
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
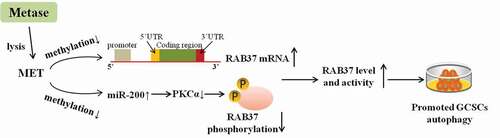
This study focused on the role of methionine (MET) in the autophagy of gastric cancer stem cells (GCSCs) and aims to elaborate its regulatory mechanism.
In the present study, the GCSCs were isolated from human gastric cancer cell lines using an anti-CD44 antibody, and then cultured in MET+ homocysteine (HCY)− or MET−HCY+ medium.
In MET+HCY–treated GCSCs, autophagy was suppressed, the methylation and phosphorylation of RAB37 were elevated, and miR-200b expression was down-regulated. Lentiviral vector (LV-) carrying methionine-γ lyase (an enzyme that could specifically lyse MET; Metase) promoted autophagy, reduced the methylation and phosphorylation of RAB37, and up-regulated miR-200b expression in MET+HCY–-treated GCSCs. Then, we found that miR-200b suppressed the expression of protein kinase C α (PKCα), a protein that could inactivate RAB37 through promoting its phosphorylation. LV-Metase down-regulated RAB37 phosphorylation via miR-200b/PKCα, thus promoting the RAB37-mediated autophagy and suppressing cell viability in MET+HCY–treated GCSCs. Finally, the in vivo study proved that LV-Metase treatment inhibited tumor growth through up-regulating RAB37 expression.
In conclusion, MET suppressed RAB37 expression via enhancing its methylation and suppressed RAB37 activity via miR-200b/PKCα axis, thus repressing RAB37-mediated autophagy in GCSCs. The supplementation of Metase lysed MET, thus inducing the autophagy of GCSCs and inhibiting tumor growth.
GRAPHICAL ABSTRACT
Introduction
Cancer stem cells (CSCs) are a subtype of cells in the tumor cell population with the characteristics of self-renewal and tumor-initiating [Citation1]. Currently, gastric cancer stem cells (GCSCs) are regarded as the major cause of the invasion, metastasis, and resistance to chemotherapy of gastric cancer [Citation2].
Autophagy, a process of resolving and recycling proteins and damaged cellular organs, has been considered to play a dual role in tumor formation and progression [Citation3,Citation4]. Recent evidence suggests that autophagy contributes to stemness loss of CSCs [Citation5]. Besides, it has been proved that autophagy reduces the chemoresistance of GCSCs via the Notch signaling pathway [Citation6], confirming that autophagy could exert its antitumor effect on gastric cancer by targeting GCSCs.
Methionine (MET) is one of the indispensable amino acids in the body. Cao et al. [Citation7] found that human primary gastric cancer cells were MET-dependence, a MET-free environment strengthened the effect of chemotherapy on gastric cancer cells. Besides, methionine-γ lyase (Metase), an enzyme that could specifically lyse MET, suppressed the proliferation of GCSCs through lysing MET in cellular supernatant [Citation8], indicating that MET is a promoter for GCSCs proliferation and Metase exhibits its anti-tumor effect via lysing MET. In addition, through the catalysis of MET adenosine transferase, MET converts to S-adenosyl methionine (SAM) directly and provides methyl units for a variety of reactions such as methylation of proteins, DNA and RNA [Citation9]. Recently, MET has been shown to be involved in the regulation of autophagy by promoting the methylation process. Sutter et al. [Citation10] reported that in yeast cells, MET induced the methylation of protein phosphatase 2A through synthesizing SAM, thus inhibiting autophagy and promoting cell growth. While, whether MET inducing GCSCs proliferation via regulating autophagy is still unknown.
RAB37 is a small GTPase protein and responsible for intracellular signal transduction and vesicle trafficking. Recent research revealed that RAB37 directly bound to autophagy-related gene 5 (ATG5) and promoted autophagosome formation in a GTP-dependent manner, thus inducing autophagy and suppressing tumor growth [Citation11]. The expression and activity of RAB37 can be regulated by many pathways. In nasopharyngeal carcinoma and lung cancer, RAB37 has been proved to be down-regulated by the hypermethylation of its promoter [Citation12,Citation13]. While whether MET plays a role in the hypermethylation of RAB37 remains to be further explored. Meanwhile, as a GTPase protein, RAB37 was inactive when bound to GDP and switch to an active form when bound to GTP [Citation14]. Tzeng et al. [Citation15] reported that protein kinase C α (PKCα) phosphorylated RAB37 on threonine-172, abrogates RAB37 GTP binding site, thus repressing the activity of RAB37. An online bioinformatics database (starbase) forecasted there was an interplay between the mRNA of PKCα and miR-200b, a miRNA that has been proved to be hypermethylated under high glucose environment and its hypermethylation can be regulated by SAM [Citation16], hinting that MET could mediate the activity of RAB37 via miR-200b/PKCα axis.
Herein in this study, we explored the role of MET in GCSCs autophagy and elaborated on the regulatory mechanism of MET on RAB37 in the light of methylation and phosphorylation, aiming to get new insight into the pathogenesis of gastric cancer.
Materials and methods
Cell culture and GCSCs isolation
Human gastric cancer cell lines, BGC-823 and SGC-7901 (Cell Bank of Chinese Academy of Sciences, China) were cultured in RPMI 1640 medium (Gibco, USA) containing 10% fetal bovine serum (FBS; Gibco, USA) at 37°C with 5% CO2.
For the isolation of GCSCs, the BGC-823 and SGC-7901 cells were cultured in a conditioned medium as previously reported [Citation17]. Seven days later, the BGC-823 and SGC-7901 formed sphere-like cell aggregates. Then, cells were harvested, stained with anti-CD44-FITC (BD Biosciences, USA) at 4°C for 1 hour, and sorted by BD FACSAria III cell sorter. After sorting, the CD44− and CD44+ BGC-823 and SGC-7901 cells were cultured in the MET-free medium, and the expression levels of miR-200b, RAB37, PKCα, microtubule-associated protein light chain 3 (LC3) I, LC3II, and P62 were measured.
To explore the effect of MET on GCSCs, CD44+ BGC-823 and CD44+ SGC-7901 cells were cultured in MET− homocysteine (HCY, 100 μM)+ or MET(100 μM)+HCY− medium. Ninety-six hours later, the cells were harvested for the following tests.
Cell transfection
When the cells were cultured to 80% confluence, cells were transfected with RNAi-vector (si-PKCα) or micro RNA inhibitor (miR-200b inhibitor) or micro RNA mimic (miR-200b mimic) or their relative negative controls (si-RNC, NC inhibitor, and NC mimic) using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s instructions.
To supplement Metase, the lentivirus expressed Metase (LV-Metase) and its negative control (LV-control) were synthesized by Ribobio (China). The appropriate volume of virus particles was calculated by the multiplicity of infection (MOI) and added in the cell culture medium.
Quantitative RT-PCR
Total RNAs were isolated from cells using an RNA Isolation Kit (Merck, USA). Complementary DNA was obtained with a One-step RNA Reverse Transcription kit (Hai Gene, China). Quantitative Real-time PCR was performed in triplicate with SYBR Premix Ex Taq II kit (TaKaRa, USA). Gene expressions were calculated by the 2−∆∆CT method and the relative expressions of them were normalized to β-actin or U6.
Western blot
The determination of protein levels of PKCα, P-RAB37, RAB37, LC3I, LC3II, P62 were done by western blot with total protein purified from cell lysate or tumor xenografts by RIPA lysis buffer (Cwbio, China). Proteins were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to PDVF membrane (ThermoFisher Scientific, USA). After being blocked with 5% bull serum albumin for 30 min, membranes were incubated with primary antibody (against PKCα, P-RAB37, RAB37, LC3B, and P62, all purchased from Abcam, UK). After 12 h, the membranes were incubated with the secondary antibodies (ab205718, Abcam, UK). Immunoblots were visualized in IBright FL1500 Intelligent Imaging System (ThermoFisher, USA) and GAPDH was used as an internal control.
Cell viability assay
The cell viability was assessed by methyl thiazolyl tetrazolium (MTT) assay. Briefly, the cells of each well were incubated with 10 μl MTT (Cwbio, China) for 4 h at 37°C in the dark. Then, the supernatant was removed and formazan crystals were dissolved by dimethyl sulfoxide (DMSO, 150 µl/well) at 37°C for 15 min. The absorbance intensity was measured at 490 nm and the cell viability (%) = (mean absorbance in test wells)/(mean absorbance in control wells) ×100.
Methylation-specific PCR
For methylation-specific PCR (MSP), genomic DNA was isolated from CD44+ BGC-823 and SGC-7901 cells using PureLink™ Genomic DNA Mini Kit (Thermo Fisher, USA). 2 μg of genomic DNA was modified by bisulfite using Bisulfite DNA Modification Kit (Yantuo biotechnology co. LTD, China). Primers specific for the unmethylated sequence (U) and primers specific for the methylated sequence (M) were designed near the promoter region of RAB37 and miR-200b, and the bisulfite-modified genomic DNA was used as the template for PCR analysis.
Quantification of apoptosis
CD44+ BGC-823 and SGC-7901 cells were seeded in 6-well plates at 4105 cells/well. When the cells were cultured to 80% confluence, cells were collected and washed with PBS. Apoptosis was measured using the Annexin V-FITC Apoptosis Detection Kit (Univ-bio, China). In brief, cells were resuspended in 300 μl Binding Buffer and incubated with 5 μl Annexin V-FITC for 15 min at room temperature in the dark. Then, 5 μl Propidium Iodide Staining Solution was added to the cells. Five minutes later, cell apoptosis was measured by the CytoFLEX flow cytometer (Beckman Coulter) immediately.
Dual-luciferase reporter gene assay
To verify the combination of miR-200b and PKCα, the sequences of PKCα were amplified and inserted into pGL3-basic plasmids. 0.5 μg plasmid and 20 nM miR-200b mimic or its negative control (NC mimic) were co-transfected in well-grown 293 T cells by using lipofectamine 2000 (ThermoFisher, USA). Forty-eight hours after transfection, the cells were lysed and the activities of Renilla luciferase and firefly luciferase were measured with Dual-luciferase Reporter Assay Kit (Promega, China) following the manufacturer’s protocol.
Mouse xenograft models
BALB/c-nu/nu nude mice (4–6 weeks old, female) were purchased from Laboratory Animal Resources, Chinese Academy of Sciences (Beijing, China). For the establishment of the xenograft model, 32 mice were randomly divided into 4 groups. In the LV-Metase group (n = 8) or LV-control group (n = 8), CD44+BGC-823 cells were infected with LV-Metase or LV-control. Then, 5 × 106 cells were diluted in 200 μl of RPMI-1640 and injected subcutaneously into mice. In the LV-Metase+LV-sh-RAB37 group (n = 8) or LV-Metase+LV-sh-RNA (the negative control of -sh-RAB37) group (n = 8), CD44+BGC-823 cells were infected with LV-Metase+LV-sh-RAB37 or LV-Metase+LV-sh-RNA. Then, 5 × 106 cells were diluted in 200 μl of RPMI-1640 and injected subcutaneously into the same position of mice. The tumor volumes of mice were measured at 5-day intervals. After 35 days, the mice were sacrificed and the tumor xenografts were collected for follow-up experiments. All procedures during the experiment were approved by the Ethical Committee of The Second Affiliated Hospital of Nanchang University.
Statistical analysis
Experimental results were expressed as mean ± standard deviation (SD) and analyzed using GraphPad 7.0 Prism. The differences were analyzed by Student t-test . Results were considered statistically significant when P < 0.05.
Results
Autophagy was elevated in GCSCs
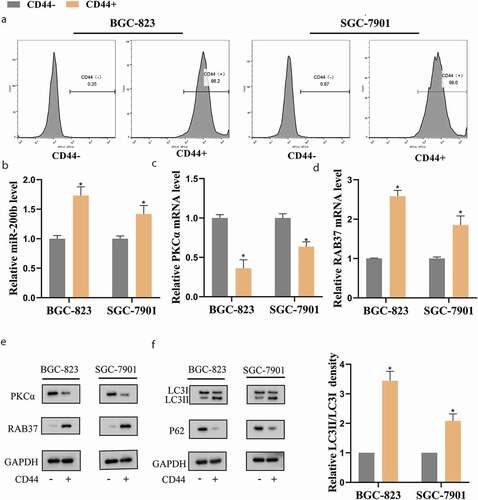
Firstly, GCSCs were isolated from human gastric cancer cell lines (BGC-823 and SGC-7901) using an antibody against CD44, a surface marker of stem cells [Citation18] ()). Subsequently, the CD44− and CD44+ cells were separated. As shown in , compared with CD44− BGC-823 and CD44− SGC-7901 cells, the expressions of miR-200b and RAB37 were increased while the expression of PKCα was decreased in CD44+ BGC-823 and CD44+ SGC-7901 cells. Besides, the expression of P62 (a selective substrate for autophagy [Citation19]) was decreased and the LC3II/LC3I ratio was up-regulated ()) in CD44+ cells, indicating an increase of autophagy in GCSCs.
Figure 1. Autophagy was elevated in gastric cancer stem cells (GCSCs)
MET increased the methylation and phosphorylation of RAB37 and suppressed autophagy in GCSCs
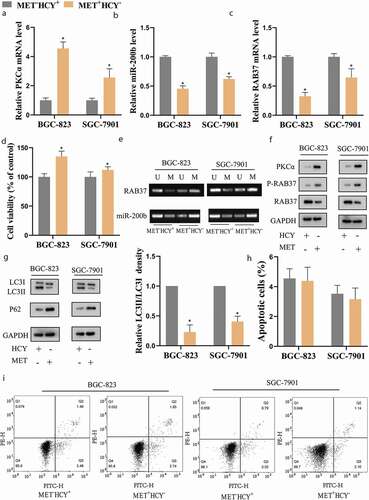
To explore the effect of MET on GSCSs, the CD44+ BGC-823 and CD44+SGC-7901 (hereinafter referred to as BGC-823 and SGC-7901) were cultured in MET−HCY+ or MET+HCY− medium. The methylation level of RAB37 and miR-200b, measured by MSP, showing that MET supplementation increased the methylation level of RAB37 and miR-200b ()), resulted in clear declines in their expressions () in BGC-823 and SGC-7901 cells. Besides, MET supplementation up-regulated the expressions of PKCα ((a,f)) and P-RAB37 ((f)) in BGC-823 and SGC-7901 cells. Meanwhile, compared with MET−HCY+-treated GCSCs, MET+HCY–-treated GCSCs exerted higher cell viability ()), lower autophagy ()), while the cell apoptosis didn’t change (,i)). Further experiments showed that the protective effect of MET on cell viability could be eliminated by autophagy activator rapamycin (Supplemental Figure 1b–d), indicating that MET preserved cell viability of GCSCs by suppressing autophagy.
Figure 2. MET increased the methylation and phosphorylation of RAB37 and suppressed autophagy in GCSCs
Metase supplementation reduced the methylation and phosphorylation of RAB37 and promoted autophagy in GCSCs
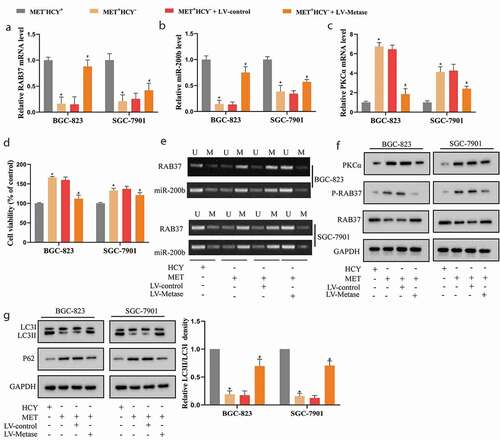
To further confirm the effect of MET on GCSCs, we overexpressed Metase (an enzyme that could specifically lyse MET) in BGC-823 and SGC-7901 cells by LV-Metase transfection. As shown in (a,b,e,f), Metase supplementation restored the expression of RAB37 and miR-200b via repressing their promoter methylation. What’s more, Metase supplementation down-regulated the expressions of PKCα (,f)) and P-RAB37 ()). Then, compared with LV-control, LV-Metase effectively elevated LC3II/LC3 ratio in MET+HCY–-treated GCSCs ()), indicating that LV-Metase reversed the inhibitory effect of MET on autophagy of GCSCs, thus reducing the cell viability of GCSCs in the presence of MET (Figure 3(d)).
Figure 3. Metase supplementation reduced the methylation and phosphorylation of RAB37 and promoted autophagy in GCSCs
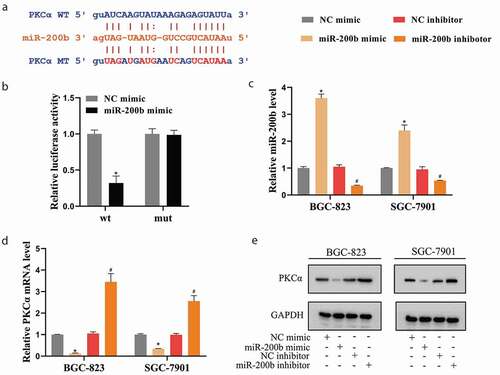
miR-200b negatively regulated PKCα expression in GCSCs
An online bioinformatics database (starbase) predicted there were several potential binding sites between miR-200b and PKCα ()). The following Dual-luciferase Reporter Assays showed that miR-200b mimic significantly reduced the luciferase activity of PKCα-wt, while neither miR-200b mimic nor NC mimic affected the luciferase activity of PKCα-mut ()). Furthermore, miR-200b was overexpressed or silenced in GCSCs utilizing the transfections of miR-200b mimic or miR-200b inhibitor, respectively ()). The results of ,e) depicted that the expression of PKCα was down-regulated by miR-200b mimic and up-regulated by miR-200b inhibitor.
Figure 4. miR-200b negatively regulated PKCα expression in GCSCs
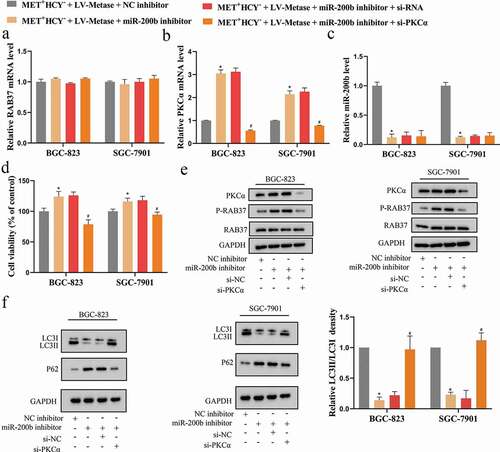
miR-200b/PKCα axis was involved in the promoting effect of Metase on autophagy of GCSCs by regulating RAB37 phosphorylation
In the previous experiments, we found that the supplementation of Metase elevated autophagy of GCSCs ()), to explore whether miR-200b and PKCα took part in this process, a series of follow-up experiments were carried out. The BGC-823 and SGC-7901 cells were transfected with LV-Metase+miR-200b inhibitor or LV-Metase+miR-200b inhibitor+si-PKCα or LV-Metase+corresponding controls (NC inhibitor or si-RNA) and then all cells were cultured in MET+HCY− medium. The transfection of miR-200b inhibitor effectively silenced miR-200b expression ()) and increased PKCα expression (,e)). The transfection of si-PKCα effectively silenced PKCα expression (,e)). While neither the miR-200b inhibitor nor si-PKCα could affect RAB37 expression (,e)). Furthermore, si-PKCα down-regulated P-RAB37 expression in the presence of miR-200b inhibitor ()), thus restoring the RAB37 activity, eliminating the inhibitory effect of the miR-200b inhibitor on autophagy ()), and removing the promoting effect of the miR-200b inhibitor on cell viability (). These above data indicating that miR-200b suppressed the phosphorylation of RAB37 via negatively regulated PKCα expression, thus inducing the autophagy of GCSCs. Considering that Metase could up-regulate miR-200b expression by reducing its methylation ()), we concluded that Metase suppressed the phosphorylation level of RAB37 via miR-200b/PKCα axis, thus promoting the RAB37-mediated autophagy of GCSCs.
Figure 5. miR-200b/PKCα axis was involved in the promoting effect of Metase on autophagy of GCSCs by regulating the phosphorylation level of RAB37
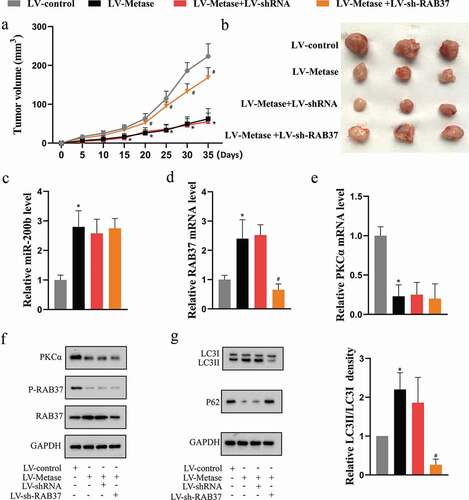
Metase suppressed tumor growth by enhancing the expression and activity of RAB37 in vivo
We further validated our in vitro findings in the xenograft mouse model. As the results showed, on the one hand, Metase supplementation up-regulated the miR-200b expression ()) and down-regulated the PKCα expression (,f)), thus reducing the phosphorylation level of RAB37 ()), on the other hand, Metase supplementation could directly elevate RAB37 expression (,f)). What’s more, the LV-Metase significantly increased autophagy ()), resulting in a decrease in tumor volume () without affecting body weight in mice (P > 0.05; Supplemental Figure 1(a)). On the contrary, the RAB37 slicing abolished thepromoting effect of LV-Metase on autophagy ()) and the inhibitory effect of LV-Metase on tumor growth ()), indicating that the anti-tumor effect of Metase depended on RAB37-mediated autophagy.
Figure 6. Metase suppressed tumor growth by enhancing the expression and activity of RAB37 in vivo.
Discussion
Compelling evidences demonstrate GCSCs as a strong driving force of tumorigenesis and a key mechanism of therapeutic resistance [Citation20]. Therefore, elucidating the molecular mechanism regulating the stemness of GCSCs is of great significance for the clinical treatment of gastric cancer. The present study, performed in GCSCs (isolated from human gastric cancer cell lines) and the xenograft mouse model, discovered that MET suppressed the autophagy of GCSCs via suppressing the expression and activity of RAB37, while the supplementation of Metase could inducing the autophagy of GCSCs and inhibited the tumor growth via lysing MET.
MET is an essential amino acid required by all cells. HCY is the direct precursor of MET [Citation7]. In vitro, MET and HCY could be converted into each other. Normal cells can grow well in the environment containing HCY instead of MET. However, in MET−HCY+-cultured tumor cells, though MET could be synthesized from HCY by an active MET synthase, the MET level not adequate to both sustain growth and meet their high transmethylation requirements, resulting in the suppression of cell proliferation [Citation21,Citation22]. This phenomenon is called MET-dependence. Several researchers used MET−HCY+ as the control of MET+HCY− medium in vitro [Citation7,Citation21,Citation22]. In the present study, compared with MET−HCY+ medium-cultured GCSCs, the MET+HCY− medium-cultured GCSCs exhibited higher cell viability and reduced autophagy (,g)), indicating that GCSCs was MET-dependence. So we wondered whether LV-Metase could inhibit GCSCs viability through lysing MET. As expected, LV-Metase suppressed cell viability via inducing autophagy in MET+HCY− medium-treated GCSCs ()) and inhibited the tumor growth in the xenograft mouse model ((a)). In line with our study, Kreis et al. [Citation23] found that Metase supplementation could effectively suppress the Walker-256 sarcoma tumor growing in rats. These data confirming the potential anti-tumor effects of Metase on several types of cancer. Then, the following experiments revealed that the inhibitory effect of Metase on cell viability relied on its regulatory effect on the methylation and phosphorylation of RAB37.
According to the latest research, RAB37 function as an organizer for autophagosome biogenesis by interacting with ATG5 [Citation24], indicating that RAB37 may take part in the regulation of cancer progression via promoting autophagy. As an enzyme, the expression and activity of RAB37 are essential for the maintenance of its normal function. In our study, we found that MET directly reduced RAB37 expression via enhancing its methylation ()) and indirectly reduced RAB37 activity via regulating the miR-200b/PKCα axis () and ), thus suppressing RAB37-mediated autophagy and promoting cell viability of GCSCs (). Through elevating RAB37 expression and reducing RAB37 phosphorylation, Metase efficiently inhibited the growth of GCSCs in vivo (). In the past, researchers investigated the anti-tumor effect of RAB37 from the perspective of regulating vesicular transport [Citation25,Citation26]. Herein in our study, we firstly reported the inhibitory effect of RAB37 on tumor growth via promoting the autophagy of GCSCs.
Several studies have indicated miR-200b as a tumor suppressor in gastric cancer. For example, Kurashige et al. [Citation27] demonstrate that miR-200b repressed cell proliferation, invasion, and migration by targeting E-box-binding homeobox 2 in gastric carcinoma. Tang et al. [Citation28] reported that the low miR-200b expression was a marker of poor gastric cancer prognosis. Besides, Zhao et al. [Citation29] found that autophagy was activated by miR-200b inhibitor in the rat cardiac fibroblast, indicating miR-200b could function as a regulator of autophagy. However, whether miR-200b could regulate the progression of gastric cancer via modulating autophagy is still unknown. In the present study, we found that the miR-200b inhibitor suppressing RAB37-mediated autophagy via promoting PKCα-induced RAB37 phosphorylation, thus eliminating the inhibitory effect of Metase on cell viability of GCSCs ( and ). In line with our study, Liu et al [Citation30] also reported that miR-200b overexpression inhibited the stemness properties and division of the glioma stem cells, suggesting that miR-200b may be a promising target for modulating stemness of cancer stem cells.
In conclusion, the current study revealed that MET suppressed the autophagy of GCSCs through RAB37, and elucidated the regulation effect of MET on RAB37 from two aspects: on the one hand, MET down-regulated RAB37 expression by promoting its methylation, on the other hand, MET could inhibit the activity of RAB37 via miR-200b/PKCα axis. Besides, our study clarified that the supplementation of Metase could induce the autophagy of GCSCs via lysing MET, providing novel perspectives for regulating the stemness of GCSC and the clinical treatment of gastric cancer.
Supplemental Material
Download Zip (757.1 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23(10):1124–1134.
- Ni SJ, Zhao L-Q, Wang X-F, et al. CBX7 regulates stem cell-like properties of gastric cancer cells via p16 and AKT-NF-kappaB-miR-21 pathways. J Hematol Oncol. 2018;11(1):17.
- Eritja N, Chen B-J, Rodríguez-Barrueco R, et al. Autophagy orchestrates adaptive responses to targeted therapy in endometrial cancer. Autophagy. 2017;13(3):608–624.
- Zhang H, McCarty N. Tampering with cancer chemoresistance by targeting the TGM2-IL6-autophagy regulatory network. Autophagy. 2017;13(3):627–628.
- Lei Y, Zhang D, Yu J, et al. Targeting autophagy in cancer stem cells as an anticancer therapy. Cancer Lett. 2017;393:33–39.
- Li LQ, Pan D, Zhang S-W, et al. Autophagy regulates chemoresistance of gastric cancer stem cells via the Notch signaling pathway. Eur Rev Med Pharmacol Sci. 2018;22(11):3402–3407.
- Cao WX, Ou J-M, Fei X-F, et al. Methionine-dependence and combination chemotherapy on human gastric cancer cells in vitro. World J Gastroenterol. 2002;8(2):230–232.
- Li YF, Zhang HT, Xin L. Hyaluronic acid-modified polyamidoamine dendrimer G5-entrapped gold nanoparticles delivering METase gene inhibits gastric tumor growth via targeting CD44+ gastric cancer cells. J Cancer Res Clin Oncol. 2018;144(8):1463–1473.
- Bauerle MR, Schwalm EL, Booker SJ. Mechanistic diversity of radical S-adenosylmethionine (SAM)-dependent methylation. J Biol Chem. 2015;290(7):3995–4002.
- Sutter BM, Wu X, Laxman S, et al. Methionine inhibits autophagy and promotes growth by inducing the SAM-responsive methylation of PP2A. Cell. 2013;154(2):403–415.
- Sheng Y, Song Y, Li Z, et al. RAB37 interacts directly with ATG5 and promotes autophagosome formation via regulating ATG5-12-16 complex assembly. Cell Death Differ. 2018;25(5):918–934.
- Wu CY, Tseng R-C, Hsu H-S, et al. Frequent down-regulation of hRAB37 in metastatic tumor by genetic and epigenetic mechanisms in lung cancer. Lung Cancer. 2009;63(3):360–367.
- Li Y, Yang X, Du X, et al. RAB37 Hypermethylation Regulates Metastasis and Resistance to Docetaxel-Based Induction Chemotherapy in Nasopharyngeal Carcinoma. Clin Cancer Res. 2018;24(24):6495–6508.
- Pfeffer SR. Structural clues to Rab GTPase functional diversity. J Biol Chem. 2005;280(16):15485–15488.
- Tzeng H-T, Li T-H, Tang Y-A, et al. Phosphorylation of Rab37 by protein kinase C alpha inhibits the exocytosis function and metastasis suppression activity of Rab37. Oncotarget. 2017;8(65):108556–108570.
- Singh K, Pal D, Sinha M, et al. Epigenetic Modification of MicroRNA-200b Contributes to Diabetic Vasculopathy. Mol Ther. 2017;25(12):2689–2704.
- Xin L, Liu L, Liu C, et al. DNA-methylation-mediated silencing of miR-7-5p promotes gastric cancer stem cell invasion via increasing Smo and Hes1. J Cell Physiol. 2020;235(3):2643–2654.
- Yan Y, Zuo X, Wei D. Concise Review: emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl Med. 2015;4(9):1033–1043.
- Ichimura Y, Kominami E, Tanaka K, et al. Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy. 2008;4(8):1063–1066.
- Toh TB, Lim JJ, Chow EK-H. Epigenetics in cancer stem cells. Mol Cancer. 2017;16(1):29.
- Cellarier E, Durando X, Vasson MP, et al. Methionine dependency and cancer treatment. Cancer Treat Rev. 2003;29(6):489–499.
- Booher K, Lin D-W, Borrego SL, et al. Downregulation of Cdc6 and pre-replication complexes in response to methionine stress in breast cancer cells. Cell Cycle. 2012;11(23):4414–4423.
- Kreis W, Hession C. Biological effects of enzymatic deprivation of L-methionine in cell culture and an experimental tumor. Cancer Res. 1973;33(8):1866–1869.
- Song Y, Shang D, Cheng H, et al. The small GTPase RAB37 functions as an organizer for autophagosome biogenesis. Autophagy. 2018;14(4):727–729.
- Tsai C-H, Cheng H-C, Wang Y-S, et al. Small GTPase Rab37 targets tissue inhibitor of metalloproteinase 1 for exocytosis and thus suppresses tumour metastasis. Nat Commun. 2014;5(1):4804.
- Wang Y-S, Tzeng H-T, Tsai C-H, et al. VAMP8, a vesicle-SNARE required for RAB37-mediated exocytosis, possesses a tumor metastasis suppressor function. Cancer Lett. 2018;437:79–88.
- Kurashige J, Kamohara H, Watanabe M, et al. MicroRNA-200b regulates cell proliferation, invasion, and migration by directly targeting ZEB2 in gastric carcinoma. Ann Surg Oncol. 2012;19(Suppl S3):S656–64.
- Tang H, Deng M, Tang Y, et al. miR-200b and miR-200c as prognostic factors and mediators of gastric cancer cell progression. Clin Cancer Res. 2013;19(20):5602–5612.
- Zhao XD, Qin RH, Yang JJ, et al. DNMT3A controls miR-200b in cardiac fibroblast autophagy and cardiac fibrosis. Inflamm Res. 2018;67(8):681–690.
- Liu A, Yu Q, Peng Z, et al. miR-200b inhibits CD133+ glioma cells by targeting the AKT pathway. Oncol Lett. 2017;13(6):4701–4707.