
ABSTRACT
Organ development is precisely guided by spatiotemporal cross-talks between a variety of signaling pathways regulating cell differentiation, proliferation, growth arrest and physiological cell death. Aberrant signaling inputs invariably lead to tissue dysfunction and to certain conditions, even malignant transformation. In this review, we focus on the functional interplay between the Hippo signaling pathway and autophagy in normal tissue homeostasis and in malignant tumor progression. Mounting experimental evidence for the regulation of cancer cell malignancy and therapy resistance by the functional cross-talk between Hippo signaling and autophagy highlights this signaling axis as a suitable therapeutic target to combat cancer.
Introduction
Control of organ size is a critical challenge during animal development which requires tightly regulated tissue homeostasis at both the cellular and the molecular level [Citation1]. Cell populations derived from different germinal layers cooperate to form a functional organ via direct and indirect physiological intercellular interactions. This inter-cellular communication is subsequently translated into intra-cellular outputs, including signals impacting on the differentiation and proliferation status of cells and as well as on degradative pathways. Rapid cell proliferation critically depends on the availability of cellular building blocks, such as lipids, amino acids and nucleotides [Citation2]. In addition to de novo synthesis, a critical source of building blocks comes from the degradation of existing organelles and macromolecules, in particular when cells are exposed to limited nutrient supply.
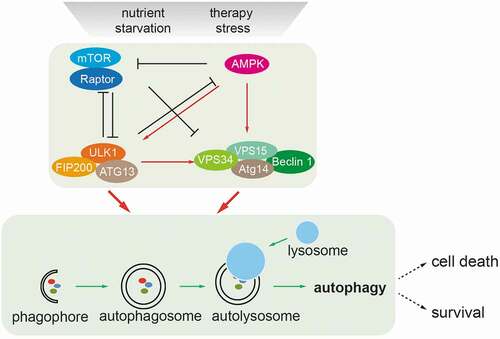
Autophagy is a self-digestion process which targets cytoplasmic components to the lysosomes for the recycling of building blocks in response to cellular stress [Citation3]. Autophagy induction in response to nutrient stress is tightly controlled by cellular signals which promote autophagy initiation and autophagic flux [Citation4]. Autophagy starts from an isolation membrane (phagophore), followed by the formation of double-membraned autophagosomes, which engulf intra-cellular proteins/subcellular organelles. Autophagosomes mature by fusion with lysosomes, leading to the degradation of the autophagosomal contents (). Thus, autophagy serves as a degradative machinery for the recycling of building blocks. Autophagy core machineries consist of a variety of components with distinct molecular components. For example, the Ser/Thr-protein kinase ULK1 and the class III PI3K kinase VPS34 form a complex with Beclin-1 (BECN1), VPS15, ATG14 or UVRAG, and the E3-ligase-like ubiquitin-conjugating ATG proteins to exert the autophagic process [Citation5]. Autophagy has been reported to play a dual role in tumorigenesis by either promoting early tumor initiation or restricting late tumor progression [Citation6,Citation7]. Likewise, autophagy represents a double-edged sword in cancer therapy, in particular in response to molecularly targeted therapies [Citation8]. While most acute therapeutic treatments induce lethal autophagy, survival autophagy appears to mediate an adaptive response and to promote cell viability in therapy-resistant cancer cells [Citation8]. Thus, the biological consequences of autophagy are context-dependent, and a deeper understanding of the regulation of autophagy in the process of multistage carcinogenesis and during cancer therapy is warranted for an optimal exploitation in therapeutic interventions.
Figure 1. Nutrient-induced regulation of autophagy. Nutrient deprivation and therapy stress activate the metabolic sensors mTOR and AMP-activated kinase (AMPK) which act as major molecular switches of autophagy in response to nutrients. Under conditions of sufficient nutrients, mTOR complex 1 (mTORC1) is activated by upstream regulators, such as amino acid-sensing pathways. Active mTORC1 negatively regulates autophagy induction by phosphorylating and inactivating the ULK1 complex (ULK1/FIP200/ATG13) and the VPS34/BECN1complex (VPS34/BECN1/ATG14/VPS15). When nutrients are limiting, such as under glucose starvation, activated AMPK directly promotes autophagy by phosphorylating and activating the ULK1 complex and the VPS34/BECN1 complex. Activated AMPK and ULK1 negatively regulate mTORC1 activity, thereby relieving its inhibitory effect on autophagy. Autophagy starts from an isolation membrane (phagophore), followed by the formation of a double-membraned autophagosome, which engulfs intra-cellular proteins and subcellular organelles. Autophagosomes then mature by fusion with lysosomes, leading to the degradation of autophagosomal contents. Cells survive in the case of limited autophagy, such as in therapy-resistant cancer cells, whereas with high levels of autophagy cells will succumb to cell death
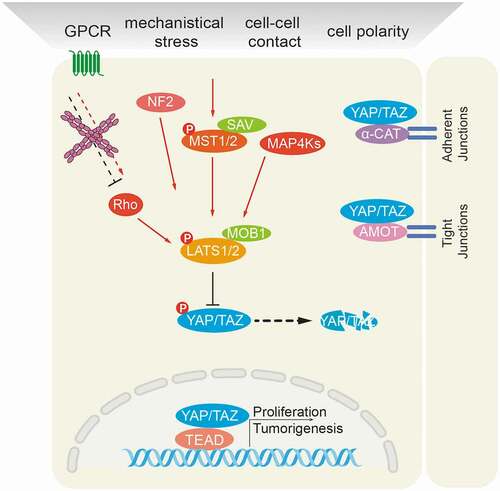
The Hippo signaling pathway is a master regulator of organ size control. Activated mammalian Hippo kinases (MST1 and 2 in mammals) or other upstream kinases, such as MAP4Ks, phosphorylate and activate Large Tumor Suppressor kinases 1 and 2 (LATS1/2), which in turn, phosphorylate and inactivate yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ). YAP/TAZ are transcriptional co-activators which, when not phosphorylated by LATS1/2, translocate to the nucleus and interact with DNA-binding transcription factors (TFs), such as TEADs, to modulate the expression of specific target genes [Citation9] (). Deregulated Hippo components have been reported in many cancer types in patients and also in experimental mouse models of cancer [Citation10]. For instance, loss of upstream Hippo signaling inputs, such as a deletion of MST1/2 or of LATS1/2 or a deficiency of the MST co-factors NF2 or Salvador, invariably leads to hepatomegaly and the development of liver cancer. Likewise, gain of function of the downstream effector YAP results in hepatomegaly as well.
Figure 2. Schematic representation of the mammalian Hippo signaling pathway. Large Tumor Suppressor kinases (LATS1/2) can be activated by a variety of signals, including by mammalian Hippo kinases MST1/2 or other upstream kinases (MAP4Ks), by G-protein coupled receptor (GPCR)-mediated modulation of Rho family GTPase activities and/or actin cytoskeleton remodeling, and by NF2-mediated membrane recruitment. In turn, activated LATS1/2 phosphorylate and inactivate YES-associated protein (YAP) on Ser127 and transcriptional co-activator with PDZ-binding motif (TAZ) on Ser89. YAP/TAZ are transcriptional co-activators that interact with DNA binding transcription factors (TFs), such as TEADs, to initiate gene expression programs. Cell junctional α-catenin (α-CAT) or angiomotin (AMOT) inactivate YAP/TAZ by activating LATS1/2 and by localizing YAP/TAZ to cell junctions. Conversely, loss of cell-cell contacts activates Yap/TAZ-mediated pro-tumorigenic gene expression
In addition to its role in tumorigenesis, mammalian Hippo signaling has been widely implicated in acute responses to cancer therapies as well as in the development of therapy resistance [Citation11]. For instance, genetic screening has identified a driving role of YAP in mediating resistance to RAF and MEK inhibitor-based therapies in BRAF-mutant melanoma cells [Citation12,Citation13]. Mechanistically, therapy-induced actin cytoskeleton remodeling promotes therapy resistance of melanoma cells via activation of YAP/TAZ, likely via TESK1-mediated actin stress fiber formation [Citation13]. Besides its cell autonomous roles, Hippo signaling also affects non-cell-autonomous processes in cancer progression. In fact, YAP/TAZ/TEAD transcriptional complexes induce the expression of soluble growth factors which can function in both autocrine and paracrine modes [Citation14]. In addition, E-cadherin-mediated adhesion between cancer cells prevents ferroptosis [Citation15], a form of cell death induced by lipid peroxidation, via LATS-YAP signaling, highlighting a therapeutic strategy to target Hippo signaling in metastatic disease [Citation16]. Likewise, TAZ exerts a similar ferroptosis-regulating function in renal cell carcinoma [Citation17]. In addition to its role in cancer cells, YAP also plays an important role in promoting helper T cell differentiation to regulatory T cells [Citation18], suggesting that the Hippo pathway may also promote cancer progression by suppressing immune surveillance [Citation19].
Emerging evidence also suggests that Hippo signaling governs tissue homeostasis by directly cross-talking to signaling pathways which are activated by cellular responses to starvation or stress, such as nutrient-regulated mTOR signaling and autophagy pathways. While the function of the Hippo-mTOR signaling axis has been widely appreciated [Citation20], we here summarize recent advances in delineating the functional connections between Hippo signaling and autophagy in tissue homeostasis and during cancer therapy response.
Nutrients regulate Hippo signaling and autophagy
Nutrient availability is a determinant factor balancing general cell proliferation and growth arrest, and also Hippo-mediated growth control. For instance, YAP/TAZ transcriptional activity is dynamically regulated by extracellular glucose levels [Citation21–23]. Specifically, energy stress induced by glucose deprivation inhibits YAP activity by promoting AMP-dependent kinase (AMPK)-mediated phosphorylation of YAP at serine residue S94 [Citation21,Citation22] ()). In fact, AMPK additionally activates LATS kinases via AMOTL1 or Rho GTPase [Citation23], but interestingly has limited effect on MST kinases. Conversely, YAP transcriptionally regulates the expression of glucose transporter 3 (GLUT3), suggesting a positive feedback regulation in glucose metabolism [Citation21]. Likewise, the availability of amino acids dynamically regulates YAP transcriptional activity [Citation24]. Conversely, YAP induces the expression of miR-29 which represses the expression of the tumor suppressor PTEN, leading to the activation of the amino acid sensor mTOR [Citation25]. Recent studies also suggest that YAP affects amino acid metabolism by direct transcriptional regulation of amino acid transporters, such as LAT1 [Citation24].
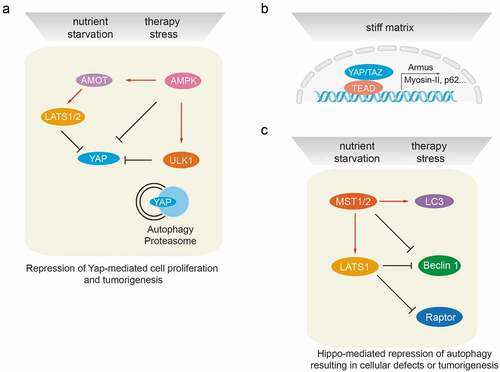
Figure 3. Cross-talk between autophagy and the Hippo signaling pathway at the molecular level. (a) Autophagy regulates Hippo signaling activity. Under starvation or therapy stress conditions, AMPK inactivates YAP by direct phosphorylation at Ser94 or by angiomotin (AMOT)/LATS1/2-mediated indirect regulation, thereby inhibiting YAP-driven oncogenic events. ULK1 is also able to phosphorylate YAP at serine residues S74/97 and to inactivate YAP. Under physiological conditions, YAP protein homeostasis is achieved by proteasomal and autophagy-mediated degradation. Hence, autophagy or proteasome deficiencies lead to an accumulation of YAP and promote YAP-driven tumorigenesis. (b) Hippo signaling mediates transcriptional regulation of autophagy. The YAP/TAZ/TEAD transcriptional complex is able to induce the expression of Armus, Myosin-II, miR-29 and p62 mRNA in stiff culture environments, thereby promoting autophagosome maturation and autophagic flux. YAP/TAZ-induced transcriptional regulation of autophagy core-components is essential to drive the cellular plasticity required for somatic stem cells as well as tumor-initiating cells. (c) Hippo signaling mediates post-transcriptional regulation of autophagy effectors. Besides activating LATS1/2, MST kinases can directly phosphorylate LC3B and promote autophagy induction, resulting in the elimination of intra-cellular bacteria. During myocardial infarction, MST1 is also able to phosphorylate BECN1 and inactivate BECN1 by inducing BECN1/BCL2 complex formation. Such inhibition of autophagy promotes cardiac dysfunction. Under cell-cell contact conditions, LATS kinases can phosphorylate Raptor at Ser606 and inhibit mTORC1 activity, leading to reduced organ size. During cancer therapy stress, in a kinase activity-independent fashion LATS1, but not LATS2, can induce K27-linked polyubiquitination of BECN1, thereby promoting BECN1 homodimer formation and inhibiting autophagic cell death. The loss of autophagy results in cellular defects and/or in tumorigenesis
Induction of autophagy in response to metabolic stress is an essential mechanism to maintain tissue homeostasis. In addition to impacting on Hippo signaling, AMPK is an essential transducer from nutrient starvation to autophagy activation [Citation26] (). Mechanistically, AMPK represses mTOR-mediated autophagy suppression via phosphorylation of key modulators of mTOR activity, such as tumor sclerosis complex 2 (TSC2) on Ser1387 and Raptor on Ser792 [Citation27–29]. In addition, AMPK can directly regulate autophagy initiation via phosphorylating key autophagy components, such as ULK1 (Ser317/Ser555/Ser777) [Citation30,Citation31], VPS34 (Thr163/Ser165) [Citation32] and BECN1 (Ser91/Ser94) [Citation32]. Interestingly, mTOR is also able to directly phosphorylate and inactive ULK1 (Ser757) [Citation30], suggesting a counterbalancing regulation of autophagy by dynamically shifting ULK1 between complexes with mTOR and with AMPK [Citation30]. Hence, nutrient-induced metabolism simultaneously regulates Hippo signaling activities and autophagy. However, reciprocal functional interactions between Hippo signaling and autophagic activities have only recently been uncovered, as summarized in the following chapters.
Autophagy regulating Hippo signaling
Inductive cues of autophagy, such as glucose deprivation, robustly induce the activation of the Hippo signaling pathway and, consequently, the phosphorylation-induced inactivation and degradation of the YAP/TAZ transcriptional co-factors. Proteasomal degradation is predominantly responsible for the clearance of inactive YAP and TAZ. Given the general nature of autophagy as another stress-induced protein-degradation system, it has been reasonable to explore potential roles of autophagy in controlling the levels of Hippo components. Indeed, impairment of autophagy by loss of ATG7, one of the main regulators of the autophagic process, leads to YAP hyperactivation and subsequently to hepatomegaly and hepatic carcinogenesis in mice [Citation33]. These results strongly indicate that YAP protein levels are restrained by autophagy ()). Likewise, autophagy repression mediated by TSC mutations and subsequent mTOR activation leads to YAP accumulation and transcriptional hyperactivation in a perivascular epithelioid tumor model [Citation34]. Together, these studies suggest that YAP might represent a therapeutic target, specifically in tumors which are driven by deregulated mTOR signaling or by a loss of lethal autophagy. The latter appears increasingly important in the development of therapy resistance in cancer cells.
In addition to nutrients, cancer therapies, such as the treatment of hepatocellular carcinoma (HCC) with the multi-kinase inhibitor sorafenib, promote autophagy in patients, suggesting a potential role of autophagy-mediated regulation of Hippo signaling in therapy response [Citation35]. Indeed, sorafenib induces down-regulation of YAP, partially via autophagy induction [Citation35]. Moreover, sorafenib-induced autophagy degrades LATS1 as well. Conversely, LATS1 restricts sorafenib-induced autophagy induction in HCC cells, indicating a tight reciprocal control between autophagy and Hippo signaling during therapy response [Citation35]. Furthermore, Aurora A kinase has been shown to promote YAP activation by suppressing autophagy in lung cancer cells [Citation36], highlighting the option of targeting Aurora A kinase to repress YAP signaling.
Autophagy is dynamically regulated via a large number of core autophagy factors which by forming distinct complexes execute the autophagic process. A recent study has demonstrated a direct protein-protein interaction and phosphorylation of fly YAP (Yorkie) by one of the autophagy complex components, the autophagy initiation kinase Atg1 [Citation37] ()). This functional interaction appears to be conserved from fly to humans. Interestingly, stress-activated phosphorylation of YAP by ULK1 at S74 and S97 inhibits YAP activity, but not its subcellular localization [Citation38], indicating a functional divergence between ULK1- and LATS-mediated YAP phosphorylation. Of note, YAP serine residue S94 phosphorylated by ULK1 is the same site phosphorylated by AMPK [Citation22]. Hence, it will be worthwhile to investigate how YAP is concomitantly controlled by the nutrient deprivation and stress-induced activities of AMPK and ULK1 in a spatiotemporal fashion.
Hippo signaling regulating autophagy
Hippo signaling is regulated via soluble growth factors, via inter-cellular interactions, such as cell density, as well as by intra-cellular stresses. YAP/TAZ, transcriptional effectors of Hippo signaling, cooperate with DNA-binding TEAD family members to initiate transcriptional outputs which coordinate growth control and stress cues, including autophagy induction. In fact, several autophagy key components have been reported to be regulated at the transcriptional level by YAP/TAZ. For instance, the expression of Armus, a protein of the RAB-GAP family, is regulated by YAP/TAZ, thereby promoting the fusion of autophagosomal vesicles with lysosomes in a stiff extracellular matrix (ECM) [Citation39] ()). Likewise, YAP/TAZ can potently promote autophagy through transcriptional upregulation of myosin-II in response to stiff ECM [Citation40] ()). These studies indicate that the cellular plasticity promoted by YAP/TAZ in response to varying substrate stiffness is at least in parts mediated by a regulation of autophagy. These studies have also demonstrated that Hippo signaling is regulated by the actin cytoskeleton and, conversely, that YAP transcriptional activity affects F-actin dynamics. Interestingly, YAP is also able to regulate autophagic flux in a TEAD-dependent transcriptional response to nutrient deprivation and therapy stress, most likely via controlling the expression of the autophagy substrate p62 [Citation41].
Comparable to most signaling pathways, Hippo signaling is initiated and amplified by kinase-mediated phosphorylation cascades. Recent studies also suggest that Hippo kinases regulate autophagy by directly phosphorylating key components of the autophagic machinery. For example, the Hippo/MST kinases are evolutionary conserved key regulators of autophagy induction from yeast, fly, worms to mammals. Lack of MST kinases or their homologues results in autophagy blockade [Citation42]. Mechanistically, MST kinases phosphorylate the autophagy effector protein microtubule-associated protein 1A/1B-light chain 3 (LC3B) at Thr50, thereby promoting autophagic flux [Citation42] ()). Importantly, MST-mediated LC3B phosphorylation is functionally coupled to the autophagic clearance of bacteria, highlighting a potential strategy of targeting MST-LC3B for treatment of infectious disease [Citation42].
MST1 has been identified as an apoptotic kinase by its phosphorylation and proteolysis-induced activation [Citation43]. For instance, MST1 promotes myocardial damage via enhancing cardiomyocyte apoptosis in response to ischemia and reperfusion stress [Citation44]. However, the role of MST1 in autophagy regulation in cardiomyocytes seems context-dependent. While MST-mediated phosphorylation of LC3B promotes cardiomyocyte autophagy, MST1 conversely represses autophagy by phosphorylating the autophagic effector protein BECN1 under pathological stresses [Citation44] ()). BECN1 itself plays a central role in autophagy by orchestrating the various autophagy-regulating protein complexes [Citation45]. BECN1 interacts with the class III PI3K lipid kinase VPS34 and the PI3K kinase regulator VPS15 to form the autophagy core complex BECN1/VPS34/VPS15. By interacting with other co-factors, this core complex shifts between autophagy-inducing (i.e. BECN1/VPS34/ATG14L and BECN1/VPS34/UVRAG) and autophagy-inhibiting states (i.e. BECN1/BECN1 homodimer and BECN1/BCL2, BECN1/BCL-xL and BECN1/VPS34/Rubicon). Importantly, MST1-mediated BECN1 phosphorylation at Thr108 promotes complex formation between BECN1 and BCL-2 family proteins and concomitantly decreases VPS34 kinase activity. As a consequence, MST1-induced phosphorylation of BECN1 suppresses autophagy and enhances apoptosis in cardiomyocytes, thus provoking a deterioration of cardiac function. Interestingly, MST1-induced myocardial injury also partially relies on the MST1-dependent phosphorylation of BCL-xL at Ser14 site [Citation46]. Targeting MST1 kinase activity-induced apoptosis may thus represent a therapeutic option in myocardial injury and heart failure.
In addition to its regulation by phosphorylation and the formation of divergent sub-complexes, BECN1’s autophagic activity is also tightly controlled by acetylation and ubiquitination [Citation47]. Ubiquitination of BECN1 generally leads either to its proteasomal degradation or to its activation, depending on the type of linkage of ubiquitin molecules to BECN1. Recent studies suggest that such differential ubiquitination seems to play a key role in BECN1-mediated autophagy regulation [Citation47].
As transducers of the Hippo kinase signaling cascade toward the transcriptional effectors YAP/TAZ, LATS1/2 kinases are well-depicted tumor suppressors [Citation48]. LATS1 and LATS2 display high sequence identity and share the same upstream activators and activation mechanisms. While it is generally believed that LATS1 and LATS2 exert redundant functions in regulating YAP/TAZ transcriptional activities, recent studies suggest a functional difference between them. In the liver, LATS2, but not LATS1, restricts hepatic cholesterol accumulation by directly binding to and quenching the transcriptional activity of sterol regulatory element-binding proteins 1 and 2 (SREBP1/2) [Citation49]. Conversely, LATS1 kinase, but not LATS2 kinase, seems to regulate autophagy via ubiquitination of BECN1 in a kinase activity-independent manner [Citation35] ()). Mechanistically, LATS1 interacts with and stabilizes BECN1 at the protein level by promoting K27-linked polyubiquitination of BECN1 at lysine residues K32 and K263. This type of ubiquitination results into BECN1 homodimer formation and thus in a repression of its autophagic activity. This autophagy-repressing function mediated by LATS1 is of medical importance, since it restricts therapy-induced autophagic cell death in cancer cells and thus contributes to the development of therapy resistance. Interestingly, an autophagy-regulatory role of LATS homologues has been also reported in fly and worms. While mutation of fly LATS (Warts) promotes autophagic cell death during salivary gland development [Citation50], RNAi-mediated depletion of Warts in worms leads to degradation of p62 [Citation51], suggesting evolutionary conserved, yet context-dependent roles of LATS in autophagy regulation. In addition, a kinase-dependent role of LATS1 has been reported recently in inhibiting mTOR complex 1 activity by directly phosphorylating Raptor at Ser606 [Citation52]. Supporting this notion, knock-in mice carrying a phospho-mimetic Raptor-S606D mutation exhibit smaller livers and hearts as a consequence of impaired mTORC1 activation, highlighting a cross-talk between Hippo signaling and mTORC1 activity in organ size control. Together with a previous report showing that YAP regulates mTORC1 by transcriptional upregulating PTEN-repressing miR-29 [Citation25], these studies indicate a potential role of Hippo signaling in fine-tuning mTORC1-instructed autophagy regulation.
In line with these observations, NDR1, a protein kinase closely related to LATS1/2 [Citation53], can also interact with BECN1 and promote survival autophagy in response to starvation [Citation54]. Furthermore, NDR1 phosphorylates and activates nuclear exporter XPO1 and promotes XPO1-dependent nuclear export of BECN1 and YAP1 [Citation55]. Likewise, NDR1 has been reported to inhibit chaperone-assisted selective autophagy (CASA) by directly interacting with BAG3. Here, the physical binding of NDR1 to BAG3 competes with the interaction of BAG3 with the CASA core promoting components HSPB8 and SYNPO2 [Citation56].
Depending on its molecular pathway activation and its extent, autophagy activation can lead to two diametrically opposite cell fates, namely cell survival or cell death. In fact, Hippo signaling-mediated autophagy regulation impacts on these two cell fates in a cell context-dependent manner. Of note, ferroptosis has been reported to mediate autophagic cell death under nutrient stress [Citation57]. Emerging evidence suggests that Hippo signaling also regulates ferroptosis induced by cell-cell interactions as well as by responses to therapeutic stress [Citation15,Citation17]. The molecular interaction between ferroptosis and Hippo-mediated autophagy regulation thus warrants further investigations, in particular during stress responses induced by reactive oxygen species (ROS) and lipid peroxidation.
Concluding remarks
Divergent nutrient and stress-induced inputs collaboratively dictate cell fate and cell plasticity during normal organ development but also in various pathological states. Recent experimental insights have highlighted thus far unknown connections between autophagy and Hippo signaling in response to a variety of stresses. Current studies pinpoint the negative regulation of the oncogenic Hippo transcriptional effector YAP by phosphorylation-induced de-activation or via protein degradation, opening new potential therapeutic avenues to combat YAP-hyperactivated tumors. On the other hand, the Hippo-mediated regulation of autophagy seems to exert a critical role in demarcating cell context-dependent outcomes, either survival autophagy or lethal autophagy, thus representing a double-edged sword in regulating autophagy in development and tumorigenesis. Hence, for the therapeutic targeting of the Hippo pathway we need to fully understand the biology underlying its various regulatory functions in different pathologies and cell contexts. Specifically, a systemic and comprehensive exploration of the molecular switches controlling divergent outcomes of autophagy is warranted to functionally delineate the interactions between Hippo signaling and autophagy in tissue homeostasis and during the response to cancer therapies.
Acknowledgments
The work described here was supported by the European Research Council (ERC) Synergy Project MERiC and the Swiss National Science Foundation Sinergia project MERiC. We apologize to all colleagues whose important work could not be cited due to space constraints.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Stanger BZ. Organ size determination and the limits of regulation. Cell Cycle. 2008;7(3):318–324.
- Zhu J, Thompson CB. Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol. 2019;20(7):436–450.
- Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330(6009):1344–1348.
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–2873.
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–131.
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401.
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364.
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11(9):709–730.
- Yu F-X, Zhao B, Guan K-L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163(4):811–828.
- Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13:246.
- Calses PC, Crawford JJ, Lill JR, et al. Hippo pathway in cancer: aberrant regulation and therapeutic opportunities. Trends Cancer. 2019;5(5):297–307.
- Lin L, Sabnis AJ, Chan E, et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet. 2015;47:250.
- Kim MH, Kim J, Hong H, et al. Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. Embo J. 2016;35(5):462–478.
- Moya IM, Halder G. Hippo–YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol. 2019;20(4):211–226.
- Wu J, Minikes AM, Gao M, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2–YAP signalling. Nature. 2019;572(7769):402–406.
- Diepenbruck M, Christofori G. Epithelial–mesenchymal transition (EMT) and metastasis: yes, no, maybe? Curr Opin Cell Biol. 2016;43:7–13.
- Yang W-H, Ding C-KC, Sun T, et al. The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. 2019;28(10):2501–8.e4.
- Ni X, Tao J, Barbi J, et al. YAP is essential for Treg-mediated suppression of antitumor immunity. Cancer Discov. 2018;8(8):1026–1043.
- Moroishi T, Hayashi T, Pan -W-W, et al. The Hippo pathway Kinases LATS1/2 suppress cancer immunity. Cell. 2016;167(6):1525–39.e17.
- Tumaneng K, Russell Ryan C, Guan K-L. Organ size control by Hippo and TOR pathways. Curr Biol. 2012;22(9):R368–R79.
- Wang W, Xiao Z-D, Li X, et al. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol. 2015;17(4):490–499.
- Mo J-S, Meng Z, Kim YC, et al. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat Cell Biol. 2015;17(4):500–510.
- DeRan M, Yang J, Shen C-H, et al. Energy stress regulates Hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep. 2014;9(2):495–503.
- Hansen CG, Ng YLD, Lam W-LM, et al. The Hippo pathway effectors YAP and TAZ promote cell growth by modulating amino acid signaling to mTORC1. Cell Res. 2015;25(12):1299–1313.
- Tumaneng K, Schlegelmilch K, Russell RC, et al. YAP mediates crosstalk between the Hippo and PI(3)K–TOR pathways by suppressing PTEN via miR-29. Nat Cell Biol. 2012;14(12):1322–1329.
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13(9):1016–1023.
- Inoki K, Ouyang H, Zhu T, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126(5):955–968.
- Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–226.
- Inoki K, Zhu T, Guan K-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590.
- Kim J, Kundu M, Viollet B, et al. and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141.
- Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein Kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–461.
- Kim J, Kim Young C, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152(1):290–303.
- Lee YA, Noon LA, Akat KM, et al. Autophagy is a gatekeeper of hepatic differentiation and carcinogenesis by controlling the degradation of Yap. Nat Commun. 2018;9(1):4962.
- Liang N, Zhang C, Dill P, et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J Exp Med. 2014;211(11):2249–2263.
- Tang F, Gao R, Jeevan-Raj B, et al. LATS1 but not LATS2 represses autophagy by a kinase-independent scaffold function. Nat Commun. 2019;10(1):5755.
- Wang P, Gong Y, Guo T, et al. Activation of Aurora A kinase increases YAP stability via blockage of autophagy. Cell Death Dis. 2019;10(6):432.
- Tyra LK, Nandi N, Tracy C, et al. Yorkie growth-promoting activity is limited by Atg1-mediated phosphorylation. Dev Cell. 2020;52(5):605–16.e7.
- Zhao B, Wei X, Li W, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21(21):2747–2761.
- Totaro A, Zhuang Q, Panciera T, et al. Cell phenotypic plasticity requires autophagic flux driven by YAP/TAZ mechanotransduction. Proc Nat Acad Sci. 2019;116(36):17848–17857.
- Pavel M, Renna M, Park SJ, et al. Contact inhibition controls cell survival and proliferation via YAP/TAZ-autophagy axis. Nat Commun. 2018;9(1):2961.
- Song Q, Mao B, Cheng J, et al. YAP enhances autophagic flux to promote breast cancer cell survival in response to nutrient deprivation. Plos One. 2015;10(3):e0120790.
- Wilkinson Deepti S, Jariwala Jinel S, Anderson E, et al. Phosphorylation of LC3 by the Hippo Kinases STK3/STK4 is essential for autophagy. Mol Cell. 2015;57(1):55–68.
- Tang F, Zhang L, Xue G, et al. hMOB3 modulates MST1 apoptotic signaling and supports tumor growth in glioblastoma multiforme. Cancer Res. 2014;74(14):3779–3789.
- Maejima Y, Kyoi S, Zhai P, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19:1478.
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1–VPS34 complex – at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20(6):355–362.
- Nakamura M, Zhai P, Del Re DP, et al. Mst1-mediated phosphorylation of Bcl-xL is required for myocardial reperfusion injury. JCI Insight. 2016;1(5). DOI:https://doi.org/10.1172/jci.insight.86217
- Boutouja F, Brinkmeier R, Mastalski T, et al. Regulation of the tumor-suppressor BECLIN 1 by distinct ubiquitination cascades. Int J Mol Sci. 2017;18(12):2541.
- Furth N, Aylon Y. The LATS1 and LATS2 tumor suppressors: beyond the Hippo pathway. Cell Death Differ. 2017;24:1488.
- Aylon Y, Gershoni A, Rotkopf R, et al. The LATS2 tumor suppressor inhibits SREBP and suppresses hepatic cholesterol accumulation. Genes Dev. 2016;30(7):786–797.
- Dutta S, Baehrecke EH. Warts is required for PI3K-regulated growth arrest, autophagy, and autophagic cell death in drosophila. Curr Biol. 2008;18(19):1466–1475.
- Guo B, Huang X, Zhang P, et al. Genome-wide screen identifies signaling pathways that regulate autophagy during Caenorhabditis elegans development. EMBO Rep. 2014;15(6):705–713.
- Gan W, Dai X, Dai X, et al. LATS suppresses mTORC1 activity to directly coordinate Hippo and mTORC1 pathways in growth control. Nat Cell Biol. 2020;22:246–256.
- Tang F, Gill J, Ficht X, et al. The kinases NDR1/2 act downstream of the Hippo homolog MST1 to mediate both egress of thymocytes from the thymus and lymphocyte motility. Sci Signal. 2015;8(397):ra100–ra.
- Joffre C, Dupont N, Hoa L, et al. The pro-apoptotic STK38 Kinase is a new Beclin1 partner positively regulating autophagy. Curr Biol. 2015;25(19):2479–2492.
- Martin AP, Jacquemyn M, Lipecka J, et al. STK38 kinase acts as XPO1 gatekeeper regulating the nuclear export of autophagy proteins and other cargoes. EMBO Rep. 2019;20(11):e48150.
- Klimek C, Jahnke R, Wördehoff J, et al. The Hippo network kinase STK38 contributes to protein homeostasis by inhibiting BAG3-mediated autophagy. Biochim Biophys Acta Mol Cell Res. 2019;1866(10):1556–1566.
- Liu J, Kuang F, Kroemer G, et al. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. 2020;27(4):420–435.