citation78]. However, the administration of siramesine failed to curb proliferation and survival of GBM cells in organotypic spheroid-brain slice culture and in xenograft mouse models [Citation78]. Thus, lysosomotropic drugs showed promising results mainly in in vitro studies, poisoning a large proportion of cancerous cells without any major effects on normal neural cells, but may require further evaluation to determine their in vivo efficacy.
In addition to lysosome destabilization, targeting lipid homeostasis emerged as a potential strategy to eradicate tumor cells in GBM. Indeed, lysosomal integrity relies on the lipid membrane composition and organization, while these organelles play several roles in lipid catabolism and transfer [Citation79,Citation80]. Thus, any rupture in this fine-tuned process might prove to efficiently induce LMP and subsequent lysosomal cell death in GBM. An original treatment, developed by the group of Balveen Kaur, uses nanovesicles formed by the coupling of saposin C, an activator of sphingosine production, to dioleoylphosphatidylserine (DOPS). These engineered nanovesicles foster ceramide followed by sphingosine production, culminating in LMP-dependent cell death of GBM cells in vitro and in vivo [Citation81]. Furthermore, glioma cell lines were eliminated upon treatment with a combination of tumor necrosis factor alpha TNFα, lipopolysaccharide LPS, and interferon gamma IFNγ (TLI). TLI provokes ceramide accumulation in the lysosomes, causing the destabilization of the lysosomal membrane and subsequent LMP. The inhibition of the sphingosine kinase (SK), an enzyme required for lipid recycling outside of lysosomes was also proposed to induce LMP specifically in glioma cells [Citation82]. More recently, Le Joncour et al. discovered MDGI (Mammary-Derived Growth Inhibitor) as an important regulator of lysosomal membrane composition. In fact, its down-regulation impairs fatty acid transport in patient-derived GBM cells, abetting dramatic changes in lysosomal membrane composition. MDGI deficiency prompts LMP and subsequent death of GBM cells. In this vein, the CAD compound, clemastine, engendered lysosomal cell death in vitro and reduced tumor growth in vivo, in agreement with the susceptibility of GBM cells to lysosomal dysfuntions [Citation8].
While investigating intrinsic mechanisms for autonomous survival of GSCs, our group unmasked another previously unknown mechanism by which GSCs regulate lysosomal biogenesis, and described a family of drugs directing lysosomal-dependent cell death in patient-derived cells in vitro, as well as to tumor growth reduction in xenograft mouse models [Citation7]. The expression of the paracaspase MALT1, an arginine protease, involved in antigen-signaling in immune cells, NF-kB signaling, and development of certain forms of aggressive lymphoma [Citation83–Citation87] was negatively correlated with the probability of survival in GBM patients. MALT1 was found basally active in GSCs [Citation7]. Pharmacological inhibition of MALT1 using phenothiazines such as mepazine (MPZ) [Citation85,Citation88], previously used in clinics in order to treat psychic disorders, specifically exterminates patient-derived cells, while sparing neural resident cell types [Citation7]. This effect was recapitulated with the competitive peptide MALT1 antagonist z-VRPR-fmk, MALT1 silencing with RNA interference, and the expression of a protease-dead version of MALT1. Altogether, several means deployed to halt MALT1 in GSCs converge on increased cell death, reduction of proliferation, and loss of stemness markers [Citation7]. Further, transmission electron microscopy (TEM) unveiled an accumulation of vacuoles and lysosomes in MALT1-inhibited cells. The intensification of the Lysotracker and LAMP2 staining confirmed an increased abundance of lysosomes. While MALT1 inhibition provokes cathepsin release, blocking these lysosomal enzymes rescued GSCs, at least partially, from MPZ-dependent cell death. Given the previously described role of QKI in regulating lysosome biogenesis [Citation9], the functional interaction between QKI and MALT1 was explored in-depth. In fact, QKI binds to MALT1 in proliferating GSCs and was released upon MALT1 inhibition [Citation7]. Thus, the inhibition of MALT1 frees QKI, which is then permitted to dictate the translation of lysosomal genes, independently of TFEB. Conversely, the knockdown of QKI opposes MPZ-induced cell death in GSCs, underlying the major roles of MALT1 and QKI in the regulation of lysosomal homeostasis in GSCs.
In addition to the roles of MALT1 and QKI, our recent work also linked mTOR to the maintenance of a low lysosome load in GSCs [Citation7]. In MALT1-inhibited cells, the constitutive mTOR activity was dampened and the kinase was dispersed from lysosome foci [Citation7,Citation89]. This might also contribute to diminished GSC expansion. Because mTOR activity is intimately linked to lysosomal stability [Citation90,Citation91], inhibiting mTOR, by destabilizing lysosomes, might prove beneficial to downsize cancer cell reservoir. Our work thus identified an unexpected mechanism by which GSC control lysosomal homeostasis, and defined phenothiazines as potential therapeutic options against GBM, inducing LMP-dependent cell death.
GBM is a deadly cancer, characterized by massive infiltration and heterogeneity [Citation92,Citation93]. Finding new weapons to fight this disease is a real challenge, because of the delicate environment and the blood-brain barrier acting as a shield to many therapeutic molecules. Numerous recent studies point toward lysosomes as an Achille’s heel for GBM, and many tested molecules show impressive results in vitro, despite poor outcome for in vivo studies. Translation to clinics will require an efficient molecule to target GSCs, while safely penetrating the brain.
Acknowledgments
We thank SOAP team members (Nantes, France).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Lathia JD, Mack SC, Mulkearns-Hubert EE, et al. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–1217.
- Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996.
- Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466.
- Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760.
- Chen J, Li Y, Yu T-S, et al. a restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526.
- Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401.
- Jacobs KA, André‐Grégoire G, Maghe C, et al. Paracaspase MALT1 regulates glioma cell survival by controlling endo‐lysosome homeostasis. Embo J. 2020;39:e102030.
- Le Joncour V, Filppu P, Hyvönen M, et al. Vulnerability of invasive glioblastoma cells to lysosomal membrane destabilization. EMBO Mol Med. 2019;11:e9034.
- Shingu T, Ho AL, Yuan L, et al. Qki deficiency maintains stemness of glioma stem cells in suboptimal environment by downregulating endolysosomal degradation. Nat Genet. 2017;49:75–86.
- de Duve C, Pressman BC, Gianetto R, et al. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J. 1955;60:604–617.
- Conus S, Simon H-U. Cathepsins: key modulators of cell death and inflammatory responses. Biochem Pharmacol. 2008;76:1374–1382.
- Eskelinen E-L, Tanaka Y, Saftig P. At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 2003;13:137–145.
- Kornfeld S, Mellman I. The biogenesis of lysosomes. Annu Rev Cell Biol. 1989;5:483–525.
- Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009;10:623–635.
- Sardiello M, Palmieri M, Di Ronza a, et al. a gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477.
- Settembre C, Ballabio a. TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy. 2011;7:1379–1381.
- Napolitano G, Ballabio a. TFEB at a glance. J Cell Sci. 2016;129:2475–2481.
- Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914.
- Roczniak-Ferguson a, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42–ra42.
- Sakamaki J, Wilkinson S, Hahn M, et al. Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol Cell. 2017;66:517–532.e9.
- Dahms NM. Insulin-like growth factor II/cation-independent mannose 6-phosphate receptor and lysosomal enzyme recognition. Biochem Soc Trans. 1996;24:136–141.
- Gary-Bobo M, Nirdé P, Jeanjean a, et al. Mannose 6-phosphate receptor targeting and its applications in human diseases. Curr Med Chem. 2007;14:2945–2953.
- Kornfeld S. Structure and function of the mannose 6-phosphate/insulinlike growth factor II receptors. Annu Rev Biochem. 1992;61:307–330.
- Munier-Lehmann H, Mauxion F, Hoflack B. Function of the two mannose 6-phosphate receptors in lysosomal enzyme transport. Biochem Soc Trans. 1996;24:133–136.
- Kirchhausen T, Owen D, Harrison SC. Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb Perspect Biol. 2014;6:a016725.
- Ross E, Ata R, Thavarajah T, et al. AMP-activated protein kinase regulates the cell surface proteome and integrin membrane traffic. Plos One. 2015;10:e0128013.
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–364.
- Settembre C, Fraldi a, Medina DL, et al. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14:283–296.
- Ganley IG, Lam DH, Wang J, et al. ULK1·ATG13·FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–12305.
- Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–1991.
- Jung CH, Jun CB, Ro S-H, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003.
- Puente C, Hendrickson RC, Jiang X. Nutrient-regulated phosphorylation of ATG13 inhibits starvation-induced autophagy. J Biol Chem. 2016;291:6026–6035.
- Huynh C, Roth D, Ward DM, et al. Defective lysosomal exocytosis and plasma membrane repair in Chediak-Higashi/beige cells. Proc Natl Acad Sci U S A. 2004;101:16795–16800.
- Jaiswal JK, Andrews NW, Simon SM. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J Cell Biol. 2002;159:625–635.
- Reddy a, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. 2001;106:157–169.
- Machado E, White-Gilbertson S, Vlekkert D, et al. Regulated lysosomal exocytosis mediates cancer progression. Sci Adv. 2015;1:e1500603.
- Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976.
- Brunk UT, Ericsson JL. Cytochemical evidence for the leakage of acid phosphatase through ultrastructurally intact lysosomal membranes. Histochem J. 1972;4:479–491.
- Firestone RA, Pisano JM, Bonney RJ. Lysosomotropic agents. 1. Synthesis and cytotoxic action of lysosomotropic detergents. J Med Chem. 1979;22:1130–1133.
- Aits S, Jäättelä M. Lysosomal cell death at a glance. J Cell Sci. 2013;126:1905–1912.
- Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. 2015;11:1437–1438.
- Nakashima S, Hiraku Y, Tada-Oikawa S, et al. Vacuolar H+-ATPase inhibitor induces apoptosis via lysosomal dysfunction in the human gastric cancer cell line MKN-1. J Biochem (Tokyo). 2003;134:359–364.
- Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435–1455.
- Zhou J, Tan S-H, Nicolas V, et al. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013;23:508–523.
- Loison F, Zhu H, Karatepe K, et al. Proteinase 3–dependent caspase-3 cleavage modulates neutrophil death and inflammation. J Clin Invest. 2014;124:4445–4458.
- Zhao K, Zhao X, Tu Y, et al. Lysosomal chymotrypsin B potentiates apoptosis via cleavage of Bid. Cell Mol Life Sci. 2010;67:2665–2678.
- Boya P, Andreau K, Poncet D, et al. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J Exp Med. 2003;197:1323–1334.
- Kågedal K, Zhao M, Svensson I, et al. Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem J. 2001;359:335–343.
- Ostenfeld MS, Høyer-Hansen M, Bastholm L, et al. Anti-cancer agent siramesine is a lysosomotropic detergent that induces cytoprotective autophagosome accumulation. Autophagy. 2008;4:487–499.
- Castino R, Peracchio C, Salini a, et al. Chemotherapy drug response in ovarian cancer cells strictly depends on a cathepsin D-Bax activation loop. J Cell Mol Med. 2009;13:1096–1109.
- Groth-Pedersen L, Ostenfeld MS, Høyer-Hansen M, et al. Vincristine induces dramatic lysosomal changes and sensitizes cancer cells to lysosome-destabilizing siramesine. Cancer Res. 2007;67:2217–2225.
- Gabandé-Rodríguez E, Boya P, Labrador V, et al. High sphingomyelin levels induce lysosomal damage and autophagy dysfunction in Niemann Pick disease type a. Cell Death Differ. 2014;21:864–875.
- Kirkegaard T, Roth AG, Petersen NHT, et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010;463:549–553.
- Gulbins E, Kolesnick RN. It takes a CAD to kill a tumor cell with a LMP. Cancer Cell. 2013;24:279–281.
- Petersen NHT, Olsen OD, Groth-Pedersen L, et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell. 2013;24:379–393.
- Granato M, Lacconi V, Peddis M, et al. HSP70 inhibition by 2-phenylethynesulfonamide induces lysosomal cathepsin D release and immunogenic cell death in primary effusion lymphoma. Cell Death Dis. 2013;4:e730.
- Nylandsted J, Wick W, Hirt UA, et al. Eradication of glioblastoma, and breast and colon carcinoma xenografts by Hsp70 depletion. Cancer Res. 2002;62:7139–7142.
- Papadopoulos C, Meyer H. Detection and clearance of damaged lysosomes by the endo-lysosomal damage response and lysophagy. Curr Biol CB. 2017;27:R1330–R1341.
- Raben N, Puertollano R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol. 2016;32:255–278.
- Lu S, Sung T, Lin N, et al. Lysosomal adaptation: how cells respond to lysosomotropic compounds. Plos One. 2017;12:e0173771.
- Radulovic M, Schink KO, Wenzel EM, et al. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. Embo J. 2018;37:e99753.
- Skowyra ML, Schlesinger PH, Naismith TV, et al. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science. 2018;360:eaar5078.
- Chauhan S, Kumar S, Jain a, et al. TRIMs and galectins globally cooperate and TRIM16 and Galectin-3 Co-direct autophagy in endomembrane damage homeostasis. Dev Cell. 2016;39:13–27.
- Jia J, Abudu YP, Claude-Taupin a, et al. Galectins control MTOR and AMPK in response to lysosomal damage to induce autophagy. Autophagy. 2019;15:169–171.
- Jia J, Bissa B, Brecht L, et al. AMPK, a regulator of metabolism and autophagy, is activated by lysosomal damage via a novel galectin-directed ubiquitin signal transduction system. Mol Cell. 2020a;77:951–969.e9.
- Jia J, Claude-Taupin a, Gu Y, et al. Galectin-3 coordinates a cellular system for lysosomal repair and removal. Dev Cell. 2020b;52:69–87.e8.
- Thurston TLM, Wandel MP, von Muhlinen N, et al. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482:414–418.
- Zhitomirsky B, Assaraf YG. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget. 2017;8:45117–45132.
- Ferranti CS, Cheng J, Thompson C, et al. Fusion of lysosomes to plasma membrane initiates radiation-induced apoptosis. J Cell Biol. 2020;219:e201903176.
- Villegas F, Lehalle D, Mayer D, et al. Lysosomal signaling licenses embryonic stem cell differentiation via inactivation of Tfe3. Cell Stem Cell. 2019;24:257–270.e8.
- Leeman DS, Hebestreit K, Ruetz T, et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science. 2018;359:1277–1283.
- Young JS, Chmura SJ, Wainwright DA, et al. Management of glioblastoma in elderly patients. J Neurol Sci. 2017;380:250–255.
- Lee JH, Lee JE, Kahng JY, et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature. 2018;560:243–247.
- Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828.
- Kaufmann AM, Krise JP. Lysosomal sequestration of amine-containing drugs: analysis and therapeutic implications. J Pharm Sci. 2007;96:729–746.
- Enzenmüller S, Gonzalez P, Karpel-Massler G, et al. GDC-0941 enhances the lysosomal compartment via TFEB and primes glioblastoma cells to lysosomal membrane permeabilization and cell death. Cancer Lett. 2013;329:27–36.
- Zhou W, Guo Y, Zhang X, et al. Lys05 induces lysosomal membrane permeabilization and increases radiosensitivity in glioblastoma. J Cell Biochem. 2020;121:2027–2037.
- Jensen SS, Petterson SA, Halle B, et al. Effects of the lysosomal destabilizing drug siramesine on glioblastoma in vitro and in vivo. BMC Cancer. 2017;17:178.
- Jaishy B, Abel ED. Lipids, lysosomes, and autophagy. J Lipid Res. 2016;57:1619–1635.
- Lawrence RE, Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol. 2019;21:133–142.
- Wojton J, Meisen WH, Jacob NK, et al. SapC-DOPS-induced lysosomal cell death synergizes with TMZ in glioblastoma. Oncotarget. 2014;5:9703–9709.
- Mora R, Dokic I, Kees T, et al. Sphingolipid rheostat alterations related to transformation can be exploited for specific induction of lysosomal cell death in murine and human glioma: sphingosine kinase and lysosomal cell death. Glia. 2010;58:1364–1383.
- Hailfinger S, Lenz G, Ngo V, et al. Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proc Natl Acad Sci. 2009;106:19946–19951.
- Jaworski M, Thome M. The paracaspase MALT1: biological function and potential for therapeutic inhibition. Cell Mol Life Sci. 2016;73:459–473.
- Nagel D, Spranger S, Vincendeau M, et al. Pharmacologic Inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell. 2012;22:825–837.
- Thys a, Douanne T, Bidère N. Post-translational Modifications of the CARMA1-BCL10-MALT1 Complex in Lymphocytes and Activated B-Cell Like Subtype of Diffuse Large B-Cell Lymphoma. Front Oncol. 2018;8:498.
- Weil R, Israel a. Deciphering the pathway from the TCR to NF-kB. Cell Death Differ. 2006;13:826–833.
- Schlauderer F, Lammens K, Nagel D, et al. Structural analysis of phenothiazine derivatives as allosteric inhibitors of the MALT1 paracaspase. Angew Chem Int Ed. 2013;52:10384–10387.
- Galan-Moya EM, Le Guelte a, Lima-Fernandes E, et al. Secreted factors from brain endothelial cells maintain glioblastoma stem-like cell expansion through the mTOR pathway. EMBO Rep. 2011;12:470–476.
- Puertollano R. mTOR and lysosome regulation. F1000Prime Rep. 2014;6:52.
- Zhitomirsky B, Yunaev a, Kreiserman R, et al. Lysosomotropic drugs activate TFEB via lysosomal membrane fluidization and consequent inhibition of mTORC1 activity. Cell Death Dis. 2018;9:1191.
- Cuddapah VA, Robel S, Watkins S, et al. a neurocentric perspective on glioma invasion. Nat Rev Neurosci. 2014;15:455–465.
- Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401.