
ABSTRACT
Sepsis is a major condition caused by an overwhelming inflammatory response to an infection. Sepsis-induced myocardial dysfunction (SIMD) is a common complication in septic patients and a major predictor of morbidity and mortality. Here, we investigated the role of tripartite motif 31 (TRIM31) protein in sepsis progression in vitro and in vivo. Quantitative real-time PCR and western blot were used to detect the expression levels of relative genes and proteins. Cell proliferation and apoptosis were evaluated to determine cell viability. H&E and IHC staining were performed to examine morphological and pathological changes in mice. ELISA assay was used to detect inflammatory factors. TRIM31 was upregulated in septic patients compared with normal people. TRIM31 depletion reduced LPS-induced apoptosis whereas TRIM31 overexpression-elevated LPS-induced apoptosis. Furthermore, TRIM31 interacted with and ubiquitinated transforming growth factor-β-activated kinase-1 (TAK1), resulting in TAK1 activation and subsequent induction of NF-κB signaling. Of note, Trim31 depletion or blockade by PDTC treatment inhibited LPS-induced apoptosis in vivo. In conclusion, TRIM31 played an important role in SIMD by activating TAK1-mediated NF-κB signaling pathway.
Introduction
Sepsis is a life-threatening condition caused by an overwhelming inflammatory response to an infection, which can result in serious complications, making it a major clinical issue worldwide [Citation1]. Septic shock and multiple organ dysfunction, including endothelial damage, acute lung injury, liver injury, and myocardial dysfunction are highly linked to sepsis [Citation2,Citation3]. The cardiovascular system is the most easily affected organ by sepsis and almost 40–50% of patients exhibit myocardial dysfunction [Citation4]. Sepsis-induced myocardial dysfunction (SIMD) is a major predictor of morbidity and mortality in septic patients. Despite numerous studies on SIMD, little is known regarding the underlying mechanisms, and prognosis remains poor [Citation5,Citation6]. Early fluid resuscitation has been the main treatment for sepsis in recent years, but prevention of myocardial injury may present a novel strategy for sepsis treatment [Citation7,Citation8]. Currently, researchers find that mitochondrial dysfunction, nitric oxide, intracellular energetics, complements, and cellular adhesion molecules may serve as mediators of SIMD [Citation5].
Nuclear factor kappa B (NF-κB), which resides in cell cytoplasm in an inactive form, translocates to the nucleus when activated and functions as a transcription factor. NF-κB proteins form homo- or hetero-dimers and regulate expression of distinct but overlapping genes involved in innate and adaptive immunity, inflammation, apoptosis, proliferation, stress response, and cancer progression [Citation9]. NF-κB consists of p50 and RelA, and is sequestered in the cytoplasm, where it associates with one of several inhibitory molecules, including IkBα, IkBβ, IkBγ, p105, and p100, among which IkBα is most abundant. Inactive NF-κB/IkB complex is activated by phosphorylation on two conserved serine (S) residue within the N-terminal domain of IkB proteins. Phosphorylation then leads to immediate polyubiquitination of IkB proteins by the SCF-β-TrCP complex [Citation9]. NF-κB activation is induced by a wide variety of signals including stress, cigarette smoke, viruses, bacteria, inflammatory stimuli, cytokines, free radicals, carcinogens, tumor promoters, and endotoxins. Lipopolysaccharide (LPS), tumor necrosis factor-α (TNF-a) and interleukin 1β (IL-1β) are among the most potent inducers of NF-κB signaling, all resulting in activation of a specific mitogen-activated protein kinase called NF-κB inducing kinase, or NIK [Citation10]. In response to TNF-α or IL-1β, signaling intermediaries, such as TNF receptor-associated factors (TRAFs) and receptor-interacting protein (RIP), are rapidly modified with K63-linked polyubiquitin chains, facilitating recruitment and activation of TGF-β-activated kinase1 (TAK1) and I kappa B kinase (IKK) complexes by binding to TAK1-binding proteins (TABs) and NF-κB essential modulator (NEMO), respectively [Citation11–13].
Tripartite motif (TRIM) is a family of proteins that play critical roles in inflammation and innate immunity. TRIM proteins act as E3 ubiquitin ligases and are characterized by the presence of RBCC motif (a RING finger, B-box, and coiled-coil domains) [Citation14]. Among the TRIM family members, TRIM8 has been previously demonstrated to regulate NF-κB activation by mediating K63-linked polyubiquitination of TAK1 [Citation15]. TRIM31, which has almost the same structure as TRIM8 and both belong to subgroup V [Citation16], has recently been implicated in the inflammatory response to infection. Specifically, TRIM31 has been shown to attenuate NLR family pyrin domain containing 3 (NLRP3) inflammasome activation by promoting proteasomal degradation of NLRP3 and the aggregation and activation of the signaling adaptor mitochondrial antiviral-signaling protein (MAVS) through Lys63-linked polyubiquitination [Citation17,Citation18]. These studies suggest that TRIM31 plays a vital role in the inflammatory response and viral infection. Importantly, TRIM31 shows rapid kinetics of induction during retinoid-induced growth arrest of breast carcinoma cells and is upregulated in gastric adenocarcinoma, suggesting a role in carcinogenesis [Citation19–21]. In addition, TRIM31 confers gemcitabine resistance in pancreatic cancer by activating the NF-κB pathway [Citation22]. However, the precise role and molecular mechanism of TRIM31 in malignancy and treatment failure of pancreatic cancer remain ambiguous.
As reported, LPS is an effective method to induce septic models in vivo and in vitro [Citation23]. Here, we probed the role of TRIM31 in LPS-induced sepsis using AC16 cells and c57 mice as in vitro and in vivo models, respectively. Interestingly, we observed a positive correlation between NF-κB and TRIM31 in sepsis. Moreover, TRIM31 depletion-blocked apoptosis induced by LPS, and similar effects were observed in mice treated with LPS in vivo. Together, our findings suggest a novel, potential mechanism of TRIM31 in sepsis.
Materials and methods
Patient tissues
Peripheral Blood Mononuclear Cell (PBMC) samples from 35 SIMD patients and 25 normal persons were collected from Shanghai Seventh People’s Hospital. Informed consent was signed by the patients. This study was approved by the ethics committee of the Shanghai Seventh People’s Hospital.
Cell culture and treatment
Human cardiomyocyte cell line AC16 was obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured in Dulbecco’s modified Eagle medium (DMEM, Gibco, Carlsbad, CA, USA) medium supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA) and 100 units of penicillin/streptomycin (Gibco, Carlsbad, CA, USA) under 5% CO2 at 37°C.
LPS (Sigma, St. Louis, MO, USA) was used to treat AC16 cells at varying concentrations (0, 0.1, 0.2, 0.5, 1, 2, 5 μg/mL) for 24 hours. For other experiments, cells were pre-treated for 30 mins with 50 μM PDTC (Pyrrolidine dithiocarbamate, Selleck, Houston, TX, USA), an inhibitor of NF-κB, or with 20 μM 5Z-7-OX, an inhibitor of TAK1 (5Z-7-oxozeaenol, Sigma, St. Louis, MO, USA).
Lentivirus construction and transfection
Human TRIM31 overexpression and knockdown lentiviruses were purchased from Genechem company (Shanghai, CHINA). The following sequences were used:
ShTRIM31-1: 5ʹ- TTCCCGTCAAAGGAAGTTTGG −3ʹ;
ShTRIM31-2: 5ʹ- TATGATGGACTCATGCCTTGC −3ʹ;
ShTRIM31-3: 5ʹ- GGAAGAACGCAATCAGGTT −3ʹ;
AC16 cells were seeded into 6-well plates and cultured overnight. TRIM31 overexpression or knockdown lentivirus were added into the each well, according to lentivirus colony-forming unit (CFU). After a 48 hours transduction, parts of the cells were collected to measure transfection efficiency by real-time PCR and Western blot, while the remaining cells were cultured for other experiments.
RNA extraction and Quantitative real-time PCR
Total RNA was extracted from cells using Trizol reagent kit (Invitrogen, Carlsbad, CA, USA) and reverse transcribed using cDNA synthesis kit (Takara Biotechnology Co. Ltd., Japan). Real-time PCR was performed with SYBR Green qPCR Mix Kit (BioRad, Hercules, California, USA). GAPDH was used as internal control. The following primers were used in this assay:
Human NF-κBforward primer, 5ʹ GTCACCGGATTGAGGAGAAAC 3ʹ,
Reverse primer, 5ʹ GATCTGCCCAGAAGGAAACAC 3ʹ
Human TRIM31for”ʹ;
Human TRIM31forʹ ward primer, 5ʹ AAGGAAGAACGCAATCAG 3ʹ
Reverse primer, 5ʹ TCGCAGAAATAGTGGAAC 3ʹ
Human GAPDHforward primer, 5ʹ AATCCCATCACCATCTTC 3ʹ,
Reverse primer, 5ʹ AGGCTGTTGTCATACTTC 3ʹ.
Western blot
Total protein was extracted using RIPA lysis buffer (Solarbio, Beijing, China). Protein concentration was determined using BCA kit (Thermo, Rockford, lL, USA). Twenty-five microgram of protein was separated by 10% SDS-PAGE gel and then transferred to PVDF membranes. Membranes were blocked with 5% nonfat milk in TBST and incubated with primary antibody overnight at 4°C, followed by incubation with horseradish peroxidase (HRP) linked secondary antibody for 1 h. Membranes were developed by enhanced chemiluminescence (ECL) kit (Millipore, Bollerica, MA, USA).
Co‐immunoprecipitation (co‐IP) assay
Cells were lysed in lysis buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP‐40, 1 mg/ml each of pepstatin A and aprotinin. Cell lysates were incubated with the indicated antibodies and protein G-agarose beads (Roche Ltd.) at 4°C for 4 h. The beads were washed three times with 1 mL of lysis buffer. The precipitates were subjected to SDS-PAGE, and subsequent immunoblot analysis was performed using the indicated antibodies.
Cell apoptosis and proliferation assay
Cell apoptosis and proliferation were analyzed using Annexin V Apoptosis Detection Kit (BD Biosciences, San Jose, CA, USA) and MTT assay (Sigma, Missouri, USA), respectively. AC16 cells were seeded and treated as previously described. For apoptosis, treated cells were harvested and stained with Annexin V and PI at dark. After 15 min, apoptosis was analyzed by flow cytometry. For proliferation, 10 μL MTT were added to treated cells, and cells were incubated at 37°C for 2 hours, followed by vibration for 10 minutes with 190 μL DMSO. A microplate reader was used to detect cell proliferation at 570 nm.
Animal model and treatment
Male C57BL/6 J pathogen-free mice (weighing 18 to 22 g) were purchased from Shanghai SLAC Laboratory Animal Co. Ltd (Shanghai China). All animals were housed at 20–23°C in a 12 hours light/dark cycle with food and tap water supplied ad libitum. All animal experiments were authorized by the Animal Care Committee of Shanghai Seventh People’s Hospital.
The mice were randomly divided into 4 groups (n = 6): (1) Control group, which was given physiological saline, (2) LPS group, which was intraperitoneally injected with 10 mg/kg LPS, (3) LPS + shTrim31 group, which was given LPS and shTrim31 lentivirus (1 × 107 PFU/mL), (4) LPS + PDTC group, which was intraperitoneally injected with LPS and 120 mg/kg PDTC. According to preliminary experiment, mice were euthanatized at 12 hours after LPS injection. Heart tissues were harvested for other experiments.
Histologic examination, immunohistochemistry (IHC) and TUNEL
Heart tissues were embedded in paraffin for sectioning (4 mm; Leica RM2125, Germany) after fixing in a 10% formalin solution, followed by staining with hematoxylin and eosin (H&E) according to standard methods. Briefly, after putting in the oven for 30 min, tissue slides were dipped in different concentrations of ethyl alcohol (100%, 95%, 85%, 70%, and 50%). The slides were then stained with hematoxylin solution for 5 min, and then sequentially dipped in water, 70% and 90% ethyl alcohol. Next, the slides were stained with eosin solution for 1 min and dipped in 100% ethyl alcohol and xylene. Lastly, the slides were blocked with neutral balsam and baked in 65°C oven for 15 min. Light microscopy (Olympus, Tokyo, Japan) was used to collect images at ×200 magnification. IHC analysis was performed to detect protein expression of NF-κB. Tissue slides were incubated with anti-NF-κB (Abcam, 1:500) antibody, and images were captured using an optical microscope (NIKON ECLIPSE Ni). Positive area was analyzed using Image J software. Cell apoptosis was examined using TUNEL Kit (Roche, Indianapolis, IN, USA) following the manufacturer’s instructions.
Enzyme-linked immunosorbent assay (ELISA)
ELISA kits (Meso Scale Discovery, Rockville, MD, USA) were used to measure IL-1, IL-6, IL-11, IL-15, TNF-α, and TNF-β concentrations from ileum samples according to manufacturer’s instructions. Absorbance was measured at 450 nm on a microplate reader (Thermo, Rockford, lL, USA).
Statistical analysis
All data are shown as mean ± SD. Statistical significance was determined using one-way analysis of variance (ANOVA) and Student’s t-test. Differences with p < 0.05 were considered statistically significant. *, p < 0.05, **, p < 0.01, ***, p < 0.001.
Results
LPS-induced TRIM31 expression, suppressed cell proliferation and promoted apoptosis
To study the role of TRIM31 in sepsis, we first checked transcription in peripheral blood samples from 35 septic patients and 25 normal persons. As shown by quantitative RT-PCR ()), TRIM31 mRNA expression was dramatically elevated in septic patient samples (p < 0.001). Next, we treated myocardial cells with different concentrations of LPS and examined cell proliferation by MTT assay. As shown in ), LPS markedly inhibited cell growth in AC16 cells in a dose-dependent manner (The inhibition rates in 72 hours were 9.54 ± 1.2, 19.36 ± 2.59, 33.63 ± 0.17, and 48.34 ± 0.7%). Likewise, we evaluated the effects of LPS on cell apoptosis by flow cytometry. As shown in ), LPS-induced cell apoptosis in a dose-dependent manner (The apoptosis rates were 4.47 ± 0.42, 15.07 ± 0.31, 28.3 ± 0.1, and 44.8 ± 0.36%), consistent with the result in ). Intriguingly, LPS treatment induced TRIM31 mRNA expression in AC16 cells (,)). Taking together, 0.5 µg/ml LPS exhibited the better cell growth inhibition rate (33.63 ± 0.17%), apoptosis rate (28.3 ± 0.1%) and cell morphology (parts of the cells turned to round), while the cells treated with 1 µg/ml LPS turned to round with unclear border, so that 0.5 µg/ml LPS was used for further study.
TRIM31 depletion attenuated LPS-induced apoptosis
To further investigate the role of TRIM31 in sepsis, we performed lentiviral-mediated knockdown of TRIM31 in AC16 cells. As shown by quantitative RT-PCR and Western blot, shTRIM31 lentivirus significantly decreased the mRNA expression and protein levels of TRIM31, indicating efficient knockdown of the gene (,)). Stable AC16 cells were treated with 0.5 µg/ml LPS and apoptosis was subsequently assessed by flow cytometry. As shown in ), LPS stimulation induced approximately 22.7% apoptosis in AC16 cells. Interestingly, depletion of TRIM31 significantly reduced apoptosis to as low as 7.6% to 8.3%. TRIM31 is known to activate NF-κB signaling [Citation24], and is also involved in the production of chemokines and inflammatory cytokines. To understand the mechanism pathway which inflammation contributes to sepsis, we examined inflammatory factors such as IL-1β and TNF-α in vitro by ELISA. As observed, LPS treatment dramatically induced IL-1β and TNF-α secretion, which was suppressed by depletion of TRIM31 ()). Moreover, IKK complex, containing IKKα, IKKβ, and IKKγ, are important molecules that initiate the NF-κB cascade activation by promoting IκB phosphorylation [Citation25]. We found that 0.5 µg/ml LPS alone induced TRIM31 expression, as well as that of cleaved-caspase 3, p-IKKβ, and p-IκBα. Interestingly, depletion of TRIM31 significantly attenuated the increase in cleaved-caspase 3, p-IKKβ, and p-IκBα expression induced by LPS treatment ()). The time course of p-IKKβ and p-IκBα are shown in Supplementary figure 1. Taken together, our results demonstrated that TRIM31 depletion attenuated LPS-induced apoptosis.
TRIM31 elevated LPS-induced apoptosis by activating the NF-κB pathway
To further verify our results, we transfected AC16 cells with lentivirus to overexpress TRIM31. Indeed, TRIM31 overexpression in AC16 cells resulted in elevated TRIM31 mRNA expression and protein level (,)). We then used PDTC to block NF-κB signaling and examined effects upon LPS treatment. Flow cytometry data indicated that AC16 cells without treatment only displayed 5% apoptosis, whereas LPS treatment alone induced apoptosis up to 26.6%. Intriguingly, overexpression of TRIM31 increased apoptosis up to 43.9%, which was attenuated by PDTC (from 43.9% to 25.1%) ()). Further, we found that TRIM31 overexpression stimulated secretion of IL-1β and TNF-α, and that these effects were blocked by PDTC ()). As shown in ), TRIM31 overexpression elevated the increase of cleaved-caspase 3, p-IKKβ and p-IκBα expression induced by LPS stimulation, while PDTC counteracted the effects caused by TRIM31 overexpression. Taken together, these results indicated that TRIM31 increased LPS induced-apoptosis via activation of NF-κB signaling.
TRIM31 triggered NF-κB signaling by activating TAK1
It has been suggested that TAK1 can phosphorylate IKKβ and increase its enzymatic activity [Citation12]. Cells derived from TAK1-deficient mice consistently display reduced production of inflammatory cytokines and impaired NF-κB and MAPK activation in response to multiple TLR ligands [Citation26,Citation27], suggesting that TAK1 plays an essential and non-redundant role in the activation of both NF-κB and MAPK pathways. Thus, we hypothesized that TRIM31 may activate NF-κB signaling pathway via TAK1. To test the hypothesis, 0.5 µg/mL LPS was used to treat AC16 cells which were transduced with TRIM31 depletion or overexpression lentivirus. Subsequently, phosho-TAK1 and total TAK1 protein levels were detected by Western blot. Interestingly, LPS alone induced the expression of p-TAK1 while LPS-induced increase in p-TAK1 was attenuated by TRIM31 depletion but elevated by TRIM31 overexpression ()). These results suggested that TRIM31 mediated LPS-induced TAK1 phosphorylation. To further explore the role of TAK1, we used 5Z-7-OX, an inhibitor of TAK1, and examined its effects upon LPS treatment. As shown in ), LPS significantly increased apoptosis compared with no treatment control. LPS, together with TRIM31 and 5Z-7-OX, exhibited higher apoptotic rate than LPS but lower than TRIM31 group. Hence, we tested whether 5Z-7-OX may reverse the effects caused by TRIM31. To do so, we performed ELISA and Western blot assays to determine the levels of IL-1β, TNF-α, cleaved-caspase-3, p-IKKβ, and p-IκBα. Indeed, TRIM31 overexpression resulted in upregulation of IL-1β, TNF-α, cleaved-caspase-3, p-IKKβ, and p-IκBα. Moreover, TRIM31-induced upregulation of these factors was alleviated by 5Z-7-OX ().
Next, we explored potential interaction between TRIM31 and TAK1. As shown in ), we observed binding of TRIM31 to TAK1. Phosphorylation of MKK6 by TAK1, which subsequently activates the JNK–p38 kinase pathway, is known to be directly regulated by K63-linked polyubiquitination, suggesting that ubiquitination plays an important regulatory role in stress response pathways, including those of IKK and JNK [Citation12]. To test whether TRIM31-induced activation of NF-κB signaling is TAK1 ubiquitination-dependent, ubiquitinated TAK1 levels were determined after transduction of AC16 cells with TRIM31 overexpression lentivirus. As observed in ), there was a slight, spontaneous auto-ubiquitination in AC16 cells without any treatment. However, ectopic expression of TRIM31 resulted in prominent TAK1 ubiquitination. Collectively, these data suggested that TRIM31 activated the NF-κB signaling pathway via TAK1 ubiquitination in LPS-induced sepsis in vitro model.
TRIM31-induced apoptosis by activating NF-κB signaling pathway in vivo
To further explore the role of TRIM31 in sepsis, we used in vivo mice model by intraperitoneal (I.P.) injection of 10 mg/kg LPS to mimic sepsis. Heart tissues were harvested from mice that received different treatments as shown in . Morphological and pathological changes were determined by a qualified pathologist. Compared with control group, mice treated with LPS alone displayed severe edema and rupture. Both Trim31 depletion and PDTC treatment reduced edema and rupture in myocardial fibers ()). IHC results showed that NF-κB ()), p-IKKβ ()), and p-IκBα ()) were significantly upregulated after treatment with LPS, but depletion of Trim31 or NF-κB inhibition inhibited LPS-induced increase of NF-κB ()), p-IKKβ ()), and p-IκBα ()). Furthermore, much more apoptosis was observed in LPS mono-treatment group compared with control group, as indicated by TUNEL staining. On the other hand, Trim31 depletion and PDTC treatment reduced apoptosis ratio, respectively ()). Of note, pretreatment of Trim31 and PDTC could not protect structural alterations and seemed to only reduce the effects induced by LPS. We next investigated IL-1β and TNF-α secretion in vivo by ELISA. LPS treatment dramatically induced IL-1β and TNF-α secretion, and these effects were suppressed by depletion of Trim31 or inhibition of NF-κB ()). As shown in ), LPS dramatically elevated the protein levels of TRIM31, p-IKKβ, p-IκBα, cleaved-caspase 3 and p-TAK1. Trim31 depletion slightly suppressed p-IKKβ, p-IκBα, cleaved-caspase 3 and p-TAK1. Taken together, our results demonstrated that TRIM31 promoted apoptosis and inflammation in LPS-induced sepsis by activating the NF-κB signaling pathway.
Discussion
TRIM31 has been shown to play important roles in pancreatic cancer [Citation22], gallbladder cancer [Citation28], colorectal cancer [Citation29], hepatocellular carcinoma [Citation30], ovarian cancer [Citation31], non-small cell lung cancer [Citation32], gastric cancer [Citation33] and breast cancer [Citation20]. Additionally, TRIM31 has been shown to play roles in immunity and inflammation [Citation34–36].
Sepsis arises from an overwhelming immune response to invading microorganisms, which occasionally leads to multiple organ failure. Recent studies have demonstrated that the host immune system recognizes infection through recognition of pathogen-associated molecular patterns (PAMPs), such as LPS, RNA in viruses, flagellin and DNA in bacteria, mannan in fungi, and lipoteichoic acid [Citation37].
In this study, we started by examining the transcription level of TRIM31 in septic patients. Strikingly, we found a dramatic increase in TRIM31 transcription in septic patients compared to normal persons ()), suggesting that TRIM31 may play a role in sepsis progression. To study the mechanisms contributing to sepsis, we then generated an in vitro model using the AC16 cell line. LPS was used to mimic sepsis in AC16 cells. To determine optimal conditions, different LPS concentrations were used to treat AC16 cells followed by cell viability assay. We chose a moderate concentration of 0.5 µg/ml for optimal use for following studies (,)). TRIM31 expression was reported to be induced by LPS [Citation18]. Consistently, we observed that LPS-induced TRM31 expression in AC16 cells (,)).
NF-κB plays critical roles in various biological processes, including immune regulation, inflammation, and cell survival. Previous report has documented the existence of two distinct pathways of LPS-induced NF-κB activation and cytokine production in human myeloid and non-myeloid cells defined by selective utilization of TLR4, MyD88, Mal/TIRAP, and IKK2, and reveal a layer of complexity not previously expected [Citation38]. Accompanied with these findings, we proposed our hypothesis that TRIM31 may function through NF-κB signaling. To test our hypothesis, we examined the effects of TRIM31 depletion on LPS-induced apoptosis. Indeed, TRIM31 depletion significantly inhibited LPS-induced apoptosis ()). Similarly, TRIM31 depletion alleviated the increase in IL-1β, TNF-α, p-IKKβ, p-IκBα, and cleaved-caspase-3 levels induced by LPS (). Consistently, TRIM31 overexpression displayed opposite effects (). Together, these data suggested that the effects of TRIM31 on LPS-induced apoptosis was mediated by the NF-κB signaling pathway.
TAK1 is a member of the MAPKKK family, which can be activated by TGF-β, TNF, and IL-1. TAK1 complex contains its adaptor proteins (TAB1, TAB2, and TAB3). TAB1 constitutively interacts with TAK1 and participates in the autophosphorylation of TAK1 that is essential for TAK1 kinase activity [Citation39]. Upon stimulation, TAK1 kinase complex phosphorylates and activates IKK (IκB kinase), leading to activation of NF-κB [Citation40]. To test whether TRIM31 activates NF-κB signaling pathway via TAK1, we used 5Z-7-OX, a specific TAK1 inhibitor, to block the activity of TAK1. Interestingly, we observed that TRIM31 elevated LPS-induced apoptosis, but 5Z-7-OX blocked the effects of TRIM31 (). Further, we found that TRIM31 interacted with and ubiquitinated TAK1 ()). TRIM/RBCC proteins are considered as a novel class of “single protein RING finger” ubiquitin E3 ligases implicated in ubiquitination [Citation41]. TAK1 is an ubiquitin-dependent kinase of IKK, which means that TAK1 ubiquitination plays a key role in the regulation of its phosphorylation and activation [Citation12]. In this study, we found that TRIM31 directly interacted with TAK1 and that the RING domain of TRIM31 was mainly responsible for ubiquitination of TAK1. Moreover, TAK1 ubiquitination-mediated activation of the NF-κB pathway induced apoptosis in AC16 cells. Consistent with this, Trim31 induced apoptosis via activation of NF-κB signaling in vivo (, and ).
As TRIM31 increased TAK1 ubiquitination and subsequently activated NF-κB signaling, inhibition of TRIM31 played a protective role in SIMD. However, the exact mechanism pathway which TRIM31 activates TAK1 remains to be explored. According to previous studies, TAK1 activation most likely occurs through the synthesis of K63-linked polyubiquitin chains [Citation12]. It will be interesting to determine in future studies where TRIM31 ubiquitinates TAK1 through K63-linked polyubiquitin chains.
Conclusion
This study demonstrates for the first time that TRIM31 can induce apoptosis by activating the NF-κB signaling pathway in vitro and in vivo. Activation of NF-κB signaling is mediated by TAK1 ubiquitination. Our findings suggest that TRIM31 may be a potential target for protection of myocardial dysfunction induced by sepsis.
Figure 1. LPS-induced TRIM31 upregulation and cell apoptosis. (a) Real-time PCR was performed to measure the relative mRNA expression of TRIM31 in peripheral blood samples from 35 septic patients and 25 normal patients. *, P < 0.05, ***, P < 0.001 vs normal persons. (b) Cell proliferation was analyzed after stimulation with LPS (three repetitions). (c) Cell apoptosis was analyzed after LPS treatment. (d, e): The mRNA and protein levels of TRIM31 were measured by qRT-PCR (d) and Western blot (e). ***, P < 0.001 vs 0 μg/ml LPS
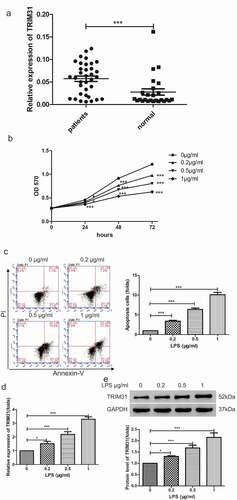
Figure 2. TRIM31 knockdown inhibited cell apoptosis induced by LPS. (a, b): The mRNA and protein levels of TRIM31 were measured by qRT-PCR (a) and Western blot (b). (c): Cell apoptosis was measured by flow cytometry. (d): ELISA was used to measure the concentrations of IL-1β and TNF-α. (e): The protein levels of NF-κB signaling related protein were measured by Western blot
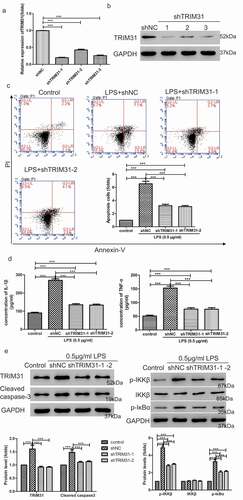
Figure 3. TRIM31 elevated cell apoptosis induced by LPS. (a, b): AC16 cells were transduced with TRIM31 overexpression or vector lentivirus. Overexpression efficiency was analyzed by qRT-PCR (a) and Western blot (b). (c): Annexin V/PI assay was used to assess cell apoptosis. (d): ELISA was used to measure the concentrations of IL-1β and TNF-α. (e): The protein levels of NF-κB signaling related protein were measured by Western blot
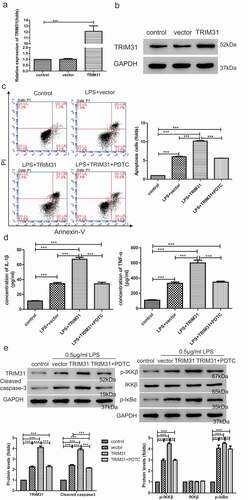
Figure 4. TAK1-mediated activation of NF-κB signaling pathway induced by TRIM31. (a): Western blot was used to determine the protein levels of TAK1 and p-TAK1. (b): Cell apoptosis was measured by flow cytometry. (c): The protein levels of cleaved-caspase-3, p-IKKβ and p-IκBα were measured by Western blot. (d): ELISA was used to measure the concentrations of IL-1β and TNF-α. (e): Co-immunoprecipitation assay was used to examine the interaction between TRIM31 and TAK1. (f): The ubiquitin level of TAK1 was measured
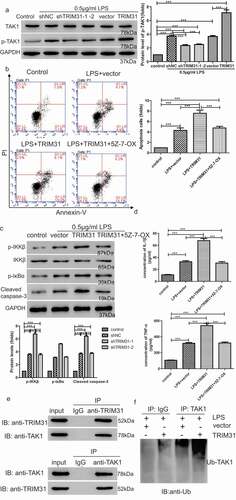
Figure 5. TRIM31 deletion suppressed apoptosis in mice model of sepsis induced by LPS. (a) H&E staining was performed on heart tissue samples of animal models. (b) IHC was used to detect the protein level of NF-κB (c) TUNEL assay was performed to measure apoptosis in heart tissue samples of animal models. a, control; b, LPS + vector; c, LPS + shTRIM31; d, LPS + PDTC
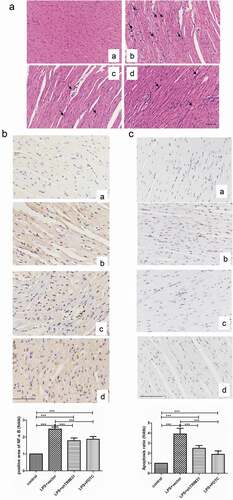
Figure 6. IHC was used to detect the protein levels of p-IKKβ (a) and p-IκBα (b). a, control; b, LPS + vector; c, LPS + shTRIM31; d, LPS + PDTC
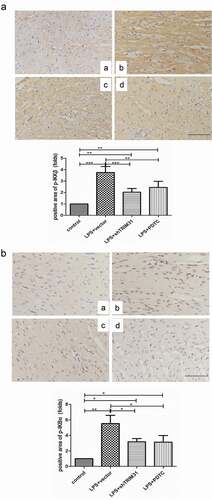
Figure 7. TRIM31 deletion blocked LPS-induced activation of NF-κB signaling pathway. (a): ELISA was used to measure the concentrations of IL-1β and TNF-α. (b) The protein levels of NF-κB signaling related protein were measured by Western blot
Supplemental Material
Download Zip (732.3 KB)Disclosure statement
The authors declare that they have no conflict of interests.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Angus DC, Linde-Zwirble WT, Lidicker J, et al. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310.
- Andriolo BN, Andriolo RB, Salomao R, et al. Effectiveness and safety of procalcitonin evaluation for reducing mortality in adults with sepsis, severe sepsis or septic shock. Cochrane Database Syst Rev. 2017;1:CD010959.
- Esper AM, Martin GS. Extending international sepsis epidemiology: the impact of organ dysfunction. Crit Care. 2009;13:120.
- Li X, Cheng Q, Li J, et al. Significance of hydrogen sulfide in sepsis-induced myocardial injury in rats. Exp Ther Med. 2017;14:2153–2161.
- Flynn A, Chokkalingam Mani B, Mather PJ. Sepsis-induced cardiomyopathy: a review of pathophysiologic mechanisms. Heart Fail Rev. 2010;15:605–611.
- Rudiger A, Singer M. Mechanisms of sepsis-induced cardiac dysfunction. Crit Care Med. 2007;35:1599–1608.
- Krishnagopalan S, Kumar A, Parrillo JE, et al. Myocardial dysfunction in the patient with sepsis. Curr Opin Crit Care. 2002;8:376–388.
- Pro CI, Yealy DM, Kellum JA, et al. A randomized trial of protocol-based care for early septic shock. N Engl J Med. 2014;370:1683–1693.
- Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013;12:86.
- Magne N, Toillon RA, Bottero V, et al. NF-kappaB modulation and ionizing radiation: mechanisms and future directions for cancer treatment. Cancer Lett. 2006;231:158–168.
- Ea CK, Deng L, Xia ZP, et al. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257.
- Wang C, Deng L, Hong M, et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351.
- Deng L, Wang C, Spencer E, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361.
- Short KM, Cox TC. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J Biol Chem. 2006;281:8970–8980.
- Li Q, Yan J, Mao AP, et al. Tripartite motif 8 (TRIM8) modulates TNFalpha- and IL-1beta-triggered NF-kappaB activation by targeting TAK1 for K63-linked polyubiquitination. Proc Natl Acad Sci USA. 2011;108:19341–19346.
- Rajsbaum R, Garcia-Sastre A, Versteeg GA. TRIMmunity: the roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J Mol Biol. 2014;426:1265–1284.
- Liu B, Zhang M, Chu H, et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat Immunol. 2017;18:214–224.
- Song H, Liu B, Huai W, et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat Commun. 2016;7:13727.
- Sugiura T, Miyamoto K. Characterization of TRIM31, upregulated in gastric adenocarcinoma, as a novel RBCC protein. J Cell Biochem. 2008;105:1081–1091.
- Dokmanovic M, Chang BD, Fang J, et al. Retinoid-induced growth arrest of breast carcinoma cells involves co-activation of multiple growth-inhibitory genes. Cancer Biol Ther. 2002;1:24–27.
- Zhang Z, Cheng L, Li J, et al. Inhibition of the Wnt/beta-catenin pathway overcomes resistance to enzalutamide in castration-resistant prostate cancer. Cancer Res. 2018;78:3147–3162.
- Pandey MKSB, Ahn KS, Kunnumakkara AB, et al. Gambogic acid, a novel ligand for transferrin receptor, potentiates TNF-induced apoptosis through modulation of the nuclear factor-kappaB signaling pathway. Blood. 2007;110(10):3517–3525. Blood 2013; 121:3778. .
- Pan P, Zhang H, Su L, et al. Melatonin balance the autophagy and apoptosis by regulating UCP2 in the LPS-induced cardiomyopathy. Molecules. 2018;23:675.
- Yu C, Chen S, Guo Y, et al. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-kappaB signaling pathway. Theranostics. 2018;8:3224–3236.
- Karin M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene. 1999;18:6867–6874.
- Sanjo H, Takeda K, Tsujimura T, et al. TAB2 is essential for prevention of apoptosis in fetal liver but not for interleukin-1 signaling. Mol Cell Biol. 2003;23:1231–1238.
- Shim JH, Xiao C, Paschal AE, et al. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005;19:2668–2681.
- Li H, Zhang Y, Hai J, et al. Knockdown of TRIM31 suppresses proliferation and invasion of gallbladder cancer cells by down-regulating MMP2/9 through the PI3K/Akt signaling pathway. Biomed Pharmacothe. 2018;103:1272–1278.
- Wang H, Yao L, Gong Y, et al. TRIM31 regulates chronic inflammation via NF-kappaB signal pathway to promote invasion and metastasis in colorectal cancer. Am J Transl Res. 2018;10:1247–1259.
- Guo P, Ma X, Zhao W, et al. TRIM31 is upregulated in hepatocellular carcinoma and promotes disease progression by inducing ubiquitination of TSC1-TSC2 complex. Oncogene. 2018;37:478–488.
- Wei Z, Liu Y, Wang Y, et al. Downregulation of Foxo3 and TRIM31 by miR-551b in side population promotes cell proliferation, invasion, and drug resistance of ovarian cancer. Med Oncol. 2016;33:126.
- Li H, Zhang Y, Zhang Y, et al. TRIM31 is downregulated in non-small cell lung cancer and serves as a potential tumor suppressor. Tumour Biol. 2014;35:5747–5752.
- Sugiura T. The cellular level of TRIM31, an RBCC protein overexpressed in gastric cancer, is regulated by multiple mechanisms including the ubiquitin-proteasome system. Cell Biol Int. 2011;35:657–661.
- Dai T, Wu L, Wang S, et al. FAF1 regulates antiviral immunity by inhibiting MAVS but is antagonized by phosphorylation upon viral infection. Cell Host Microbe. 2018;24:776–90 e5.
- Onoufriadis A, Stone K, Katsiamides A, et al. Exome sequencing and genotyping identify a rare variant in NLRP7 gene associated with ulcerative colitis. J Crohns Colitis. 2018;12:321–326.
- Kimsa MW, Strzalka-Mrozik B, Kimsa MC, et al. Differential expression of tripartite motif-containing family in normal human dermal fibroblasts in response to porcine endogenous retrovirus infection. Folia Biol (Praha). 2014;60:144–151.
- Kakihana Y, Ito T, Nakahara M, et al. Sepsis-induced myocardial dysfunction: pathophysiology and management. J Intensive Care. 2016;4:22.
- Andreakos E, Sacre SM, Smith C, et al. Distinct pathways of LPS-induced NF-kappa B activation and cytokine production in human myeloid and nonmyeloid cells defined by selective utilization of MyD88 and Mal/TIRAP. Blood. 2004;103:2229–2237.
- Roh YS, Song J, Seki E. TAK1 regulates hepatic cell survival and carcinogenesis. J Gastroenterol. 2014;49:185–194.
- Yamaguchi K, Shirakabe K, Shibuya H, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270:2008–2011.
- Meroni G, Diez-Roux G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. BioEssays. 2005;27:1147–1157.