
ABSTRACT
Autophagy, an evolutionarily conserved mechanism that promotes cell survival by recycling nutrients and degrading long-lived proteins and dysfunctional organelles, is an important defense mechanism, and its attenuation has been well documented in senescence and aging-related diseases. Abdominal aortic aneurysm (AAA), a well-known aging-related disease, has been defined as a chronic degenerative process in the abdominal aortic wall; however, the complete mechanism is unknown, and a clinical treatment is lacking. Accumulating evidence has recently revealed that numerous drugs that can induce autophagy are effective in the treatment of AAA. The purpose of this systematic review was to focus on the cross-talk between autophagy and high-risk factors and the potential pathogenesis of AAA to understand not only the host defense and pathogenesis but also potential treatments.
KEYWORDS:
1. Introduction
Autophagy, first named by de Duve [Citation1], is an evolutionarily conserved mechanism that promotes cell survival by recycling nutrients and degrading long-lived proteins and dysfunctional organelles and is an important defense mechanism in the body [Citation2]. In mammals, macroautophagy (henceforth referred to as autophagy) plays a critical role in health and disease [Citation3]. Starting with the formation of the phagophore and ending with the destruction of the autophagosome, the autophagy pathway requires a double-membrane structure and is divided into three steps: regulation, phagophore formation, and finally cargo loading and lysosomal fusion [Citation4,Citation5]. Autophagy is induced to recycle energy to sustain metabolic processes under conditions of starvation, growth factor deficiency or high-energy demand [Citation6]. The substrates targeted by autophagy mainly include protein aggregates, damaged mitochondria, unneeded peroxisomes, excess ribosomes, the endoplasmic reticulum (ER), endosomes, lipid droplets, and intracellular pathogens [Citation7,Citation8]. The failure to control the accumulation of these types of materials in defective autophagy is related to numerous diseases, including neurodegenerative diseases [Citation9], cancers [Citation10], cardiovascular diseases [Citation11], diabetes [Citation12], inflammatory bowel disease [Citation13] and various diseases that have not been entirely confirmed.
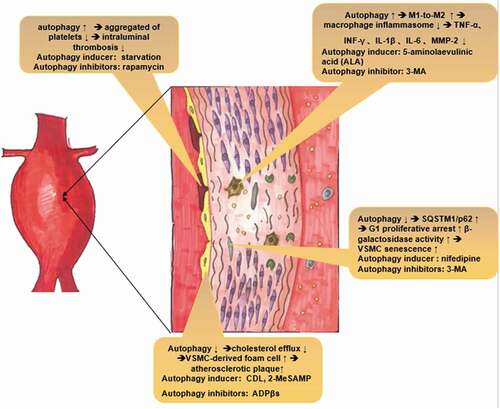
Figure 1. Potential molecular mechanism by which autophagy plays a protective role in AAA pathogenesis. Thrombosis: Enhanced autophagy could decrease intraluminal thrombosis by reducing the aggregation of platelets. Macrophages: Autophagy deficiency leads to hyperactivation of the macrophage inflammasome and enhances plaque progression, thereby releasing inflammatory cytokines, such as TNF-α, IL-1β, and MMP-2, which ultimately exacerbate AAA formation. VSMC senescence: Deficient autophagy could accumulate SQSTM1/p62 and accelerate the development of stress-induced senescence, as indicated by cellular and nuclear hypertrophy, CDKN2A-RB-mediated G1 phase arrest and senescence-associated β-galactosidase activity. Atherosclerosis: Deficient autophagy exacerbates atherosclerotic plaques by reducing cholesterol efflux and exacerbating VSMC-derived foam cell generation
The mechanism of autophagy is a complicated network of autophagy-related (ATG) proteins and ATG complexes that are involved in the induction and formation of autophagosomes [Citation14]. A full account of the extensive autophagy system is beyond the scope of this review. Briefly, under starvation conditions, ATG proteins such as Atg16L1 target phagophore assembly sites (PASs, also called preautophagosomal structures) and assemble in an orderly manner to initiate autophagy. During punctate structure formation, the protein complex, including ULK1 and FiP200, which are the most upstream molecules, is essential for the formation of the Pi3-kinase (PI3K) complex with Atg14. The Atg12-Atg5-Atg16L1 complex and microtubule-associated protein 1 light chain 3 (LC3) are downstream units. The key step in autophagosome formation is the conversion of LC3-I to LC3-II [Citation15]. LC3, the most thoroughly studied autophagy protein, is the orthologue of Atg8 in mammals, is associated with autophagosome development and maturation and is used to monitor autophagy activity [Citation16,Citation17]. The autophagosome and the lysosome merge to form autolysosomes, which are degraded and released for recycling.
Abdominal aortic aneurysm (AAA) is a focal, permanent dilatation of an artery to more than 50% of its normal diameter and is a common aging-associated disease [Citation18]. The high-risk factors for AAA include aging [Citation19], sex and smoking [Citation18]. The final outcome without intervention if rupture. Unfortunately, there is still no clinical treatment, and the cellular and molecular mechanisms underlying AAA pathogenesis are still unclear [Citation20]. In recent years, an increasing number of researchers have discovered that autophagy may play a critical role in AAA. On the one hand, the association between autophagy and the high-risk factors for AAA, including senescence, gender and cigarette smoking, has been documented [Citation21–24]; on the other hand, the ATG protein, which participates in the induction and formation of autophagosomes, has also been observed in AAA tissues [Citation25]. Furthermore, studies indicated that some drugs, such as rapamycin, could repress the pathophysiology of AAA by suppressing the activation of the mTOR pathway [Citation26]. Thus, this review aims to determine the role autophagy plays in AAA by discussing current studies demonstrating the link between AAA and autophagy.
2. Autophagy-mediated high-risk factors exacerbate AAA formation
There has been no review on the role autophagy plays in high-risk factors for AAA, as studies focused mostly on the cellular and molecular mechanisms of autophagy in AAA. To address this lack, we chose to summarize related studies that focused on high-risk factors involved in AAA, including senescence, sex and smoking.
2.1. Autophagy dysfunction in aortic wall senescence
Senescence, as the most crucial high-risk factor for AAA, is a complicated biological process that leads to progressive deterioration of vascular structure and function, mainly manifesting as vascular remodeling, endothelial defects, and the loss of vascular compliance [Citation27]. Normal and pathological aging are often associated with reduced autophagy [Citation28]. Any intervention in simple organisms resulting in the prolongation of the life span could induce an increase in autophagy, which is critical for beneficial effects on life span [Citation29]. Studies indicate that activation of antisenescence pathways mediates life span extension by inducing autophagy [Citation29]. Multiple reports indicated that ATG proteins have reduced expression in aged tissues and that autophagy diminished with aging. Additionally, defective autophagy has been shown to enhance vascular smooth muscle cell (VSMC) senescence. A previous study showed that Atg7-knockout mouse VSMCs accumulated SQSTM1/p62, which accelerated the development of stress-induced senescence, as indicated by cellular and nuclear hypertrophy, CDKN2A-RB-mediated G1 phase arrest and senescence-associated β-galactosidase (SA-β-gal) activity [Citation30]. This interesting discovery may reveal the link between autophagy and age-related changes in the vasculature. Moreover, a recent study indicated that senescence-impaired autophagic activity, which was restored by nifedipine, could alleviate HO-induced senescence in VSMCs by regulating SA-β-gal activity and the expression of senescence marker protein 30 (SMP30), p53, and p21 [Citation31]. Autophagy in the aorta may decline with age and lead to age-related endothelial function disorders and arterial stiffness [Citation32]. Moreover, trehalose or spermidine (autophagy inducers) can slow age-related increases in aortic pulse wave velocity and decrease the accumulation of aortic collagen I and AGEs [Citation33]. As a defense reaction to unfavorable conditions, autophagy seems to be dysfunctional in aortic wall during senescence.
2.2. Estrogen could reinforce autophagy via transcription factors and FOXO3
Existing evidence has shown that males have a higher incidence of AAA than females, and estrogen plays a critical role [Citation34]. In the 1970s, an association between systemic deprivation of steroid sex hormones and autophagy was reported [Citation35,Citation36]. In the past few years, numerous studies have reported that the estrogen receptor influences autophagy, although the molecular mechanisms and signaling components are still unknown. Recently, various studies have shown that E2 plays a role in promoting autophagy [Citation37–39]. In the nucleus, estrogen-specific receptors (ESRs, including ESR1 and ESR2) integrate with the estrogen responsive element (ERE) and directly or indirectly regulate ATG expression via transcription factors (TFs) that participate in the regulation of autophagy, including autophagy induction, vesicle nucleation and extension, and completion [Citation40]. Estrogen-mediated autophagy regulation via Forkhead box O3 (FOXO3) has also been well documented. FOXO3, which is considered a “longevity gene”, has been reported to regulate Atg14 gene expression [Citation41]. The activation of FOXO3 could induce ATG expression in mice via protein kinase AMP-associated catalytic subunit alpha 2 (PRKAA2) [Citation41]. E2 strongly enhances the effects of FOXO3 via ESR1 [Citation42]. However, estrogen can also mediate the inactivation of FOXO3 by G protein-coupled estrogen receptor 1 (GPER1) [Citation43]. Unfortunately, no original research has explored whether autophagy induced by estrogen is a cause of the lower morbidity in women than in men with AAA.
2.3 Cigarettes could induce autophagy
Nicotine, the main harmful component of cigarettes, has always been recognized as an important risk factor for inducing AAA formation [Citation44]. However, autophagy seems to play an ignominious role in this process. The nicotine-mediated theory of AAA has been further supported by reports showing that AAA tissues obtained from ApoE−/− mice exhibit upregulation of MMP 2 via AMP-activated protein kinase (AMPK)-α2 [Citation45]. AMPK, a classic autophagy pathway molecule that is activated by AMP or ADP, is considered to be an energy sensor that regulates energy homeostasis in the body [Citation46]. Studies showed that nicotine could exacerbate angiotensin II (AngII)-induced AAA formation via autophagy-mediated AMPK- α2 activation and MMP 2 upregulation and may reveal the pathogenic mechanism of nicotine in AAA formation [Citation45].
Moreover, Wang et al. [Citation24] showed that nicotine-mediated autophagy in VSMCs could accelerate atherosclerosis. Nicotine, which binds to nicotinic acetylcholine receptors (nAChRs), can induce the production of reactive oxygen species (ROS), which activate nuclear factor kappa B (NF-κB). IκBα and p65 phosphorylation promote p65 translocation and enhance autophagy, ultimately contributing to the phenotypic switching of VSMCs and accelerating atherosclerosis [Citation24]. In the early stage of AAA, autophagy activation may be a compensatory reaction and a self-clearance mechanism that protects VSMCs against nicotine-induced cellular distress by removing abnormal organelles and protein aggregates. However, with the advancement of AAA and the degradation and deposition of extracellular matrix, autophagy is either excessively stimulated or impaired, leading to or promoting cell death and diseases [Citation47]. This mechanism may explain why cigarettes, as a high-risk factor for AAA, could induce autophagy, which is considered a protective effect in AAA. In short, as a compensatory reaction to injury, autophagy is unable to protect against excessive damage.
3. Autophagy-mediated AAA pathophysiology
Current theories on the pathogenic features of AAA include chronic inflammation, atherosclerosis and thrombosis [Citation20]. Autophagy plays an crucial role in all of these processes. Here, we summarize the prevailing views that touch on the function of autophagy and its role in AAA formation based on the above three aspects. Moreover, the molecular pathogenesis of AAA, including the roles of macrophages, VSMCs and endothelial cells, have been included.
3.1. Autophagy and inflammation
In the past, inflammation was thought to only exist in inflammatory AAA, which accounts for 5% of all AAA cases [Citation48,Citation49]. However, more researchers have realized that inflammation plays a critical role in the pathogenesis of AAA. One of the defining characteristics of AAA is inflammation with the presence of mononuclear inflammatory cell infiltration in aneurysmal tissue. Macrophage inflammasome activation and infiltration is the classic and crucial pathway in the inflammation-mediated pathogenesis of AAA. Macrophages, induced by atherosclerosis, oxidative stress, and inflammatory cytokines, can degrade the extracellular matrix by infiltrating aortic tissue and secreting proteases [Citation50,Citation51]. On the other hand, infiltrating macrophages release inflammatory cytokines such as interleukin-1β (IL-1β) and interleukin-6 in the aneurysmal tissue to exacerbate inflammatory responses [Citation52,Citation53]. Notably, studies have thoroughly documented that autophagy plays a critical role in maintaining and differentiating hematopoietic stem cells (HSCs) while differentiating monocytes into macrophages [Citation54,Citation55]. Babak Razani et al [Citation56] showed that autophagy deficiency led to hyperactivation of the macrophage inflammasome and enhanced plaque progression in ATG5-deficient ApoE-/- mice. IL-1β expression and serum levels due to selective secretion by inflammatory macrophage in aortic tissue were also found to be elevated [Citation56]. A recent mechanistic study indicated that 5-aminolaevulinic acid (ALA) could enhance macrophage differentiation toward the M2 phenotype by activating the AMPK-mTORC1 pathway in M1 macrophages [Citation57]. These findings indicate that autophagy engages a beneficial cycle that enhances M1-to-M2 polarization anti-inflammatory reactions. This discovery might help elucidate the mechanism underlying autophagy as a potential target to prevent AAA events.
Another study showed that regulatory T cells (Tregs) play a critical role in protecting against AAA development by downregulating inflammation. Foxp3+ Tregs, which are induced by the IL-2 complex, suppress inflammatory responses in aneurysmal lesions and decrease mortality in AngII-induced AAA [Citation58]. Wei et al [Citation59] found that autophagy was a key point in regulating Tregs and restraining mTORC1 and c-Myc functions and glycolytic metabolism. Autophagy does not statically regulate Tregs. Moreover, Atg7 or Atg5 knockout in Tregs could exacerbate apoptosis and destroy aging-related stability.
There are also various pathways that have not entirely been confirmed, including cytokine-cytokine receptor interactions, leukocyte transendothelial migration, B cell and T cell signaling pathways, natural killer cell-mediated cytotoxicity and hematopoietic cell lineages [Citation60,Citation61]. Further study is needed.
3.2. Diverse effects of autophagy in atherosclerosis
Data from current studies on the role of atherosclerosis in AAA are mixed, and the relationship between atherosclerosis and AAA is still unclear. The traditional view suggests that AAA is one of the end stages of atherosclerosis, suggesting that AAA is an excessive compensatory arterial response to luminal dimensions that are altered by atherosclerotic plaque growth [Citation62,Citation63]. However, distinct susceptibility to AAA induction has also been found in different mouse strains that do not develop atherosclerosis [Citation64]. Regardless of the ultimate conclusion, it is undeniable that these processes are closely linked. Interestingly, autophagy has diverse effects in atherosclerosis. In early atherosclerosis, autophagy promotes cholesterol efflux from macrophage foam cells, thereby contributing to the regression of atherosclerotic plaques via ATM-dependent inhibition of mTOR [Citation65,Citation66]. Liao et al [Citation67] showed that macrophage autophagy (deletion of Atg5) in LDLR−/− mice exacerbated atherosclerotic plaque development after 12 and 16 weeks of a Western-style diet by increasing macrophage apoptosis and necrosis, indicating that autophagy plays an important role in early atherosclerosis. In the case of mild oxidative stress in early atherosclerosis, autophagy could degrade damaged proteins and intracellular material, contributing to cellular recovery [Citation68]. The latest study also showed that the P2RY12 receptor could reduce cholesterol efflux and exacerbate VSMC-derived foam cell generation in advanced atherosclerosis by inhibiting autophagy [Citation69].
However, autophagy becomes dysfunctional after the atherosclerotic plaque develops or oxidative stress becomes severe. Razani et al [Citation56] showed that the protein expression of SQSTM1/p62, which is considered an aortic autophagy substrate, was dramatically increased in atherosclerotic aortas and was further elevated with increasing age and plaque burden in ApoE−/− mice fed a Western-style diet, indicating that autophagy was defective. Defective autophagy causes leakage of cytochrome c from damaged mitochondria that cannot be effectively cleared, and then activation of the intrinsic apoptotic pathway may occur [Citation70].
3.3. Autophagy and thrombosis
AAA is considered a complex thrombotic and inflammatory disorder. Endothelial damage, hemodynamic changes, platelet aggregation and impaired release of coagulation antagonists play crucial roles in the development of intraluminal thrombosis (ILT), the progression of aneurysm development, and eventual aneurysm rupture [Citation71–73]. ILT, a three-dimensional fibrin structure composed on blood proteins, blood cells, platelets and cellular debris, is the biological source of secreted proteases, inflammatory cytokines, and fibrinolytic products within the layers of the thrombi, the aneurysm wall and infiltrates in the bloodstream [Citation74,Citation75]. ILT impedes oxygen flow, leading to hypoxia in the aortic wall, and it can also influence the proteolytic degradation of the underlying aneurysm [Citation76,Citation77]. Traditional views were that ILT within an aneurysm would not reduce the risk of AAA rupture because it did not reduce both the mean and the pulse pressure near the aneurysm wall [Citation78]. Accumulating evidence indicates that although increased ILT is associated with lower peak wall stress and mean wall stress, it is also associated with aneurysm rupture at smaller diameters and lower stress levels [Citation79,Citation80].
Autophagy has been reported to play a crucial role in regulating hemorrhagic and thrombotic diseases by influencing the number and function of platelets, which play a core role in physiological hemostasis [Citation81]. Jiang et al [Citation82] showed that rapamycin, an autophagy inhibitor, promoted platelet adhesion to endothelial cells, inducing thrombosis. Additionally, Wang et al. [Citation83] discovered that oxidative stress induced by ox‐LDL facilitated platelet activation, inducing autophagy in platelets by inhibiting the PI3K/AKT/mTOR signal pathway and ultimately deteriorating platelet aggregation and adhesion. This result reveals that autophagy plays a detrimental role in AAA formation by inducing thrombosis. However, the effect of autophagy in platelets is reversed under starvation conditions. Paul et al [Citation84] discovered that increased autophagy under starvation reduced the aggregation of platelets and that the tail‐bleeding time was increased in a starved‐rat model. In that study, enhanced autophagy and cellular energy depletion may have been the key causes of decreased platelet activation and aggregation. The true role of autophagy in thrombosis in AAA remains unknown and requires more study.
4. Current autophagy-mediated pharmacological treatments of AAA
Researchers have focused on pharmacological treatments of AAA that could potentially offer benefits to patients by effectively alleviating the expansion of aneurysms. Current studies have demonstrated that pharmacological intervention could suppress an already developed AAA in small animal models by regulating proinflammatory mediators (including some antibiotics), autophagy, immunosuppression, and prostaglandin metabolism [Citation85]. Notably, treatment with metformin after the establishment of AAA could cause a reduction in the aneurysmal diameter [Citation86]. Interventions with calorie restriction (CR) have also been effective in halting AAA progression in mice [Citation87]. Interestingly, accumulating evidence has revealed that various pharmacological interventions have been effective in the treatment of AAA via autophagy in recent years (). Moreover, the success of gene interventions in AAA was also achieved through modulating the autophagy pathway (), which strongly demonstrated that autophagy deficiency participated in the progression of AAA formation.
Table 1. Pharmacological treatment of AAA via autophagy
Table 2. Autophagy-related MicroRNA in AAA
Rapamycin, a classic autophagy inducer, has been reported to halt AAA progression in intraluminal elastase perfusion-induced C57BL/6 J mice [Citation26]. Considering that mTOR signaling is overactivated in aortic smooth muscle cells (SMCs) in AAA mice, Li demonstrated that rapamycin could limit the expansion of the abdominal aorta and reduce the aortic diameter following intraluminal elastase perfusion by inhibiting the mTOR pathway, which could downregulate macrophage infiltration, MMP expression, and inflammatory cytokine production [Citation26].
CR was also effective in limiting AAA formation in AngII-induced ApoE−/− mice [Citation87]. CR was the only nonpharmacological treatment to slow the aging process in a wide variety of organisms, ranging from yeast to primates, by decreasing caloric intake without causing malnutrition [Citation88]. CR modulates several key molecular players involved in the regulation and execution of autophagy, nutrient signaling, and energy metabolism via the AMPK and Sirtuin (SIRT) pathways [Citation89]. Liu showed that CR alleviated the incidence of AAA formation and attenuated elastin degradation in mice. Moreover, the activity of SIRT1 was found to be upregulated in VSMCs upon CR, which strongly demonstrated that CR-induced autophagy plays a vital role in preventing AAA [Citation87].
Another interesting discovery in recent years is that metformin, a traditional antidiabetic agent, is able to induce autophagy by activating AMPK. Therefore, metformin is considered a potential hormesis-inducing agent with life span-promoting and longevity-associated properties [Citation90]. Metformin inhibits gluconeogenesis in the liver by activating AMPK, which is a sensor of cellular energy levels and responds to increases in the ratio of AMP/ATP [Citation90,Citation91]. Metformin has also been reported to be a potential therapeutic target in AAA via inflammation, ECM remodeling, and VSMC homeostasis [Citation92].The cross-talk between metformin and autophagy in AAA still needs further study.
PI3K acts as an inflammatory initiating factor and regulates immune cell signaling and migration. PI3K inhibitors or the genetic loss of PI3K causes a reduction in the chemotactic responses of lymphocytes, neutrophils, macrophages, and eosinophils in migration assays [Citation93]. Yu showed that treatment with wortmannin, a classic PI3K inhibitor, could prevent the formation and progression of AAA by attenuating inflammatory cell infiltration [Citation94]. The role autophagy plays in this process is not well understood and needs further research.
5. Summary and perspective
In summary, AAA is an important cause of death in adults due to aortic rupture, and there is a lack of effective medical therapies to limit the growth of small AAAs. Various potential mechanisms have been reported, such as inflammation, thrombosis, atherosclerosis and innate factors. To explore the relationship between AAA and autophagy, we discussed how autophagy-mediated high-risk factors exacerbate AAA formation and the potential pathogenesis of AAA. We also listed current evidence that pharmacological treatment could prevent AAA formation by inducing autophagy. Most results emphasized the protective role that autophagy plays in AAA formation, but more research is needed. Regarding senescence, aging people, who lack effective autophagy, seem to be more susceptible to developing AAA via the accumulation of SQSTM1/p62 and the development of stress-induced VSMC senescence through various pathways. On the other hand, females have also been consistently associated with reductions in the growth rates of established AAAs, as enhanced autophagy induced by estrogen may play a critical role via TFs and FOXO3.
Overall, although the specific pathogenesis of AAA formation remains unclear, the protective role of autophagy in potential mechanisms, including atherosclerosis, thrombosis and inflammation via various autophagy-mediated pathways, has been gradually accepted. Older people with defective autophagy seem to be more susceptible to AAA than younger people. Moreover, females with increased autophagy induced by estrogen may be more resistant to AAA than males. These findings support that autophagy plays a protective role in AAA development but deteriorates because of aging or other unknown reasons.
6. Inadequacies and future directions
Despite the increasing knowledge about the role of autophagy in AAA development, this field is still only in its beginning. The latest study suggested that nicotine could induce an ischemic-sensitive phenotype in the heart by inducing autophagy [Citation100]. This finding seems to be opposite of we have known thus far about autophagy in AAA. Moreover, the diverse roles that autophagy plays in atherosclerosis remain unclear. Little is known about the involvement of autophagy in the modulation of AAA induced by nicotine, although some initial data have suggested important associations. Ongoing and future clinical trials, as well as gene studies, will ultimately determine the actual beneficial impact of autophagy on AAA.
Disclosure statement
The authors declare that they have no conflicts of interest.
Additional information
Funding
References
- de Duve D. The peroxisome: a new cytoplasmic organelle[J]. Proc R Soc Lond B Biol Sci. 1969;173(1030):71–83.
- Mizushima N, Levine B, AM C, et al. Autophagy fights disease through cellular self-digestion[J]. Nature. 2008;451(7182):1069–1075.
- Bento CF, Renna M, Ghislat G, et al. Mammalian autophagy: how does it work?[J]. Annu Rev Biochem. 2016;85:685–713.
- Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms[J]. Autophagy. 2018;14(2):207–215.
- Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes[J]. Embo J. 2017;36(13):1811–1836.
- Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism[J]. Nat Rev Mol Cell Biol. 2015;16(8):461–472.
- Zaffagnini G, Martens S. Mechanisms of selective autophagy[J]. J Mol Biol. 2016;428(9 Pt A):1714–1724.
- Shaid S, Brandts CH, Serve H, et al. Ubiquitination and selective autophagy[J]. Cell Death Differ. 2013;20(1):21–30.
- Wu MY, Song JX, Wang SF, et al. Selective autophagy: the new player in the fight against neurodegenerative diseases?[J]. Brain Res Bull. 2018;137:79–90.
- Yan Y, Chen X, Wang X, et al. The effects and the mechanisms of autophagy on the cancer-associated fibroblasts in cancer[J]. J Exp Clin Cancer Res. 2019;38(1):171.
- Ren J, Zhang Y. Targeting autophagy in aging and aging-related cardiovascular diseases[J]. Trends Pharmacol Sci. 2018;39(12):1064–1076.
- Gonzalez CD, Lee MS, Marchetti P, et al. The emerging role of autophagy in the pathophysiology of diabetes mellitus[J]. Autophagy. 2011;7(1):2–11.
- Haq S, Grondin J, Banskota S, et al. Autophagy: roles in intestinal mucosal homeostasis and inflammation[J]. J Biomed Sci. 2019;26(1):19.
- Behrends C, Sowa ME, Gygi SP, et al. Network organization of the human autophagy system[J]. Nature. 2010;466(7302):68–76.
- Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins[J]. Autophagy. 2010;6(6):764–776.
- Schaaf MB, Keulers TG, Vooijs MA, et al. LC3/GABARAP family proteins: autophagy-(un)related functions[J]. Faseb J. 2016;30(12):3961–3978.
- Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing[J]. Embo J. 2000;19(21):5720–5728.
- Golledge J, Muller J, Daugherty A, et al. Abdominal aortic aneurysm: pathogenesis and implications for management[J]. Arterioscler Thromb Vasc Biol. 2006;26(12):2605–2613.
- Liao S, Curci JA, Kelley BJ, et al. Accelerated replicative senescence of medial smooth muscle cells derived from abdominal aortic aneurysms compared to the adjacent inferior mesenteric artery[J]. J Surg Res. 2000;92(1):85–95.
- Golledge J. Abdominal aortic aneurysm: update on pathogenesis and medical treatments[J]. Nat Rev Cardiol. 2019;16(4):225–242.
- Ren J, Sowers JR, Zhang Y, et al. Autophagy, and cardiovascular aging: from pathophysiology to therapeutics[J]. Trends Endocrinol Metab. 2018;29(10):699–711.
- Ma Y, Qi M, An Y, et al. Autophagy controls mesenchymal stem cell properties and senescence during bone aging[J]. Aging Cell. 2018;17:1.
- Tyutyunyk-Massey L, Gewirtz DA. Roles of autophagy in breast cancer treatment: target, bystander or benefactor[J]. Semin Cancer Biol. 2019. DOI:10.1016/j.semcancer.2019.11.008
- Wang Z, Liu B, Zhu J, et al. Nicotine-mediated autophagy of vascular smooth muscle cell accelerates atherosclerosis via nAChRs/ROS/NF-κB signaling pathway[J]. Atherosclerosis. 2019;284:1–10.
- Zheng YH, Tian C, Meng Y, et al. Osteopontin stimulates autophagy via integrin/CD44 and p38 MAPK signaling pathways in vascular smooth muscle cells[J]. J Cell Physiol. 2012;227(1):127–135.
- Li G, Qin L, Wang L, et al. Inhibition of the mTOR pathway in abdominal aortic aneurysm: implications of smooth muscle cell contractile phenotype, inflammation, and aneurysm expansion[J]. Am J Physiol Heart Circ Physiol. 2017;312(6):H1110–H1119.
- Alfaras I, Di Germanio C, Bernier M, et al. Pharmacological strategies to retard cardiovascular aging[J]. Circ Res. 2016;118(10):1626–1642.
- Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging[J]. Cell. 2011;146(5):682–695.
- Madeo F, Zimmermann A, Maiuri MC, et al. Essential role for autophagy in life span extension[J]. J Clin Invest. 2015;125(1):85–93.
- Grootaert MO, da Costa Martins PA, Bitsch N, et al. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis[J]. Autophagy. 2015;11(11):2014–2032.
- Kim SG, Sung JY, Kim JR, et al. Nifedipine-induced AMPK activation alleviates senescence by increasing autophagy and suppressing of Ca(2+) levels in vascular smooth muscle cells[J]. Mech Ageing Dev. 2020;190:111314.
- Salabei JK, Hill BG. Implications of autophagy for vascular smooth muscle cell function and plasticity[J]. Free Radic Biol Med. 2013;65:693–703.
- LaRocca TJ, Gioscia-Ryan RA, Hearon CM Jr., et al. The autophagy enhancer spermidine reverses arterial aging[J]. Mech Ageing Dev. 2013;134(7–8):314–320.
- Li X, Zhao G, Zhang J, et al. Prevalence and trends of the abdominal aortic aneurysms epidemic in general population–a meta-analysis[J]. PLoS ONE. 2013;8(12):e81260.
- Park J, Shin H, Song H, et al. Autophagic regulation in steroid hormone-responsive systems[J]. Steroids. 2016;115(undefined):177–181.
- Schiebler TH, Danner KG. The effect of sex hormones on the proximal tubules in the rat kidney[J]. Cell Tissue Res. 1978;192(3):527–549.
- Totta P, Busonero C, Leone S, et al. Dynamin II is required for 17β-estradiol signaling and autophagy-based ERα degradation[J]. Sci Rep. 2016;6(undefined):23727.
- Yang YH, Chen K, Li B, et al. Estradiol inhibits osteoblast apoptosis via promotion of autophagy through the ER-ERK-mTOR pathway[J]. Apoptosis. 2013;18(11):1363–1375.
- Kimura A, Ishida Y, Nosaka M, et al. Exaggerated arsenic nephrotoxicity in female mice through estrogen-dependent impairments in the autophagic flux[J]. Toxicology. 2016;339(undefined):9–18.
- Luo H, Liu M, Luo S, et al. Dynamic monitoring of GPER-mediated estrogenic effects in breast cancer associated fibroblasts: an alternative role of estrogen in mammary carcinoma development[J]. Steroids. 2016;112(undefined):1–11.
- Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo[J]. Cell Metab. 2007;6(6):458–471.
- Sisci D, Maris P, Grazia Cesario M, et al. The estrogen receptor α is the key regulator of the bifunctional role of FoxO3a transcription factor in breast cancer motility and invasiveness[J]. Cell Cycle. 2013;12(21):3405–3420.
- Zekas E, Prossnitz ER. Estrogen-mediated inactivation of FOXO3a by the G protein-coupled estrogen receptor GPER[J]. BMC Cancer. 2015;15(undefined):702.
- Sugamura K, Keaney JF Jr. Nicotine: linking smoking to abdominal aneurysms[J]. Nat Med. 2012;18(6):856–858.
- Wang S, Zhang C, Zhang M, et al. Activation of AMP-activated protein kinase alpha2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo[J]. Nat Med. 2012;18(6):902–910.
- Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis[J]. Nat Rev Mol Cell Biol. 2018;19(2):121–135.
- Levine B, Yuan J. Autophagy in cell death: an innocent convict?[J]. J Clin Invest. 2005;115(10):2679–2688.
- JL C, CL S, HJ S, et al. Inflammatory aneurysms of the aorta[J]. J Vasc Surg. 1985;2(1):113–124.
- Aiello MR, Cohen WN. Inflammatory aneurysm of the abdominal aorta[J]. J Comput Assist Tomogr. 1980;4(2):265–267.
- Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis[J]. Circulation. 2002;105(9):1135–1143.
- Curci JA, Liao S, Huffman MD, et al. Expression and localization of macrophage elastase (matrix metalloproteinase-12) in abdominal aortic aneurysms[J]. J Clin Invest. 1998;102(11):1900–1910.
- KG J, DJ B, LC B, et al. Interleukin-6 (IL-6) and the prognosis of abdominal aortic aneurysms[J]. Circulation. 2001;103(18):2260–2265.
- Harrison SC, Smith AJ, Jones GT, et al. Interleukin-6 receptor pathways in abdominal aortic aneurysm[J]. Eur Heart J. 2013;34(48):3707–3716.
- Jacquel A, Obba S, Boyer L, et al. Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions[J]. Blood. 2012;119(19):4527–4531.
- Chen P, Cescon M, Bonaldo P. Autophagy-mediated regulation of macrophages and its applications for cancer[J]. Autophagy. 2014;10(2):192–200.
- Razani B, Feng C, Coleman T, et al. Autophagy links inflammasomes to atherosclerotic progression[J]. Cell Metab. 2012;15(4):534–544.
- Yang Y, Wang J, Guo S, et al. Non-lethal sonodynamic therapy facilitates the M1-to-M2 transition in advanced atherosclerotic plaques via activating the ROS-AMPK-mTORC1-autophagy pathway[J]. Redox Biol. 2020;32:101501.
- Yodoi K, Yamashita T, Sasaki N, et al. Foxp3+ regulatory T cells play a protective role in angiotensin II-induced aortic aneurysm formation in mice[J]. Hypertension. 2015;65(4):889–895.
- Wei J, Long L, Yang K, et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis[J]. Nat Immunol. 2016;17(3):277–285.
- Rush C, Nyara M, JV M, et al. Whole genome expression analysis within the angiotensin II-apolipoprotein E deficient mouse model of abdominal aortic aneurysm[J]. BMC Genomics. 2009;10(undefined):298.
- JM S, Hsu M, J A, et al. Transcriptional profiling and network analysis of the murine angiotensin II-induced abdominal aortic aneurysm[J]. Physiol Genomics. 2011;43(17):993–1003.
- Golledge J, Norman PE. Atherosclerosis and abdominal aortic aneurysm: cause, response, or common risk factors?[J]. Arterioscler Thromb Vasc Biol. 2010;30(6):1075–1077.
- BR K, Bäck M, ML B-P, et al. Biomechanical factors in atherosclerosis: mechanisms and clinical implications[J]. Eur Heart J. 2014;35(43):3013–3020, 3020a-3020d.
- MA D, MK S, Zhao S, et al. Background differences in baseline and stimulated MMP levels influence abdominal aortic aneurysm susceptibility[J]. Atherosclerosis. 2015;243(2):621–629.
- Brichkina A, DV B. WIP-ing out atherosclerosis with autophagy[J]. Autophagy. 2012;8(10):1545–1547.
- Ouimet M, Marcel YL. Regulation of lipid droplet cholesterol efflux from macrophage foam cells[J]. Arterioscler Thromb Vasc Biol. 2012;32(3):575–581.
- Liao X, JC S, Wang Y, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis[J]. Cell Metab. 2012;15(4):545–553.
- Filomeni G, De ZD, Cecconi F, et al. Oxidative stress and autophagy[J]. Antioxid Redox Signal. 2006;8(null):152–162.
- Pi S, Mao L, Chen J, et al. The P2RY12 receptor promotes VSMC-derived foam cell formation by inhibiting autophagy in advanced atherosclerosis[J]. Autophagy. 2020;1741202:1–21.
- De Meyer GR, Grootaert MO, Michiels CF, et al. Autophagy in vascular disease[J]. Circ Res. 2015;116(3):468–479.
- Siasos G, Mourouzis K, Oikonomou E, et al. The role of endothelial dysfunction in aortic aneurysms[J]. Curr Pharm Des. 2015;21(28):4016–4034.
- Arzani A, GY S, RLA D, et al. A longitudinal comparison of hemodynamics and intraluminal thrombus deposition in abdominal aortic aneurysms[J]. Am J Physiol Heart Circ Physiol. 2014;307(12):H1786–1795.
- Tong J, GAJ H. Structure, mechanics, and histology of intraluminal thrombi in abdominal aortic aneurysms[J]. Ann Biomed Eng. 2015;43(7):1488–1501.
- Fontaine V, Touat Z, EMV M, et al. Role of leukocyte elastase in preventing cellular re-colonization of the mural thrombus[J]. Am J Pathol. 2004;164(6):2077–2087.
- LP H, BH G, PW C, et al. Ultrasonic evaluation of abdominal aortic thrombus[J]. J Ultrasound Med. 1982;1(8):315–318.
- Kazi M, Thyberg J, PM R, et al. Influence of intraluminal thrombus on structural and cellular composition of abdominal aortic aneurysm wall[J]. J Vasc Surg. 2003;38(6):1283–1292.
- DA V, PC L, DH W, et al. Association of intraluminal thrombus in abdominal aortic aneurysm with local hypoxia and wall weakening[J]. J Vasc Surg. 2001;34(2):291–299.
- GW S, NJ A, JM VB, et al. Experimental study of the influence of endoleak size on pressure in the aneurysm sac and the consequences of thrombosis[J]. Br J Surg. 2000;87(1):71–78.
- SJ H, JD C, KM C, et al. Intraluminal thrombus is associated with early rupture of abdominal aortic aneurysm[J]. J Vasc Surg. 2018;67(4):1051–1058.e1051.
- Koole D, HJ Z, Schoneveld A, et al. Intraluminal abdominal aortic aneurysm thrombus is associated with disruption of wall integrity[J]. J Vasc Surg. 2013;57(1):77–83.
- Luo XL, Jiang JY, Huang Z, et al. Autophagic regulation of platelet biology[J]. J Cell Physiol. 2019;234:14483–14488.
- Jiang P, Lan Y, Luo J, et al. Rapamycin promoted thrombosis and platelet adhesion to endothelial cells by inducing membrane remodeling[J]. BMC Cell Biol. 2014;15(undefined):7.
- Wang X, YF F, Liu X, et al. ROS promote Ox-LDL-induced platelet activation by up-regulating autophagy through the inhibition of the PI3K/AKT/mTOR pathway[J]. Cell Physiol Biochem. 2018;50(5):1779–1793.
- Paul M, Hemshekhar M, Kemparaju K, et al. Aggregation is impaired in starved platelets due to enhanced autophagy and cellular energy depletion[J]. Platelets. 2019;30(4):487–497.
- Yoshimura K, Morikage N, SK N-F, et al. Current status and perspectives on pharmacologic therapy for abdominal aortic aneurysm[J]. Curr Drug Targets. 2018;19(11):1265–1275.
- Wang Z, Guo J, Han X, et al. Metformin represses the pathophysiology of AAA by suppressing the activation of PI3K/AKT/mTOR/autophagy pathway in ApoE(-/-) mice[J]. Cell Biosci. 2019;9:68.
- Liu Y, Wang TT, Zhang R, et al. Calorie restriction protects against experimental abdominal aortic aneurysms in mice[J]. J Exp Med. 2016;213(11):2473–2488.
- Piper MD, Bartke A. Diet and aging[J]. Cell Metab. 2008;8(2):99–104.
- Madeo F, Carmona-Gutierrez D, SJF H, et al. Caloric restriction mimetics against age-associated disease: targets, mechanisms, and therapeutic potential[J]. Cell Metab. 2019;29(3):592–610.
- Piskovatska V, Stefanyshyn N, KB S, et al. Metformin as a geroprotector: experimental and clinical evidence[J]. Biogerontology. 2019;20(1):33–48.
- Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis[J]. Curr Opin Cell Biol. 2015;33:1–7.
- Raffort J, Hassen-Khodja R, Jean-Baptiste E, et al. Relationship between metformin and abdominal aortic aneurysm[J]. J Vasc Surg. 2019. DOI:10.1016/j.jvs.2019.08.270
- Foster JG, Blunt MD, Carter E, et al. Inhibition of PI3K signaling spurs new therapeutic opportunities in inflammatory/autoimmune diseases and hematological malignancies[J]. Pharmacol Rev. 2012;64(4):1027–1054.
- Yu J, Liu R, Huang J, et al. Inhibition of Phosphatidylinositol 3-kinease suppresses formation and progression of experimental abdominal aortic aneurysms[J]. Sci Rep. 2017;7(1):15208.
- Zhang S, Kan X, Li Y, et al. Deficiency of gammadeltaT cells protects against abdominal aortic aneurysms by regulating phosphoinositide 3-kinase/AKT signaling[J]. J Vasc Surg. 2018;67(3):899–908 e891.
- Hao Q, Chen X, Wang X, et al. Curcumin attenuates angiotensin ii-induced abdominal aortic aneurysm by inhibition of inflammatory response and ERK signaling pathways[J]. Evid Based Complement Alternat Med. 2014;2014(undefined):270930.
- Peng J, He X, Zhang L, et al. MicroRNA‑26a protects vascular smooth muscle cells against H2O2‑induced injury through activation of the PTEN/AKT/mTOR pathway[J]. Int J Mol Med. 2018;42(3):1367–1378.
- Zhao L, Huang J, Zhu Y, et al. miR-33-5p knockdown attenuates abdominal aortic aneurysm progression via promoting target adenosine triphosphate-binding cassette transporter A1 expression and activating the PI3K/Akt signaling pathway[J]. Perfusion. 2019. DOI:10.1177/0267659119850685:267659119850685
- Ma X, Yao H, Yang Y, et al. miR-195 suppresses abdominal aortic aneurysm through the TNF-α/NF-κB and VEGF/PI3K/Akt pathway[J]. Int J Mol Med. 2018;41(4):2350–2358.
- Zhang P, Li Y, Fu Y, et al. Inhibition of autophagy signaling via 3-methyladenine rescued nicotine-mediated cardiac pathological effects and heart dysfunctions[J]. Int J Biol Sci. 2020;16(8):1349–1362.