
ABSTRACT
Chromatin plays a pivotal role in regulating the DNA damage response and during DNA double-strand break repair. Upon the generation of DNA breaks, the chromatin structure is altered by post-translational modifications of histones and chromatin remodeling. How the chromatin structure, and the epigenetic information that it carries, is reestablished after the completion of DNA break repair remains unclear though. Also, how these processes influence recovery of the cell cycle remains poorly understood. We recently performed a reverse genetic screen for novel chromatin regulators that control checkpoint recovery after DNA damage. Here we discuss the implications of PHD finger protein 6 (PHF6) and additional candidates from the NuA4 ATPase-dependent chromatin-remodeling complex and the Cohesin complex, required for sister chromatid cohesion, in DNA repair and checkpoint recovery in more detail. In addition, the potential role of this novel function of PHF6 in cancer development and treatment is reviewed.
Introduction
Genome instability is a hallmark of cancer cells and a driving force in tumorigenesis [Citation1,Citation2] and is additionally linked to a variety of other human diseases and aging [Citation3–Citation5]. Of the various forms of DNA damage, DNA double-strand breaks (DSBs) are particularly deleterious as they can lead to a significant loss of genetic material or chromosomal translocations and therefore are a threat to genome integrity [Citation1,Citation6]. Cells fortunately possess an accurate response to DNA damage that facilitates the repair of DNA and thereby prevents genomic alterations.
The cellular DNA damage response (DDR) is directed by the activation of several protein kinases, in which ATR and ATM are master regulators. These kinases phosphorylate numerous downstream targets including the checkpoint kinases Chk1 and Chk2, resulting in a transient cell cycle arrest that allows time for DNA repair [Citation7,Citation8]. Once the damage is repaired, cell cycle progression is resumed in a regulated manner in a process known as checkpoint recovery [Citation9]. Critical during this process in G2 phase is the re-activation of Cyclin B1-Cdk1 activity, which involves Plk1 and its upstream regulators Aurora A and Bora [Citation10,Citation11]. Plk1 also controls the degradation of checkpoint mediator Claspin, responsible for Chk1 activation, in a manner dependent on the βTrCP-SCF ubiquitin ligase [Citation12–Citation15]. Likewise, the phosphatase PPM1D/Wip1 reverts the phosphorylation of factors critical in DDR activation, such as histone H2AX, p53 and Chk1, thereby contributing to recovery [Citation16–Citation18]. Additional factors inhibiting DNA damage-induced post-translational modifications and enzymes regulating the protein stability of mediators of checkpoint recovery, such as ubiquitin hydrolases, are without doubt involved in checkpoint recovery, but direct evidence for the contribution of such enzymes to this process still needs to be experimentally proven.
DNA damage response in the context of chromatin
In mammalian cells, the genomic DNA is packaged into a condensed structure, the chromatin, consisting of DNA wrapped around histone proteins. Chromatin acts as a template for many cellular processes and thereby unavoidably influences DNA-associated processes like transcription, replication and repair. Post-translational modifications of histones, through the action of histone-modifying enzymes, and the displacement of histones or entire nucleosomes by ATP-dependent chromatin-remodeling complexes and histone chaperones contribute to the highly dynamic nature of chromatin [Citation19]. During the DDR and DNA repair, chromatin functions as an intrinsic barrier and therefore plays a critical role by controlling access to DNA and serving as a docking or signaling site for proteins involved in these processes [Citation20,Citation21]. Indeed, upon the generation of DSBs, the local chromatin structure is altered at different levels [Citation22].
One of the earliest modifications after the induction of a DSB is the phosphorylation of histone H2AX at serine 139 (named γH2AX) by the central DDR kinase ATM, on either side of the lesion [Citation23]. H2AX phosphorylation triggers an extensive cascade of other post-translational modifications of both histone and non-histone proteins that recruit a multitude of DDR and repair proteins to the site of the DNA lesion. Among these are 53BP1 and BRCA1, critical for the DSB repair pathway choice. In fact, the recruitment of 53BP1 to the site of the DNA lesion depends on the methylation of histone H4 on Lys20 (H4K20), the degradation of competing H4K20me readers, deacetylation of H4K16 and ubiquitination of H2AK15 [Citation24]. Such modified histones are read and interpreted by specific domains. Chromo-, PHD or Tudor domains recognize methylated residues whereas bromodomains or BRCT domains interact with acetylated or phosphorylated histones, respectively [Citation25]. It is perhaps not surprising that a growing number of proteins harboring such domains are recognized to function in the DDR.
Although most histone modifications function to recruit proteins to chromatin, some alter the packaging of nucleosomes or affect the accessibility of the chromatin [Citation26,Citation27]. For example, acetylation of histones is associated with the relaxed state of the chromatin and might, therefore, provide access for signaling and repair of DSBs [Citation26,Citation27]. Also PARylation of nucleosomes was hypothesized to induce a more relaxed chromatin state [Citation28]. Evidence suggests that chromatin disassembly and reassembly are also important during DNA repair [Citation29]. Histone proteins flanking the DNA lesion were shown to be removed during DNA repair and it was demonstrated that chromatin expands locally in an ATP-dependent fashion [Citation30,Citation31]. In accordance, several ATP-dependent chromatin remodelers, that slide nucleosomes along DNA, evict nucleosomes from chromatin or exchange histones or histone dimers in nucleosomes, were shown to be recruited to DSBs [Citation32].
The effectiveness of the DDR and DNA repair, and the cellular sensitivity to DSBs seem to be tightly related to the structure and compaction of the chromatin. Consequently, defects in damage-induced chromatin alterations are associated with genomic instability and cancer development [Citation6,Citation33]. During the later stages of the DDR, when the break has been repaired, the chromatin needs to be fully restored in order to resume cell cycle progression, which suggests that chromatin dynamics are also required to efficiently recover from a genotoxic insult [Citation27]. However, how the chromatin structure, and the epigenetic information that it carries, is reestablished after DSB repair is complete, is poorly understood.
Checkpoint recovery screen for chromatin regulators
As mentioned above, many proteins involved in the DDR use the damaged and/or modified chromatin as a template for their actions [Citation20,Citation21]. However, we know very little about how different chromatin modifications and states influence the DDR and particularly checkpoint recovery. It remains unclear whether chromatin modifications that occur after DNA damage are eventually reverted, and how this is coordinated with checkpoint recovery.
To investigate if changes in chromatin structure and modifications control checkpoint recovery, we recently performed a reverse genetic screen in human cells [Citation34]. An siRNA-based library targeting 529 genes related to the structure, maintenance or modification of chromatin was used to screen for the efficiency of cell cycle-committed cells to spontaneously recover after treatment with a non-lethal dose of ionizing irradiation (IR). Cells were synchronized in G2 and mitotic entry was determined 24 hours later either in damaged or undamaged conditions ()). Deconvolution and re-screening in a second cell line resulted in the identification of 19 genes with a reduction in recovery and 3 genes that displayed an increased recovery after DNA damage, compared to the control cells ()). Pathway analysis of the screen hits indicated a strong functional correlation between the identified genes as the majority of the hits were associated with two protein complexes: the Cohesin complex and the NuA4 complex. Nine other genes were not directly connected to each other through known functional interactions ()). To gain a deeper understanding of the involvement of the hits in checkpoint recovery, we performed several additional screens to assess p53- and ATM/ATR-dependence of checkpoint recovery, cell viability upon IR and DNA repair through γH2AX and 53BP1 IR-induced focus (IRIF) formation.
Figure 1. (a) Experimental setup used to screen for chromatin regulators involved in the IR-induced G2 checkpoint recovery. Cells, synchronized by thymidine block and release, were irradiated in G2 phase. One hour later nocodazole was added, and 16 hours later cells were collected and analyzed for mitotic progression by pHH3. (b) Hits resulting from the G2 checkpoint recovery screen. Genes associated to the NuA4 complex are depicted in green and to the Cohesin complex in blue.
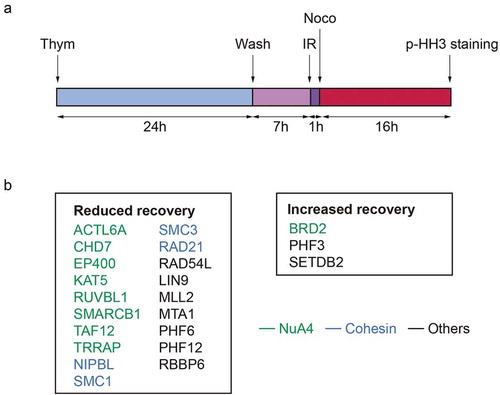
Detailed follow-up of PHF3, ACTL6A, BRD2 and PHF6 demonstrated discrepancies between different siRNA oligonucleotides regarding their knockdown efficiency and their influence on recovery, thereby making the validation of these hits challenging. Nevertheless, by generating knockout cell lines using CRISPR-mediated genome editing, PHF6 was confirmed as a bona fide mediator of checkpoint recovery [Citation34].
NuA4 protein complex
From the 22 identified hits, 9 genes (ACTL6A, KAT5, TRRAP, RUVBL1, EP400, SMARCB1, CHD7, BRD2 and TAF12) are associated with the NuA4 protein complex, a chromatin-remodeling complex that regulates nucleosome stability and thereby chromatin structure [Citation35–Citation37]. Knockdown of some other genes associated with the NuA4 complex, including MORF4L1, DMAP1, ACTB and EPC2, also hampered the ability of cells to recover. These additional genes scored close to the threshold in the primary screen, further strengthening a role for the NuA4 complex in checkpoint recovery (data not shown).
The NuA4 complex contains histone acetyltransferase (HAT) activity but also harbors additional activities, including ATPase, DNA helicase and structural DNA-binding properties [Citation36]. This complex has been linked to genome maintenance through regulation of a number of DDR-associated processes [Citation38]. For example, components of the NuA4 complex have been implicated in the activation of ATM after DNA damage [Citation39–Citation42]. We found that loss of subunits required for the HAT activity of the NuA4 complex (ACTL6A, KAT5/Tip60, TRRAP and TAF12) altered checkpoint recovery in a p53-dependent manner [Citation34]. Indeed, the NuA4 complex was shown to be involved in the acetylation and transcriptional induction of p53 [Citation43,Citation44]. We additionally showed recruitment of ACTL6A to laser-induced DNA damage [Citation34], which indicates that the HAT activity might be required at the DNA lesions, possibly for the acetylation of histones, as was suggested for H2AX [Citation42,Citation45]. Interestingly, Drosophila Tip60 (dTip60) acetylates phosphorylated H2AX from damaged nucleosomes and exchanges it with an unmodified version of the histone [Citation46]. A similar role has recently been described for NuA4 component Rvb1 in human cells [Citation47]. It remains unclear though how NuA4 exactly mediates the removal of histones after DNA damage, if it can remove all types of DNA damage-induced histone modifications and whether this activity influences checkpoint recovery. We found that loss of components involved in the ATPase activity of the NuA4 complex (TRRAP, EP400, SMARCB1 and CHD7) reduced checkpoint recovery in an ATM/ATR-dependent manner [Citation34], suggesting that NuA4-dependent chromatin remodeling, including histone eviction/replacement, is actively regulated in response to DNA damage.
Loss of NuA4 components led to a decrease in γH2AX or 53BP1 IRIF, suggesting involvement of the complex in DNA repair, possibly during the disassembly or restoration of chromatin surrounding DNA breaks. Besides, depletion of most of the identified NuA4 components reduced cell viability upon DNA damage [Citation34]. Together these results indicate that the NuA4 complex is pivotal during checkpoint recovery and thereby in preserving genome integrity.
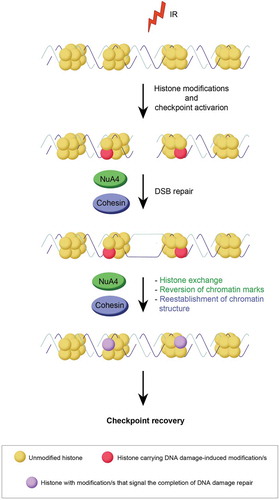
A likely possibility is that NuA4-dependent nucleosome assembly, chromatin access and nucleosome editing function in checkpoint recovery next to, or in collaboration with, the kinases Aurora A and PLK1 and the phosphatase PPM1D [Citation48]. NuA4 may facilitate the incorporation of new histone modifications, newly modified histones or histone variants at a DSB, where the nucleosomes are distorted and promote recovery in that manner [Citation49]. The NuA4 complex could facilitate restoration of the chromatin environment at the break-site, initiating the transition from the completion of DNA repair to cell cycle restart ().
Figure 2. Graphical explanation of the NuA4 and Cohesin complexes on checkpoint recovery. Potential contributions of the NuA4 complex in green, those of the Cohesin complex in blue. See text for details.
Although in recent years much was discovered about the functioning of NuA4 and its individual complex partners, it remains to be investigated what the exact consequences of its actions are in relation to checkpoint recovery, genomic stability and diseases including cancer. Inefficient NuA4 regulation could lead to inadequate epigenetic status at the DSBs, eventually leading to genome instability. Indeed, genes associated with the NuA4 complex are frequently found to be mutated in cancer [Citation50] and the NuA4 complex is investigated as a target for cancer therapy [Citation50,Citation51]. Nevertheless, to further explore this opportunity, a better understanding of the role of NuA4 in the DDR and specifically during checkpoint recovery is required.
Cohesin complex
Our recovery screen also resulted in four different components of the Cohesin complex: SMC1, SMC3, NIPBL and RAD21. Cohesin regulates the repair of DSBs through homologous recombination (HR) and is involved in checkpoint activation in S and G2 phases [Citation52,Citation53]. RAD54L, also involved in HR, was an additional hit in the screen [Citation54]. Our results indicate that Cohesin modulates recovery in a p53- and ATM/ATR-dependent manner [Citation34]. Moreover, loss of Cohesin resulted in increased IRIF of γH2AX and 53BP1, suggesting that reduced recovery could be a result of inefficient DNA repair.
Whereas the phosphorylation of SMC1 by ATM does not influence its localization upon damage [Citation55], the recruitment of the Cohesin-loader NIPBL to sites of DNA damage was shown to be dependent on ATM/ATR [Citation56], suggesting that DNA damage-dependent loading of Cohesin could be required for checkpoint recovery. In addition, Cohesin could be necessary for the reestablishment of chromatin structure after DNA repair and defects in Cohesin proteins would therefore lead to continued activation of the DNA damage checkpoint (). Future research will pinpoint the exact role of the Cohesin complex in checkpoint recovery.
PHF6 is a novel regulator of checkpoint recovery
In the screen we also identified genes without a previously described relationship to the NuA4 or Cohesin complexes, namely SETDB2, PHF3, RBBP6, PHF6, MTA1, PHF12, MLL2 and LIN9. These could be unknown interactors of the NuA4 or Cohesin complexes or mediate checkpoint recovery in a distinct manner. We focused our studies on the transcriptional regulator PHF6, which is implicated in development and tumorigenesis [Citation57]. To confirm the involvement of PHF6 in checkpoint recovery, we disrupted the gene in U2OS cells by CRISPR/Cas9-mediated genome editing. Knockout of PHF6 resulted in reduced checkpoint recovery, which was ameliorated by exogenous expression of GFP-PHF6 in these cells, thereby validating PHF6 as a recovery mediator. In addition to defective checkpoint recovery, we observed an increase in γH2AX and a reduction of 53BP1 IRIF upon loss of PHF6 [Citation34]. The increase in γH2AX foci at early and late time points after IR indicates inefficient repair of DSBs in cells lacking PHF6, while decreased 53BP1 focus formation points to an impaired cellular response to DNA damage.
The PHF6 protein does not have enzymatic activity but contains two plant homeodomain domains (PHD1 and PHD2), zinc finger-like motifs present in proteins involved in transcriptional and chromatin regulation [Citation58]. Experiments using PHF6 knockout cells complemented with PHD1 or PHD2 domain mutants indicated that both PHD domains contribute to γH2AX and 53BP1 IRIF formation and efficient checkpoint recovery, suggesting that PHF6 operates through its two PHD domains, which may act independently or in consort, on damaged chromatin [Citation34]. PHF6 has been shown to interact with histones H3, H1.2, H2B.1, H3.1, H2A.Z [Citation59,Citation60] and the PHD2 domain was reported to directly bind to double-strand DNA in vitro in a sequence-specific manner [Citation61]. The PHD1 domain was reported to interact with transcription factor UBF [Citation62,Citation63]. UBF subsequently recruits PHF6 to the nucleolus where it binds ribosomal DNA (rDNA) promoter regions to suppress transcription. In addition, PHF6-deficiency was shown to lead to the formation of DNA-RNA hybrids (R-loops) and the accumulation of R-loop-dependent rDNA damage [Citation62]. Moreover, PHF6 was reported to suppress the expression of cell cycle regulator p21, which correlated with therapy resistance in T-cell acute lymphoblastic leukemia (T-ALL) [Citation64]. PHF6 was also found to promote transcriptional elongation of genes by association with polymerase associated factor PAF1 [Citation65]. The region in between the two PHD domains is necessary for the interaction of PHF6 with retinoblastoma binding protein 4 (RBBP4), a member of the nucleosome remodeling deacetylase (NuRD) complex which supports transcription regulation, cell cycle progression and genome stability [Citation59,Citation66]. Lastly, PHF6 contains two putative ATM/ATR phosphorylation sites, suggesting that this protein might be regulated in response to DNA damage [Citation8,Citation57]. All these results together indicate that PHF6, through its PHD domains, can modulate chromatin and thereby regulate transcription, DNA repair and checkpoint recovery.
PHF6 mediates checkpoint recovery through classical NHEJ
We observed that a GFP-tagged version of PHF6 was rapidly and transiently recruited to laser-induced DNA breaks in living cells, suggesting that PHF6 might mediate its function in checkpoint recovery locally. Importantly, localization of PHF6 on laser-induced damage was dependent on PARP1/2 [Citation34]. PARP1 has recently been linked to DSB repair by the non-homologous end joining (NHEJ) pathway through CHD2, which causes the expansion of chromatin surrounding DSBs [Citation67]. By employing specific reporter assays for DSB repair by either NHEJ or HR we found that loss of PHF6 inhibits DNA repair through end-joining, whereas the effect of depletion of PHF6 on HR was ambiguous due to discrepancies between different siRNA oligonucleotides [Citation34]. Future research will need to show if PHF6 exclusively regulates NHEJ or acts as a more general DSB repair-promoting factor. Detailed analysis of DSB repair in PHF6-depleted cells through sequencing showed an increase in deletions, insertions and microhomology usage at the site of damage, thereby indicating that PHF6 is required for classical NHEJ. Knockout of PHF6 additionally resulted in a moderate sensitivity of U2OS cells to IR, similar to depletion of core-NHEJ factor XRCC4, which is consistent with an NHEJ repair defect upon PHF6 loss. Altogether, our data identified PHF6 as a novel factor in the regulation of DSB repair through classical NHEJ [Citation34]. Inefficient DNA repair by loss of PHF6 results in the accumulation of DNA lesions and therefore a reduced recovery from the G2 checkpoint after IR. However, what remains to be studied is the mechanism of how PHF6 exactly regulates NHEJ-mediated DSB repair.
Does PHF6 influence recovery through the NuRD complex?
Peptides of the nucleosome remodelers CHD1 and CHD2 were found in a PHF6 interaction study, although an in vivo interaction was not confirmed [Citation62]. Interestingly, depletion of CHD2, like PHF6, reduced the efficiency of checkpoint recovery, albeit not enough to reach the threshold in our primary screen (data not shown). PHF6 and CDH2 could be recruited by PARP1 to cooperate in opening the chromatin structure around the break in order to facilitate efficient NHEJ repair. Alternatively, PHF6 may operate through its characterized interaction with the NuRD complex [Citation59]. NuRD has both ATP-dependent chromatin remodeling and histone deacetylation activity and was shown to be essential for the DDR. NuRD subunits MTA2 and CHD4 were shown to promote ubiquitination of histones surrounding a DNA break, leading to BRCA1 focus formation, required for DSB repair, and checkpoint activation [Citation68,Citation69]. Moreover, recruitment of NuRD to DSBs by the bromodomain protein ZMYND8 represses transcription and stimulates homology-directed DNA repair [Citation70–Citation72]. Finally, deacetylation of histones H3K56 and H4K16 through its members HDAC1 and HDAC2 is critical for DSB repair through NHEJ [Citation73]. Importantly, also some other genes associated with the NuRD complex influenced checkpoint recovery in our screen. One of the positive hits, MTA1, showed almost comparable results in the secondary screens to those observed after loss of PHF6 [Citation34]. In addition, knockdown of NuRD protein RBBP4 hampered the ability of cells to recover, scoring close to the threshold in the primary screen, and depletion of CHD4, HDAC1, HDAC2 and RBBP7, also members of the NuRD complex, modestly reduced recovery (data not shown). Taking these data into account makes us hypothesize that PHF6 could facilitate the localization of NuRD to damaged chromatin, leading to enhanced HDAC1 and HDAC2 activity and promotion of DSB repair through NHEJ.
Alternatively, as PHF6 was shown to bind histones in vitro [Citation61] and our preliminary results indicate an increased association of PHF6 to histones at later time points after DNA damage (data not shown), PHF6 could collaborate with chromatin remodelers like NuRD or CHD2 and mediate checkpoint recovery by facilitating the exchange, incorporation or eviction of (modified) histones around DSBs after the completion of repair ().
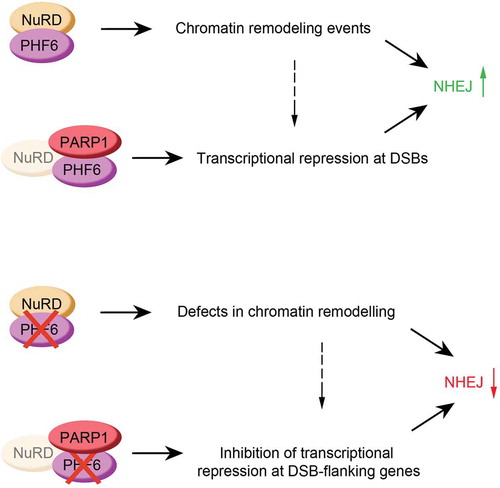
Figure 3. Graphical explanation of how PHF6 could control NHEJ (top panel), and the effect of PHF6 knock out on this process (bottom panel). See text for details.
Is the role of PHF6 in transcription linked to its DNA repair function?
Given the described role of PHF6 in transcriptional suppression, we studied the expression of a broad range of proteins involved in NHEJ after PHF6 loss but did not observe any changes [Citation34]. PHF6 could still be involved in the general repression of transcription around sites of damage though. In response to DSBs, cells trigger the rapid silencing of the genes surrounding the lesion in an ATM-, DNA-PK- and PARP1-dependent manner [Citation74,Citation75]. As already mentioned, PHF6 was shown to be recruited to the nucleolus by UBF, where it binds to rDNA promoters and represses transcription, thereby preventing the formation of genotoxic ribosomal DNA-RNA hybrid structures [Citation63]. In addition, other studies suggest that PHF6 is involved in transcriptional repression of different non-ribosomal genes, such as CDKN1A/p21 [Citation64]. Although the PHD2 domain of PHF6 was demonstrated to bind double-strand DNA in vitro in a sequence-specific way [Citation61], others suggested that PHF6 does not recognize specific DNA sequences but instead, controls transcription by interacting with histones [Citation60,Citation76]. PARP1/2-mediated recruitment of PHF6 to DNA lesions might thus contribute to the transcriptional silencing of the DSB-flanking genes, which was shown to be important for NHEJ-mediated repair [Citation77].
As mentioned before, chromatin modifications around DSBs are essential for a correct repair process [Citation26]. Through its ability to interact with histones, PHF6 could be important for the recruitment of different chromatin-modifying factors that are necessary for the silencing of transcription, such as CDYL1 [Citation78], a protein known to abolish transcription through the methylation of histone H3K27, the NELF complex, which regulates transcription by pausing the RNAPII [Citation77], or the already mentioned NuRD complex. The dependency of PHF6 localization on PARP and its previously described role in suppression of transcription suggests that PHF6 could regulate NHEJ through general transcriptional repression (). Future research should clarify the precise mechanism of PHF6 in checkpoint recovery, and if PHF6 exerts this function via the NuRD complex and/or through transcriptional repression.
The role of PHF6 in disease and therapy
The novel role of PHF6 in DNA repair and checkpoint recovery may lead to a better understanding of the development of disease(s) and also provide new therapeutic and diagnostic opportunities. The PHF6 gene is highly conserved among vertebrate species and likely essential for development [Citation79,Citation80]. Germline mutations in PHF6 can lead to the X-linked recessive Borjeson-Forssman-Lehmann Syndrome (BFLS), a rare mental retardation disorder further characterized by epilepsy, hypogonadism and obesity [Citation81,Citation82]. Recent findings from knockout mouse models reported a role for PHF6 in hematopoiesis and hematopoietic stem cell homeostasis, as loss of PHF6 led to altered numbers of hematopoietic stem and progenitor cells. Besides, PHF6 deficiency in combination with either aberrant TLX3 expression or an active NOTCH mutation resulted in leukemia, indicating that PHF6 is a tumor suppressor involved in the onset of hematological malignancies [Citation83–Citation85]. Indeed, somatic inactivating mutations in PHF6 are commonly found in T-ALL, mixed phenotype acute leukemia (MPAL) and less frequently in acute myeloid leukemia (AML) and other cancers, and have been associated with resistance to drugs in T-ALL [Citation64,Citation86–Citation88]. Such somatic mutations in PHF6 frequently result in frameshifts, nonsense (stop) mutations or deletions, leading to truncations or a complete protein loss, while germline mutations are typically hypomorphic. Enhanced expression of PHF6 was also found in several human cancer types such as breast and colorectal cancers [Citation89]. Taken together, these data indicate that PHF6 can act as a tumor suppressor as well as an oncogene.
Cancer cells are generally more susceptible to genotoxic stress because of faster proliferation and therefore a higher accumulation of DNA lesions compared to untransformed cells. In addition, mutations in DNA repair genes make tumor cells more sensitive to DNA-damaging agents. For this reason, various chemotherapeutic agents are used to treat different cancers, and tumors harboring PHF6 alterations, such as leukemia and glioblastoma, are among them. Our results showing that disruption of PHF6 in U2OS osteosarcoma led to a modest cellular sensitivity to IR are in line with this [Citation34]. We additionally demonstrated that inhibition of PARP1/2 prevents the localization of PHF6 at sites of DNA lesions. Consequently, (co)treatment with a PARP1/2 inhibitor may influence therapy outcome of tumors harboring PHF6 loss-of-function mutations.
Only half of the adult T-ALL patients display long-term survival with the current treatment strategies [Citation90,Citation91], indicating there is a clinical need for improved stratification of these patients to avoid both under- and overtreatment and treatment-related toxicities, as well as novel therapy options. PHF6 is a promising biomarker gene for personalized therapeutic strategies for the treatment of these patients. However, more insight into the sensitivity of PHF6 mutant cells and a deeper molecular understanding of the functional roles of PHF6 in cancer is needed in order to utilize its potential in the development of personalized medicine or diagnostics.
Acknowledgments
We would like to thank Rob Wolthuis, Jeroen Guikema and Raimundo Freire for helpful discussion during the preparation of the manuscript.
Disclosure statement
The authors declare that they have no competing interests.
Additional information
Funding
References
- Aguilera A, Gomez-Gonzalez B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008 Mar;9(3):204–217.
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000 Jan 7;100(1):57–70.
- McKinnon PJ, Caldecott KW. DNA strand break repair and human genetic disease. Annu Rev Genomics Hum Genet. 2007;8:37–55.
- Rass U, Ahel I, West SC. Defective DNA repair and neurodegenerative disease. Cell. 2007 Sept 21;130(6):991–1004.
- Hasty P, Campisi J, Hoeijmakers J, et al. Aging and genome maintenance: lessons from the mouse? Science. 2003 Feb 28;299(5611):1355–1359.
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009 Oct 22;461(7267):1071–1078.
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001 Sept 1;15(17):2177–2196.
- Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007 May 25;316(5828):1160–1166.
- van Vugt MA, Bras A, Medema RH. Restarting the cell cycle when the checkpoint comes to a halt. Cancer Res. 2005 Aug 15;65(16):7037–7040.
- Macurek L, Lindqvist A, Lim D, et al. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008 Sept 4;455(7209):119–123.
- van Vugt M, Medema RH. Checkpoint adaptation and recovery: back with Polo after the break. Cell Cycle. 2004 Nov;3(11):1383–1386.
- Mamely I, Vugt M, Smits VA, et al. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr Biol. 2006 Oct 10;16(19):1950–1955.
- Mailand N, Bekker-Jensen S, Bartek J, et al. Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol Cell. 2006 Aug 4;23(3):307–318.
- Peschiaroli A, Dorrello NV, Guardavaccaro D, et al. SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol Cell. 2006 Aug 4;23(3):319–329.
- Freire R, Vugt M, Mamely I, et al. Claspin: timing the cell cycle arrest when the genome is damaged. Cell Cycle. 2006 Dec;5(24):2831–2834.
- Macurek L, Lindqvist A, Voets O, et al. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene. 2010 Apr 15;29(15):2281–2291.
- Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005 May 15;19(10):1162–1174.
- Lindqvist A, de Bruijn M, Macurek L, et al. Wip1 confers G2 checkpoint recovery competence by counteracting p53-dependent transcriptional repression. Embo J. 2009 Oct 21;28(20):3196–3206.
- Papamichos-Chronakis M, Peterson CL. Chromatin and the genome integrity network. Nat Rev Genet. 2013 Jan;14(1):62–75.
- Koundrioukoff S, Polo S, Almouzni G. Interplay between chromatin and cell cycle checkpoints in the context of ATR/ATM-dependent checkpoints. DNA Repair (Amst). 2004 Aug-Sept;3(8–9):969–978.
- Warmerdam DO, Kanaar R. Dealing with DNA damage: relationships between checkpoint and repair pathways. Mutat Res. 2010 Apr-June;704(1–3):2–11.
- Deem AK, Li X, Tyler JK. Epigenetic regulation of genomic integrity. Chromosoma. 2012 Apr;121(2):131–151.
- Burma S, Chen BP, Murphy M, et al. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001 Nov 9;276(45):42462–42467.
- Zhao Y, Brickner JR, Majid MC, et al. Crosstalk between ubiquitin and other post-translational modifications on chromatin during double-strand break repair. Trends Cell Biol. 2014 July;24(7):426–434.
- Taverna SD, Li H, Ruthenburg AJ, et al. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007 Nov;14(11):1025–1040.
- van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009 May;19(5):207–217.
- Soria G, Polo SE, Almouzni G. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell. 2012 June 29;46(6):722–734.
- Smeenk G, van Attikum H. The chromatin response to DNA breaks: leaving a mark on genome integrity. Annu Rev Biochem. 2013;82:55–80.
- Linger JG, Tyler JK. Chromatin disassembly and reassembly during DNA repair. Mutat Res. 2007 May 1;618(1–2):52–64.
- Kruhlak MJ, Celeste A, Dellaire G, et al. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol. 2006 Mar 13;172(6):823–834.
- Tsukuda T, Fleming AB, Nickoloff JA, et al. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 2005 Nov 17;438(7066):379–383.
- Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304.
- Luijsterburg MS, van Attikum H. Chromatin and the DNA damage response: the cancer connection. Mol Oncol. 2011 Aug;5(4):349–367.
- Warmerdam DO, Alonso-de Vega I, Wiegant WW, et al. PHF6 promotes non-homologous end joining and G2 checkpoint recovery. EMBO Rep. 2020 Jan 7;21(1):e48460.
- Cai Y, Jin J, Tomomori-Sato C, et al. Identification of new subunits of the multiprotein mammalian TRRAP/TIP60-containing histone acetyltransferase complex. J Biol Chem. 2003 Oct 31;278(44):42733–42736.
- Doyon Y, Cote J. The highly conserved and multifunctional NuA4 HAT complex. Curr Opin Genet Dev. 2004 Apr;14(2):147–154.
- Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol. 2007 Apr;8(4):284–295.
- Price BD, D’Andrea AD. Chromatin remodeling at DNA double-strand breaks. Cell. 2013 Mar 14;152(6):1344–1354.
- Sun Y, Jiang X, Chen S, et al. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A. 2005 Sept 13;102(37):13182–13187.
- Squatrito M, Gorrini C, Amati B. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol. 2006 Sept;16(9):433–442.
- Kaidi A, Jackson SP. KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature. 2013 June 6;498(7452):70–74.
- Ikura T, Tashiro S, Kakino A, et al. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol Cell Biol. 2007 Oct;27(20):7028–7040.
- Tang Y, Luo J, Zhang W, et al. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006 Dec 28;24(6):827–839.
- Barlev NA, Liu L, Chehab NH, et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001 Dec;8(6):1243–1254.
- Tang J, Cho NW, Cui G, et al. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol. 2013 Mar;20(3):317–325.
- Kusch T, Florens L, Macdonald WH, et al. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science. 2004 Dec 17;306(5704):2084–2087.
- Jha S, Shibata E, Dutta A. Human Rvb1/Tip49 is required for the histone acetyltransferase activity of Tip60/NuA4 and for the downregulation of phosphorylation on H2AX after DNA damage. Mol Cell Biol. 2008 Apr;28(8):2690–2700.
- Shaltiel IA, Krenning L, Bruinsma W, et al. The same, only different - DNA damage checkpoints and their reversal throughout the cell cycle. J Cell Sci. 2015 Feb 15;128(4):607–620.
- Xu Y, Sun Y, Jiang X, et al. The p400 ATPase regulates nucleosome stability and chromatin ubiquitination during DNA repair. J Cell Biol. 2010 Oct 4;191(1):31–43.
- Judes G, Rifai K, Ngollo M, et al. A bivalent role of TIP60 histone acetyl transferase in human cancer. Epigenomics. 2015;7(8):1351–1363.
- Coffey K, Blackburn TJ, Cook S, et al. Characterisation of a Tip60 specific inhibitor, NU9056, in prostate cancer. PLoS One. 2012;7(10):e45539.
- Watrin E, Peters JM. The cohesin complex is required for the DNA damage-induced G2/M checkpoint in mammalian cells. Embo J. 2009 Sept 2;28(17):2625–2635.
- Watrin E, Peters JM. Cohesin and DNA damage repair. Exp Cell Res. 2006 Aug 15;312(14):2687–2693.
- Tan TL, Kanaar R, Wyman C. Rad54, a Jack of all trades in homologous recombination. DNA Repair (Amst). 2003 July 16;2(7):787–794.
- Yazdi PT, Wang Y, Zhao S, et al. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002 Mar 1;16(5):571–582.
- Bot C, Pfeiffer A, Giordano F, et al. Independent mechanisms recruit the cohesin loader protein NIPBL to sites of DNA damage. J Cell Sci. 2017 Mar 15;130(6):1134–1146.
- Todd MA, Ivanochko D, Picketts DJ. PHF6 degrees of separation: the multifaceted roles of a chromatin adaptor protein. Genes (Basel). 2015 June 19;6(2):325–352.
- Sanchez R, Zhou MM. The PHD finger: a versatile epigenome reader. Trends Biochem Sci. 2011 July;36(7):364–372.
- Todd MA, Picketts DJ. PHF6 interacts with the nucleosome remodeling and deacetylation (NuRD) complex. J Proteome Res. 2012 Aug 3;11(8):4326–4337.
- Soto-Feliciano YM, Bartlebaugh JME, Liu Y, et al. PHF6 regulates phenotypic plasticity through chromatin organization within lineage-specific genes. Genes Dev. 2017 May 15;31(10):973–989.
- Liu Z, Li F, Ruan K, et al. Structural and functional insights into the human Borjeson-Forssman-Lehmann syndrome-associated protein PHF6. J Biol Chem. 2014 Apr 4;289(14):10069–10083.
- Wang J, Leung JW, Gong Z, et al. PHF6 regulates cell cycle progression by suppressing ribosomal RNA synthesis. J Biol Chem. 2013 Feb 1;288(5):3174–3183.
- Todd MA, Huh MS, Picketts DJ. The sub-nucleolar localization of PHF6 defines its role in rDNA transcription and early processing events. Eur J Hum Genet. 2016 Oct;24(10):1453–1459.
- Xiang J, Wang G, Xia T, et al. The depletion of PHF6 decreases the drug sensitivity of T-cell acute lymphoblastic leukemia to prednisolone. Biomed Pharmacother. 2019 Jan;109:2210–2217.
- Zhang C, Mejia LA, Huang J, et al. The X-linked intellectual disability protein PHF6 associates with the PAF1 complex and regulates neuronal migration in the mammalian brain. Neuron. 2013 June 19;78(6):986–993.
- Liu Z, Li F, Zhang B, et al. Structural basis of plant homeodomain finger 6 (PHF6) recognition by the retinoblastoma binding protein 4 (RBBP4) component of the nucleosome remodeling and deacetylase (NuRD) complex. J Biol Chem. 2015 Mar 6;290(10):6630–6638.
- Luijsterburg MS, de Krijger I, Wiegant WW, et al. PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by non-homologous end-joining. Mol Cell. 2016 Feb 18;61(4):547–562.
- Smeenk G, Wiegant WW, Vrolijk H, et al. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J Cell Biol. 2010 Sept 6;190(5):741–749.
- Pan MR, Hsieh HJ, Dai H, et al. Chromodomain helicase DNA-binding protein 4 (CHD4) regulates homologous recombination DNA repair, and its deficiency sensitizes cells to poly(ADP-ribose) polymerase (PARP) inhibitor treatment. J Biol Chem. 2012 Feb 24;287(9):6764–6772.
- Gong F, Chiu LY, Cox B, et al. Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev. 2015 Jan 15;29(2):197–211.
- Spruijt CG, Luijsterburg MS, Menafra R, et al. ZMYND8 co-localizes with NuRD on target genes and regulates poly(ADP-Ribose)-dependent recruitment of GATAD2A/NuRD to sites of DNA damage. Cell Rep. 2016 Oct 11;17(3):783–798.
- Savitsky P, Krojer T, Fujisawa T, et al. Multivalent histone and DNA engagement by a PHD/BRD/PWWP triple reader cassette recruits ZMYND8 to K14ac-rich chromatin. Cell Rep. 2016 Dec 6;17(10):2724–2737.
- Miller KM, Tjeertes JV, Coates J, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010 Sept;17(9):1144–1151.
- Marnef A, Legube G. Organizing DNA repair in the nucleus: DSBs hit the road. Curr Opin Cell Biol. 2017 June;46:1–8.
- Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol. 2017 Oct;18(10):610–621.
- Miyagi S, Sroczynska P, Kato Y, et al. The chromatin-binding protein Phf6 restricts the self-renewal of hematopoietic stem cells. Blood. 2019 June 6;133(23):2495–2506.
- Awwad SW, Abu-Zhayia ER, Guttmann-Raviv N, et al. NELF-E is recruited to DNA double-strand break sites to promote transcriptional repression and repair. EMBO Rep. 2017 May;18(5):745–764.
- Abu-Zhayia ER, Awwad SW, Ben-Oz BM, et al. CDYL1 fosters double-strand break-induced transcription silencing and promotes homology-directed repair. J Mol Cell Biol. 2018 Aug 1;10(4):341–357.
- Voss AK, Gamble R, Collin C, et al. Protein and gene expression analysis of Phf6, the gene mutated in the Borjeson-Forssman-Lehmann Syndrome of intellectual disability and obesity. Gene Expr Patterns. 2007 Oct;7(8):858–871.
- Crawford J, Lower KM, Hennekam RC, et al. Mutation screening in Borjeson-Forssman-Lehmann syndrome: identification of a novel de novo PHF6 mutation in a female patient. J Med Genet. 2006 Mar;43(3):238–243.
- Visootsak J, Rosner B, Dykens E, et al. Clinical and behavioral features of patients with Borjeson-Forssman-Lehmann syndrome with mutations in PHF6. J Pediatr. 2004 Dec;145(6):819–825.
- Turner G, Lower KM, White SM, et al. The clinical picture of the Borjeson-Forssman-Lehmann syndrome in males and heterozygous females with PHF6 mutations. Clin Genet. 2004 Mar;65(3):226–232.
- McRae HM, Garnham AL, Hu Y, et al. PHF6 regulates hematopoietic stem and progenitor cells and its loss synergizes with expression of TLX3 to cause leukemia. Blood. 2019 Apr 18;133(16):1729–1741.
- Miyagi S, Iwama A. Plant homeodomain finger protein 6 in the regulation of normal and malignant hematopoiesis. Curr Opin Hematol. 2020 July;27(4):248–253.
- Hsu YC, Chen TC, Lin CC, et al. Phf6-null hematopoietic stem cells have enhanced self-renewal capacity and oncogenic potentials. Blood Adv. 2019 Aug 13;3(15):2355–2367.
- Hiddingh L, Raktoe RS, Jeuken J, et al. Identification of temozolomide resistance factors in glioblastoma via integrative miRNA/mRNA regulatory network analysis. Sci Rep. 2014 June;11(4):5260.
- Van Vlierberghe P, Patel J, Abdel-Wahab O, et al. PHF6 mutations in adult acute myeloid leukemia. Leukemia. 2011 Jan;25(1):130–134.
- Van Vlierberghe P, Palomero T, Khiabanian H, et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet. 2010 Apr;42(4):338–342.
- Hajjari M, Salavaty A, Crea F, et al. The potential role of PHF6 as an oncogene: a genotranscriptomic/proteomic meta-analysis. Tumour Biol. 2016 Apr;37(4):5317–5325.
- Marks DI, Rowntree C. Management of adults with T-cell lymphoblastic leukemia. Blood. 2017 Mar 2;129(9):1134–1142.
- Hodby KA, Marks DI. Recent advances in the management of acute lymphoblastic leukaemia. Curr Treat Options Oncol. 2020 Feb 20;21(3):23,020–0712-8.