
ABSTRACT
DNA double strand breaks (DSBs) are among the most toxic DNA lesions and can be repaired accurately through homologous recombination (HR). HR requires processing of the DNA ends by nucleases (DNA end resection) in order to generate the required single-stranded DNA (ssDNA) regions. The SWI/SNF chromatin remodelers are 10–15 subunit complexes that contain one ATPase (BRG1 or BRM). Multiple subunits of these complexes have recently been identified as a novel family of tumor suppressors. These complexes are capable of remodeling chromatin by pushing nucleosomes along the DNA. More recent studies have identified these chromatin remodelers as important factors in DNA repair. Using the DR-U2OS reporter system, we show that the down regulation of BRG1 significantly reduces HR efficiency, while BRM has a minor effect. Inactivation of BRG1 impairs DSB repair and results in a defect in DNA end resection, as measured by the amount of BrdU-containing ssDNA generated after DNA damage. Inactivation of BRG1 also impairs the activation of the ATR kinase, reduces the levels of chromatin-bound RPA, and reduces the number of RPA and RAD51 foci after DNA damage. This defect in DNA end resection is explained by the defective recruitment of GFP-CtIP to laser-induced DSBs in the absence of BRG1. Importantly, we show that BRG1 reduces nucleosome density at DSBs. Finally, inactivation of BRG1 renders cells sensitive to anti-cancer drugs that induce DSBs. This study identifies BRG1 as an important factor for HR, which suggests that BRG1-mutated cancers have a DNA repair vulnerability that can be exploited therapeutically.
Introduction
DNA double strand breaks (DSBs) are among the most cytotoxic and carcinogenic forms of DNA damage [Citation1,Citation2]. The toxicity of this type of DNA lesion is exploited by radiation therapy and the vast majority of anti-cancer agents, which generate an overwhelming number of DSBs, thus inducing cell death [Citation3–5]. DSBs must be repaired promptly in order to safeguard genomic integrity. If left unrepaired, these lesions can also lead to chromosomal aberrations and translocations that could result in cancer.
DSBs are repaired by two different pathways in human cells, non-homologous end joining (NHEJ) and homologous recombination (HR). NHEJ is the fastest and most prevalent DSB repair pathway in human cells and entails the joining of two broken DNA ends with no requirement for sequence homology [Citation6]. HR, on the other hand, is a more complex and accurate pathway for the repair of DSBs [Citation2]. HR uses a sister chromatid as a template in order to repair DSBs and therefore can only occur during late stages of S phase and the G2 phase of the cell cycle. This repair mechanism entails the initial recognition of the break by the MRE11-RAD50-NBS1 (MRN) complex and the ATM kinase [Citation7,Citation8], followed by the recruitment of the CtIP nuclease. MRE11 and CtIP initiate the nuclease-mediated degradation of the 5ʹ DNA ends in a process known as DNA end resection, which is critical for HR [Citation9]. DNA end resection generates long stretches of single-stranded DNA (ssDNA) with a free 3ʹ end that will be coated by the ssDNA binding protein RPA. These RPA-coated ssDNA regions activate the ATR kinase [Citation10,Citation11]. RPA will then be replaced by the RAD51 recombinase, which will form filaments along these ssDNA regions and catalyze the homology search and strand invasion steps of HR [Citation2]. Defects in HR result in increased sensitivity to various anti-cancer drugs and increased chromosomal aberrations likely due to the overuse of the NHEJ pathway [Citation3,Citation4,Citation12]. One specific genetic interaction identified regarding HR and chemotherapy drugs is the discovery that HR-deficient cells are particularly sensitive to the inhibition of poly (ADP-ribose) polymerase (PARP) [Citation13,Citation14]. Indeed, this lead to the use of PARP inhibitors (PARPi) to treat breast and ovarian cancers that bear mutations in the breast cancer susceptibility genes (BRCA1/2), both of which play important roles in HR [Citation14,Citation15]. The use of PARPi to target specifically HR-deficient cells has stimulated the search for novel players in the HR pathway that would display this synthetic lethality effect and that are also mutated in cancers. Identifying new genes important for HR that are mutated in cancer could lead to the expansion of the number and types of cancers that could respond to PARPi therapy.
The importance of chromatin structure in DNA repair is now widely recognized [Citation16,Citation17]. Chromatin structure seems to be particularly important in the case of HR, since it has been proposed that nucleosomes must be removed or moved out of the way in order for DNA end resection to take place [Citation18,Citation19]. SWI/SNF chromatin remodeling complexes are potential candidates for this function in mammalian cells. These complexes are composed of 10–15 subunits with one catalytic ATPase (BRG1 or BRM), three core subunits (BAF47, BAF155, BAF170), and a group of accessory factors of unknown function ()) [Citation20,Citation21]. In mammalian somatic cells there are mainly two forms of the SWI/SNF complexes: BAF (BRG1/BRM-associated factors) and PBAF (polybromo-BRG1-associated factors). The BAF complex can contain either ATPase, BRG1 or BRM, while the PBAF complex can only contain the BRG1 ATPase [Citation20]. The PBAF complex also contains two bromodomain-containing proteins, BRD7 and PBRM1, which bind to acetylated histones [Citation20]. Both of these chromatin remodeling complexes have been widely studied in the context of transcription regulation and are known as activators and repressors that push and slide nucleosomes in order to allow or block the binding of transcription factors to DNA sequences [Citation21,Citation22]. Multiple subunits of the SWI/SNF complexes have been identified as a novel family of tumor suppressors mutated at very high frequencies in various cancers [Citation23]. The mechanisms of tumor suppression for this family of chromatin remodelers in poorly understood. In a previous study we showed that BRG1 is recruited to DSBs through its interactions with a TopBP1-E2F1-RB complex and that destabilizing this complex by inactivating RB impaired the recruitment of BRG1 to DSBs. These RB-deficient cells displayed a defect in DNA end resection and HR [Citation24]. Other groups have shown that cells lacking BRG1 display a defect in DNA damage signaling in the context of UV-induced DNA damage and replication arrest [Citation25,Citation26]. Regarding DSB repair, there are reports linking BRG1 phosphorylation by the ATM kinase and its binding to the phosphorylated histone variant H2AX (γH2AX) through acetylated residues on γH2AX as important for the repair process [Citation27,Citation28]. BRG1 is mutated at very high frequencies in a variety of cancers and it is therefore of critical importance to define the function of this ATPase and the SWI/SNF complexes in the repair of DSBs, as it could have important implications in the treatment of BRG1-mutated cancers.
Figure 1. BRG1 is recruited to DSBs and stimulates HR. (a) SWI/SNF complexes in somatic mammalian cells exist in two combinations: BAF and PBAF. The BAF complex can contain either the BRG1 or BRM ATPase (red), whereas the PBAF complex contains exclusively BRG1. Both complexes contain a group of core subunits (BAF47, BAF155, BAF170, orange) and a number of accessory factors of unknown function (yellow). Subunits marked with an asterisk (*) are known to be mutated in a variety of cancers [Citation20]. (b) U2OS cells were transduced with a retrovirus expressing ER*-HA-I-PpoI. These cells were treated for 12 h with tamoxifen (2 μM, + I-PpoI) or not (- I-PpoI), crosslinked, and nuclear extracts were prepared. Chromatin immunoprecipitation was performed for the indicated proteins. Quantitative PCR was performed to determine the amount of protein recruited at the specific locus (489 bp 3ʹ to the I-PpoI cut site in the rDNA region). Fold enrichment was calculated by dividing the percentage (%) of input of the + I-PpoI by the – I-PpoI. The % of input refers to the amount of DNA obtained from the immunoprecipitation of the given factor divided by the total amount of DNA (input). (c) HR efficiency was measured using the DR-U2OS system [Citation36]. DR-U2OS cells were transfected with the indicated siRNAs and 72 h later cells were transfected with an empty plasmid (-) or a plasmid encoding the I-SceI endonuclease (+). After 48 h, the % of GFP positive cells was measured by flow cytometry to assess HR efficiency. Results were normalized as siCTRL equal to 100%. All experiments were done in triplicate and graphs represent averages of three independent experiments ± SD (* p < 0.05, ** p < 0.01 by student t test)
![Figure 1. BRG1 is recruited to DSBs and stimulates HR. (a) SWI/SNF complexes in somatic mammalian cells exist in two combinations: BAF and PBAF. The BAF complex can contain either the BRG1 or BRM ATPase (red), whereas the PBAF complex contains exclusively BRG1. Both complexes contain a group of core subunits (BAF47, BAF155, BAF170, orange) and a number of accessory factors of unknown function (yellow). Subunits marked with an asterisk (*) are known to be mutated in a variety of cancers [Citation20]. (b) U2OS cells were transduced with a retrovirus expressing ER*-HA-I-PpoI. These cells were treated for 12 h with tamoxifen (2 μM, + I-PpoI) or not (- I-PpoI), crosslinked, and nuclear extracts were prepared. Chromatin immunoprecipitation was performed for the indicated proteins. Quantitative PCR was performed to determine the amount of protein recruited at the specific locus (489 bp 3ʹ to the I-PpoI cut site in the rDNA region). Fold enrichment was calculated by dividing the percentage (%) of input of the + I-PpoI by the – I-PpoI. The % of input refers to the amount of DNA obtained from the immunoprecipitation of the given factor divided by the total amount of DNA (input). (c) HR efficiency was measured using the DR-U2OS system [Citation36]. DR-U2OS cells were transfected with the indicated siRNAs and 72 h later cells were transfected with an empty plasmid (-) or a plasmid encoding the I-SceI endonuclease (+). After 48 h, the % of GFP positive cells was measured by flow cytometry to assess HR efficiency. Results were normalized as siCTRL equal to 100%. All experiments were done in triplicate and graphs represent averages of three independent experiments ± SD (* p < 0.05, ** p < 0.01 by student t test)](/cms/asset/7610d62c-0d8d-435d-8bfa-d1ae4cf40502/kccy_a_1831256_f0001_c.jpg)
In this study we evaluated the role of BRG1 and BRM in HR. While both ATPases are recruited to DSBs, depletion of BRG1 causes a significant reduction in HR efficiency, when compared to the modest reduction observed upon depletion of the BRM ATPase. Moreover, inactivation of BRG1 using CRISPR/Cas9 resulted in slower repair of DSBs as measured by comet assay and by the clearance of γH2AX after camptothecin (CPT) treatment. Inactivation of BRG1 also impairs the activation of the ATR kinase after the induction of DSBs, while the activation of the ATM kinase was not affected. Moreover, inactivation of BRG1 results in impaired DNA end resection when measured by the generation of ssDNA by BrdU, or RPA foci-formation, or the amount of chromatin-bound RPA after CPT treatment. In agreement with a defect in DNA end resection and HR, inactivation of BRG1 also results in decreased levels of chromatin-bound RAD51 and RAD51 foci-formation after CPT treatment. This defect in DNA end resection can be explained by the finding that cells lacking BRG1 display a defect in the recruitment of GFP-CtIP to laser-induced DSBs in live cells, and reduced CtIP foci-formation after CPT treatment. Importantly, the inactivation of BRG1 does not affect MRE11 foci-formation. At the chromatin level, we observe that while control cells show decreased nucleosome density at DSBs, cells lacking BRG1 show increased nucleosome density at break sites. Finally, inactivation of BRG1 rendered cells sensitive to a variety of anti-cancer agents that induce DSBs, including: CPT, etoposide, bleomycin, and cisplatin. Our work shows that BRG1 plays an important role in HR by stimulating DNA end resection through the reduction of nucleosome density at DSBs. We propose that this reduction of nucleosome density at DSBs may be important for the recruitment and/or stabilization of the CtIP nuclease and the DNA end resection process. As BRG1 is mutated at very high frequencies in a variety of cancers, it is possible that this repair function contributes to the tumor suppressor capacity of this chromatin remodeler. It is also possible that this DNA repair vulnerability can be exploited for the treatment of tumors bearing BRG1 mutations.
Materials and methods
Cell lines
U2OS cells were obtained from ATCC and maintained in DMEM media supplemented with 10% FBS (X&Y Cell Culture) and antibiotics at 37°C in 5% CO2. BRG1 knock-down cell lines were generated by transducing U2OS cells with lentiviral particles expressing shRNA targeting the SMARCA4 gene (human BRG1, Santa Cruz Biotechnology) following by selection using puromycin (Sigma). Single cell clones were isolated by cell sorting (Bio-Rad S3e cell sorter) and knock-down efficiency was confirmed by western blot analysis. CRISPR/Cas9 knock-out (KO) cells were generated using the system designed by the Zhang lab [Citation29]. Briefly, single guide RNAs targeting exon 3 of the SMARCA4 gene (forward guide CACCGGCCGAGGAGTTCCGCCCAG, reverse guide AAACCTGGGCGGAACTCCTCGGCC) were designed using the Benchling platform (www.benchling.com), annealed and cloned into the pSpCas9(BB)-2A-Puro(PX459) V2.0 plasmid (Addgene ID# 62,988). U2OS cells were transfected with the empty plasmid (Cas9) or with the plasmid containing the single guide RNA targeting BRG1 (BRG1-KO). Puromycin selection was performed for 7 days following transfection, single clones were isolated by cell sorting (Bio-Rad S3e cell sorter), and BRG1-KO clones were identified by Western blot analysis. IR treatments were performed with RS-2000 Biological Irradiator (Rad Source). When indicated, cells were treated with the camptothecin (CPT, Cayman Chemical Co.), bleomycin (LKT Laboratories), cisplatin (Cayman Chemical Co), etoposide (Cayman Chemical Co), or olaparib (SelleckChem).
Antibodies
See supplementary table S1.
Chromatin immunoprecipitation (ChIP)
U2OS cells were transduced with a retrovirus expressing HA-ER*-I-PpoI (Addgene ID# 32,565) enzyme twice to increase transduction efficiency and treated with 2 μM tamoxifen (Sigma) for 12 h [Citation30]. Cells were harvested by adding formaldehyde (1% final concentration, 15 min, Sigma), and quenched by glycine (1.25 mM final concentration, Sigma) [Citation31]. Nuclear extracts were prepared and sonicated (Diagenode Bioruptor). Occupancy was measured by qPCR using primers for the rDNA locus [Citation30]. Each experiment was carried out in triplicate.
Homologous recombination assay
DR-U2OS cells (gift from Dr. Maria Jasin, MSKCC) were transfected with control-scrambled siRNAs smart pool (siCTRL) or siRNAs smart pool against different proteins (100 nM, Dharmacon) as indicated, using lipofectamine 2000 (Invitrogen). Seventy-two hours post transfection, cells were transfected with pCAB-ISceI plasmid (Addgene ID# 26,477) using Fugene HD (Promega). Forty-eight hours post-transfection, cells were harvested by trypsinization and resuspended in PBS and flow cytometry was performed to monitor GFP positive cells (Beckman Coulter CytoFLEX). Analysis was performed using Kaluza Analysis 2.0. A fraction of the cells was used to perform western blot analysis.
Western blot analysis
Cells were treated with drugs as indicated (or not treated) and harvested in cold PBS, followed by resuspension in cell lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 0.5 mM EGTA, protease inhibitor cocktail and phosphatase inhibitor cocktail). Protein concentration of whole cell lysates was measured and 50 µg of protein was used for SDS-PAGE. Gels were transferred to PDVF membranes, blocked for 30 min in 5% dry milk or BSA (Sigma) and incubated with the primary antibodies overnight. Secondary antibodies conjugated to HRP were used and visualized by ECL plus (PerkinElmer) and Bio-Rad Chemidoc.
Comet assay
Assay was performed following the manufacturer’s directions (Cell Biolabs). Cells were treated with the indicated drug for 1 h, and then allowed to repair the DNA damage for 2 h or 24 h. Cells were harvested at the indicated times by trypsinization, diluted in low melting point agarose, lysed in neutral buffer solution, cell electrophoresis was performed, DNA was stained and images were acquired (Leica DM5500 B microscope). Comet tails were measured (50 comets per cell line, per time point, per experiment) using ImageJ and the tail moment was calculated by using the Open Comet plugin. Tail moment is defined as the product of the tail length and the percentage of DNA in the comet tail.
Histone γH2AX, pATM, RAD51, CtIP nuclear foci
Cells were seeded on coverslips 24 h prior to the experiment. Cells were treated with the indicated drugs for the indicated times, rinsed with cold PBS once, fixed 15 min at room temperature in 3% formaldehyde (Sigma), permeabilized with 0.5% Triton X-100 (Sigma) in PBS for 5 min, and blocked in 10% FCS in PBS for 45 min. Then cells were incubated with the appropriate primary antibodies for 1 h and rinsed 3X with cold PBS, followed by incubation with the appropriate secondary antibodies conjugated to Alexa fluor 488 and Alexa fluor 596 (Invitrogen) for 1 h at room temperature, and rinsed 3X with cold PBS. Coverslips were mounted in ProLong Gold with DAPI (Invitrogen) and pictures were taken using a confocal microscope (60x oil, Nikon A1R High-Speed Confocal microscope). Foci quantification was performed using ImageJ software and at least 50 cells per time point per genotype were analyzed per experiment for data analysis.
Single-stranded DNA BrdU immunofluorescent staining and RPA foci
Briefly, cells were incubated with BrdU (100 μM, Sigma) for 36 h, treated with the indicated drug and harvested at the indicated times, as previously described [Citation32]. In situ extraction protocol was employed for the visualization of BrdU, RPA, and MRE11 foci. At the indicated times cells were rinsed with cold PBS, incubated 5 min in pre-extraction buffer (25 mM HEPES pH 7.5, 50 mM NaCl, 1 mM EDTA, 3 mM MgCl2, 300 mM Sucrose, 0.5% Triton X-100) at 4°C, incubated 5 min in CSK buffer (10 mM Tris pH 7.4,10 mM NaCl, 3 mM MgCl2, 1 mM EDTA, 0.5% Triton X-100, 0.5% Na-Deoxycholate) at 4°C, fixed 15 min at room temperature in 3% formaldehyde (Sigma), permeabilized with 0.5% Triton X-100 (Sigma) for 5 min and blocked in 10% FCS for 45 min and stained with an anti-BrdU and anti-cyclin A antibodies (to label S/G2 cells), or anti-RPA and anti-cyclin A antibodies. Images were taken on the confocal microscope (60x oil, Nikon A1R High-Speed Confocal microscope) and foci number was measured in cyclin A positive cells (late S/G2 phase cells) with ImageJ software.
Chromatin-bound RPA or RAD51 by flow cytometry
The protocol was adapted from Forment et al. [Citation33]. Briefly, cells were mock treated or treated with camptothecin (CPT, 0.5 μM for 1 h) and harvested by trypsinization. Cells were permeabilized 10 min in ice with T-PBS (PBS + 0.2%Triton X-100), fixed in 4% formaldehyde and stained with and anti-RPA antibody (1 h), rinsed and incubated with a secondary antibody conjugated to Alexa fluor 647. Cells were resuspended in and RNAse A/PI solution and FACS analysis was performed as previously described [Citation33]. The increase in chromatin-bound RPA in S/G2 cells upon CPT treatment was measured using Kaluza Analysis 2.0. Samples to monitor the increase in chromatin-bound RAD51 were treated in the same way, except that after the 1 h CPT treatment, cells were rinsed with fresh media and allowed 6 h for HR to take place and then harvested as above. Immuno staining was performed with a RAD51 antibody.
Cell cycle synchronization
Cells were synchronized in late S/G2 phase by double thymidine block. Briefly, cells were incubated with media containing 2 mM thymidine (Sigma) for 18 h (first thymidine block), rinsed twice with warm PBS and incubated with fresh media for 9 h (first thymidine release). Then cells were incubated again in media containing 2 mM thymidine (second thymidine block) for 18 h, rinsed twice with warm PBS and incubated in fresh media (second thymidine release, cells arrested at G1) for 8 h (late S/G2 phases). At this point cells were treated with the indicated drugs for the indicated times. Cell cycle synchronization was verified by flow cytometry.
Cell cycle analysis
Cell cycle analysis was performed by monitoring DNA content using flow cytometry. Cells were harvested by trypsinization at the indicated times, fixed in 70% ethanol solution and incubated overnight. Cells were then resuspended in PBS staining solution (0.5% BSA, 0.5% Tween-20, 1 µg/µL PI, 20 µg/mL RNAse A). Data collection was performed in a Beckman Coulter CytoFLEX and analysis was performed in Kaluza Analysis 2.0.
Laser micro irradiation
Cells were seeded in 35 mm glass-bottom dishes on day one. Day two cells were transfected with pCDNA5-eGFP-CtIP plasmid (0.5 µg, gift from Dr. Tanya Paull, UT Austin) using Fugene HD (Promega). Day three the media was replaced with fresh media containing 10 µM BrdU (Sigma). Day four cells were microirradiated using a 405 nm laser and images were acquired every 10 seconds for 10 min. Experiments were performed in 60x oil, Nikon A1R High-Speed Confocal microscope. A minimum of 20 cells were analyzed per genotype. Analysis was performed in ImageJ.
Cell survival assay
Cells (10,000 cells/well) were seeded in 96-well plate and 24 h after were treated with the indicated concentrations of the indicated drugs for 4 days. On day 4 cell survival was assessed using CellTiter96 (Invitrogen). Experiments were done in triplicate.
Activation of the caspase 3/7 pathway by flow cytometry
The activation of the caspase 3/7 was measured by the CellEvent Caspase 3/7 green kit (Invitrogen). Cells were treated with the indicated drug for 72 h and then harvested in cold PBS, incubated with the caspase 3/7 fluorescent substrate for 30 min and samples were analyzed by flow cytometry using Cytox (Invitrogen) as a DNA dye.
Colony forming assay
One thousand cells were seeded per well of a 6 well plate and left to attach overnight. Then, cells were treated with increasing concentrations of the indicated drug for 10–14 days. After the incubation period the media was removed and the wells were rinsed with PBS twice, followed by a 30 min incubation in 6% glutaraldehyde (v/v, Sigma) and 0.5% crystal violet (v/v, Sigma). Plates are rinsed with water 3 times and air dried. Pictures are acquired and colonies are counted using ImageJ.
Statistical analysis
All experiments were carried out in triplicates and statistical analysis was performed in GraphPad Prism 8 using the unpaired t test analysis when comparing two samples and ANOVA when comparing more than two samples. Significance was determined by p < 0.05 is significant (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****) is highly significant.
Results
BRG1 is recruited to DSBs and stimulates HR
SWI/SNF chromatin remodeling complexes are characterized by the presence of one ATPase subunit (BRG1 or BRM), which is stabilized by a group of core factors (BAF47, BAF155, BAF170) ()) [Citation20]. The BAF complex can contain either BRG1 or BRM, but the PBAF complex contains exclusively BRG1 [Citation20]. Both of these ATPases are capable of remodeling chromatin but the functions of these ATPases do not necessarily overlap. First, we monitored the recruitment of BRG1 and BRM to DSBs. We used the I-PpoI system designed by the Kastan lab to induce enzyme-mediated DSBs upon treatment of U2OS cells, which had been transduced with a retrovirus encoding the I-PpoI nuclease fused to a modified estrogen receptor (ER), with tamoxifen for 12 hours [Citation30]. After treatment with tamoxifen, we performed chromatin immunoprecipitation (ChIP) using antibodies against BRG1 and BRM, and primers flanking a break site at the rDNA locus (489 bp 3ʹ to the I-PpoI cut site). As shown in ), BRG1 and BRM are recruited to DSBs. This is in agreement with previous work in yeast and human cells [Citation24,Citation34,Citation35]. We next decided to test whether these ATPases played a role in HR. We used the DR-U2OS system designed by the Jasin Lab to test the effect of these enzymes in HR [Citation36]. We find that BRG1 stimulates HR, as down regulation of this ATPase reduced HR efficiency by ~40-50%, while down regulation of BRM had a modest effect on HR (15% reduction), in agreement with our previous work () and Supplementary Fig. S1A) [Citation24]. Importantly, the silencing of BRG1 did not have a significant effect in cell cycle progression (Supplementary Fig. S1B). It is important to note that BRM clearly cannot substitute for the function of BRG1 in HR, as the absence of BRG1 in the presence of BRM shows a significant HR defect ()). Together, these data show that while BRG1 and BRM are recruited to DSBs, BRG1 plays a more prominent role in HR, and will be the focus of this study.
Inactivation of BRG1 impairs the repair of DSBs and the activation of the ATR kinase
In order to study the function of BRG1 we inactivated this ATPase in U2OS cells using CRISPR/Cas9 technology [Citation29]. Using the CRISPR/Cas9 system designed by the Zhang lab we targeted the exon 3 of BRG1 (SMARCA4 gene) with a single guide RNA and the Cas9 nuclease [Citation29]. Cells were selected in puromycin for 7 days after the sg+Cas9 transfection and single cell clones were isolated by cell sorting. Inactivation of BRG1 was confirmed by western blot and did not result in a significant decrease in protein levels of any of the other SWI/SNF subunits we tested, including BRM ()). To confirm that the inactivation of BRG1 resulted in a defect in DSB repair we performed the comet assay in cells lacking BRG1 (BRG1-KO). U2OS cells were treated with the topoisomerase I poison camptothecin (CPT) for 1 h (1 μM), then the drug was washed off and cells were allowed to repair the damage for 2 h or 24 h. At the indicated times, cells were lysed and subjected to cell electrophoresis where their genomic material would migrate. This migration would leave a DNA trace (comet tail) that represents the unrepaired (fragmented) DNA [Citation37]. Inactivation of BRG1 resulted in a DNA repair defect shown by the higher tail moment at every time point studied after CPT treatment in cells lacking BRG1, when compared to control cells (, c) and Supplementary Fig. S2D). A similar repair defect was observed when cells were treated with the radiomimetic drug bleomycin (Bleo), which also induces DSBs (Supplementary Fig. S2A-C). We also monitored the phosphorylation of the histone variant H2AX (γH2AX) after CPT treatment. DNA damage-induced γH2AX nuclear foci are a known maker for DSBs and their disappearance is considered a measure of DSB repair [Citation38,Citation39,Citation40]. BRG1-KO cells displayed higher levels of γH2AX foci than control cells at every time point after treatment with CPT and even in the absence of CPT () and Supplementary Fig. S2E). Altogether, these data demonstrate that BRG1 plays a role in the repair of DSBs and its inactivation results in a defect in the repair of these breaks.
Figure 2. Inactivation of BRG1 impairs the repair of DSBs and the activation of the ATR kinase. (a) Western blot analysis of parental U2OS cells (U2OS) that were transfected with an empty CRISPR/Cas9-containing plasmid (Cas9) or with the CRISPR/Cas9 containing a single guide DNA sequence targeting the exon 3 of the SMARCA4 gene (BRG1-KO) [Citation29]. Cells were selected with puromycin for 7 days after transfection and two single cell clones were isolated by cell sorting. (b) Control U2OS cells (Cas9) and U2OS cells lacking BRG1 (BRG1-KO) were subjected to the comet assay after treatment with camptothecin (+CPT, 1 μM for 1 h) and allowed to repair the damage for the indicated times. Then, cells were lysed, subjected to cell electrophoresis, and their DNA was stained. Representative images of the comets at 2 h after 1 h CPT treatment are shown. (c) Comet tail moment were calculated with ImageJ using the Open comet plug-in at the indicated times after treatment with CPT (1 μM for 1 h). (d) Control U2OS cells (Cas9) and U2OS cells lacking BRG1 (BRG1-KO) were grown in cover slips and treated with CPT (1 μM for 1 h) and allowed to repair their DNA for the indicated times. Cells were fixed at the indicated times and stained with an antibody against γH2AX. γH2AX foci images were acquired and the foci were counted with ImageJ. (e) Control U2OS cells (Cas9) and U2OS cells lacking BRG1 (BRG1-KO) were treated with 0.5 μM CPT for the indicated times, whole cell lysates were prepared, and subjected to SDS-PAGE. The levels of phosphorylated Chk1 (S345) and RPA (S4/8) were monitored with the indicated antibodies. GAPDH is used as a loading control. Representative images are shown. All experiments were done in triplicate and graphs represent averages of three independent experiments ± SD (* p < 0.05, ** p < 0.01, *** p < 0.001 by student t test)
![Figure 2. Inactivation of BRG1 impairs the repair of DSBs and the activation of the ATR kinase. (a) Western blot analysis of parental U2OS cells (U2OS) that were transfected with an empty CRISPR/Cas9-containing plasmid (Cas9) or with the CRISPR/Cas9 containing a single guide DNA sequence targeting the exon 3 of the SMARCA4 gene (BRG1-KO) [Citation29]. Cells were selected with puromycin for 7 days after transfection and two single cell clones were isolated by cell sorting. (b) Control U2OS cells (Cas9) and U2OS cells lacking BRG1 (BRG1-KO) were subjected to the comet assay after treatment with camptothecin (+CPT, 1 μM for 1 h) and allowed to repair the damage for the indicated times. Then, cells were lysed, subjected to cell electrophoresis, and their DNA was stained. Representative images of the comets at 2 h after 1 h CPT treatment are shown. (c) Comet tail moment were calculated with ImageJ using the Open comet plug-in at the indicated times after treatment with CPT (1 μM for 1 h). (d) Control U2OS cells (Cas9) and U2OS cells lacking BRG1 (BRG1-KO) were grown in cover slips and treated with CPT (1 μM for 1 h) and allowed to repair their DNA for the indicated times. Cells were fixed at the indicated times and stained with an antibody against γH2AX. γH2AX foci images were acquired and the foci were counted with ImageJ. (e) Control U2OS cells (Cas9) and U2OS cells lacking BRG1 (BRG1-KO) were treated with 0.5 μM CPT for the indicated times, whole cell lysates were prepared, and subjected to SDS-PAGE. The levels of phosphorylated Chk1 (S345) and RPA (S4/8) were monitored with the indicated antibodies. GAPDH is used as a loading control. Representative images are shown. All experiments were done in triplicate and graphs represent averages of three independent experiments ± SD (* p < 0.05, ** p < 0.01, *** p < 0.001 by student t test)](/cms/asset/4cd0310c-cb00-44b4-8d6c-10a282881496/kccy_a_1831256_f0002_c.jpg)
We next wanted to test the activation of the DNA damage response in cells lacking BRG1. We first measured the activation of the ATR kinase, as our previous work and that of others in yeast suggest that BRG1 stimulates DNA end resection [Citation24,Citation35]. Inactivation of BRG1 resulted in an attenuated activation of the ATR kinase, measured by the phosphorylation of Chk1 (pChk1) and RPA (pRPA) after CPT treatment () and S2F). We also observed similar results upon depletion of BRG1 by shRNA and exposing those cells to ionizing radiation (Supplementary Fig. S2G). Interestingly, the inactivation of BRG1 did not seem to affect the activation of the ATM kinase, as observed directly by the phosphorylation of the Chk2 kinase, or the number of phosphorylated ATM (S1981, pATM) nuclear foci (Supplementary Figure S2G-I). Altogether, these data demonstrate that BRG1 is important for the repair of DSBs and for the activation of the ATR kinase, but not for the activation of the ATM kinase.
Inactivation of BRG1 impairs DNA end resection
The ATR kinase is activated by the RPA-coated ssDNA regions generated during DNA end resection, and thus a defective ATR activation often stems from a defect in DNA end resection [Citation10,Citation11]. In order to test whether the inactivation of BRG1 results in a defect in DNA end resection we measured resection first by using a BrdU incorporation-based method [Citation32]. In this method cells are incubated with BrdU for 36–48 h in order for the cells to incorporate BrdU into their DNA, then cells are treated with DNA damage and resection is allowed to take place. After resection has taken place, cells are fixed and immunofluorescence is performed under non-denaturing conditions using antibodies against BrdU and cyclin A. Under non-denaturing conditions the BrdU antibody only reacts with BrdU contained within ssDNA. Thus, following DNA damage the BrdU-containing ssDNA that will be detected arose from resection and can be used as a measure of resection in late S/G2 cells (cyclin A positive cells) [Citation32]. Inactivation of BRG1 results in decreased levels of CPT-induced ssDNA when compared to control cells (, b)). These results show that BRG1 stimulates DNA end resection. These ssDNA regions are immediately coated by RPA. CPT-induced RPA nuclear foci were measured in these cells and were also reduced in cells lacking BRG1, when compared to control cells (, d)), thus further confirming the defect in DNA end resection in the absence of BRG1.
Figure 3. Inactivation of BRG1 impairs DNA end resection. (a) Inactivation of BRG1 impairs the generation of ssDNA after DNA damage. Control U2OS cells (Cas9) and U2OS cells lacking BRG1 (BRG1-KO) were grown on coverslips, incubated with BrdU (100 μM) for 48 h, then were not treated (NT), or treated with CPT (+CPT, 1 μM for 1 h), and allowed to perform repair for 4 h [Citation32]. Cells were then incubated with pre-extraction and extraction buffers and fixed with formaldehyde. Cells were immuno-labeled with antibodies against cyclin A and BrdU. The generation of ssDNA was measured by quantifying BrdU foci in cyclin A positive cells (late S/G2 cells), as the antibody only recognizes BrdU contained within ssDNA regions, which in this case arose due to DNA end resection after CPT treatment. The DNA was stained using DAPI. Images were acquired using a confocal microscope (60X oil). Quantification of ssDNA regions was performed using ImageJ and approximately 50 cells were counted by time point, per experiment. (b) Representative images are shown of control cells (Cas9) and cells lacking BRG1 (BRG1-KO) stained with a BrdU antibody following no treatment (NT), 1 h CPT treatment (+CPT, 1 µM), and 1 h CPT treatment followed by 4 h recovery (+CPT + 4 h, 1 µM). (c) Inactivation of BRG1 also impairs RPA foci-formation after DNA damage. Cells were treated as in (A) and stained with antibodies against cyclin A and RPA. CPT-induced RPA foci in cyclin A positive cells were quantified using ImageJ and approximately 50 cells were counted by time point, per experiment. (d) Representative images of RPA foci are shown of control cells (Cas9) and cells lacking BRG1 (BRG1-KO) as in (B). (e) Inactivation of BRG1 reduces the amount of CPT-induced chromatin-bound RPA. Chromatin-bound RPA was monitored by flow cytometry as a measure of DNA end resection [Citation33]. Cells were treated with CPT (0.5 μM for 1 h), followed by extraction of non-chromatin-bound RPA by cell permeabilization, followed by fixation with formaldehyde and immuno-labeling with an antibody against RPA. The DNA content of the cells was labeled with PI. Quantification of CPT-induced chromatin-bound RPA measured by flow cytometry in control cells (Cas9) and cells lacking BRG1 (BRG1-KO). Fold-increase is the ratio of the %+CPT/%NT as shown in Supplementary Figure S3A. (f) Quantification of CPT-induced chromatin-bound RAD51. Cells were treated with CPT as in (E) and allowed to repair the damage for 6 h, followed by permeabilization to remove the non-chromatin-bound RAD51, followed by fixation with formaldehyde. Chromatin-bound RAD51 was measured by flow cytometry. (g) Inactivation of BRG1 results in a decrease in RAD51 foci after CPT treatment. Cells were synchronized at late S/G2 phase by double thymidine block and 8 h after releasing the second thymidine block, cells were treated with CPT (1 μM for 1 h) and RAD51 foci were analyzed at the indicated times. Cells were fixed and immuno-labeled with an antibody against RAD51, and nuclei were stained with DAPI. Images were acquired and RAD51 nuclear foci were counted using ImageJ and approximately 50 cells were counted by time point, per experiment. (h) Representative confocal microscopy images of RAD51 foci are shown at the indicated times. All experiments were done in triplicate and graphs represent averages of three independent experiments ± SD (* p < 0.05, ** p < 0.01, *** p < 0.001 by student t test)
![Figure 3. Inactivation of BRG1 impairs DNA end resection. (a) Inactivation of BRG1 impairs the generation of ssDNA after DNA damage. Control U2OS cells (Cas9) and U2OS cells lacking BRG1 (BRG1-KO) were grown on coverslips, incubated with BrdU (100 μM) for 48 h, then were not treated (NT), or treated with CPT (+CPT, 1 μM for 1 h), and allowed to perform repair for 4 h [Citation32]. Cells were then incubated with pre-extraction and extraction buffers and fixed with formaldehyde. Cells were immuno-labeled with antibodies against cyclin A and BrdU. The generation of ssDNA was measured by quantifying BrdU foci in cyclin A positive cells (late S/G2 cells), as the antibody only recognizes BrdU contained within ssDNA regions, which in this case arose due to DNA end resection after CPT treatment. The DNA was stained using DAPI. Images were acquired using a confocal microscope (60X oil). Quantification of ssDNA regions was performed using ImageJ and approximately 50 cells were counted by time point, per experiment. (b) Representative images are shown of control cells (Cas9) and cells lacking BRG1 (BRG1-KO) stained with a BrdU antibody following no treatment (NT), 1 h CPT treatment (+CPT, 1 µM), and 1 h CPT treatment followed by 4 h recovery (+CPT + 4 h, 1 µM). (c) Inactivation of BRG1 also impairs RPA foci-formation after DNA damage. Cells were treated as in (A) and stained with antibodies against cyclin A and RPA. CPT-induced RPA foci in cyclin A positive cells were quantified using ImageJ and approximately 50 cells were counted by time point, per experiment. (d) Representative images of RPA foci are shown of control cells (Cas9) and cells lacking BRG1 (BRG1-KO) as in (B). (e) Inactivation of BRG1 reduces the amount of CPT-induced chromatin-bound RPA. Chromatin-bound RPA was monitored by flow cytometry as a measure of DNA end resection [Citation33]. Cells were treated with CPT (0.5 μM for 1 h), followed by extraction of non-chromatin-bound RPA by cell permeabilization, followed by fixation with formaldehyde and immuno-labeling with an antibody against RPA. The DNA content of the cells was labeled with PI. Quantification of CPT-induced chromatin-bound RPA measured by flow cytometry in control cells (Cas9) and cells lacking BRG1 (BRG1-KO). Fold-increase is the ratio of the %+CPT/%NT as shown in Supplementary Figure S3A. (f) Quantification of CPT-induced chromatin-bound RAD51. Cells were treated with CPT as in (E) and allowed to repair the damage for 6 h, followed by permeabilization to remove the non-chromatin-bound RAD51, followed by fixation with formaldehyde. Chromatin-bound RAD51 was measured by flow cytometry. (g) Inactivation of BRG1 results in a decrease in RAD51 foci after CPT treatment. Cells were synchronized at late S/G2 phase by double thymidine block and 8 h after releasing the second thymidine block, cells were treated with CPT (1 μM for 1 h) and RAD51 foci were analyzed at the indicated times. Cells were fixed and immuno-labeled with an antibody against RAD51, and nuclei were stained with DAPI. Images were acquired and RAD51 nuclear foci were counted using ImageJ and approximately 50 cells were counted by time point, per experiment. (h) Representative confocal microscopy images of RAD51 foci are shown at the indicated times. All experiments were done in triplicate and graphs represent averages of three independent experiments ± SD (* p < 0.05, ** p < 0.01, *** p < 0.001 by student t test)](/cms/asset/bdd7d5f6-d43f-445d-86cb-5dec7f9b2838/kccy_a_1831256_f0003_c.jpg)
We can also monitor DNA end resection indirectly by measuring the increase in CPT-induced chromatin-bound RPA by flow cytometry [Citation33]. Cells were treated with CPT for 1 h (0.5 μM), followed by a pre-extraction of non-chromatin-bound RPA, followed by fixation and immunolabeling of RPA. As shown in ) (and Supplementary Fig. S3A), cells lacking BRG1 displayed lower levels of chromatin-bound RPA after CPT treatment, when compared to control cells, once again demonstrating a defect in DNA end resection in the absence of BRG1. Next, we adapted this method to measure the amount of chromatin-bound RAD51 after CPT treatment. During HR the RPA protein coating these ssDNA regions produced during resection is replaced by the RAD51 recombinase, which will help with the homology search and strand-invasion steps of HR [Citation2]. If DNA end resection is impaired, then a reduced amount of chromatin-bound RAD51 should be observed. Cells were treated with CPT for 1 h (0.5 μM) and allowed to recover for 6 h, then cells were permeabilized to extract the non-chromatin bound RAD51, followed by fixation and immunolabeling of RAD51. Once again, cells lacking BRG1 displayed reduced amounts of chromatin-bound RAD51 after CPT treatment, when compared to control cells ()). We next decided to analyze the formation of CPT-induced RAD51 nuclear foci in cells lacking BRG1. In order to analyze RAD51 foci-formation we synchronized cells at the G2 phase of the cell cycle by double thymidine block (Supplementary Fig. S3B). As shown in -h), inactivation of BRG1 results in a reduction of RAD51 foci-formation after CPT treatment, in agreement with a defect in DNA end resection. Altogether, these findings demonstrate that inactivation of BRG1 impairs DNA end resection, which explains the defect in HR in cells lacking BRG1. It is also likely that BRM cannot replace BRG1 for this function in DNA end resection, as BRG1-KO cells, which show a defect in resection, express normal levels of BRM ()). It should be noted that in an attempt to further confirm this defect in resection is due to BRG1, we transfected BRG1-KO cells with a plasmid expressing WT-BRG1 to correct this repair defect. Unfortunately the re-introduction of WT-BRG1 in these cells induced a strong G1 arrest, which impeded our efforts to study resection and recombination as these events occur in late S/G2 phases of the cell cycle. This cell cycle arrest has been previously documented and is mediated, at least in part, through its interaction with RB [Citation41].
BRG1 promotes the recruitment of CtIP and reduces nucleosome density at DSBs
DNA end resection is initiated by the MRE11 and CtIP nucleases [Citation9,Citation42]. In order to further define the DNA end resection defect observed in BRG1-KO cells, we tested whether the recruitment of these nucleases was affected by the inactivation of BRG1. Cas9 and BRG1-KO cells were treated with CPT for 1 h and then MRE11 nuclear foci were analyzed at different times after treatment (Supplementary Fig. S4A-B). Inactivation of BRG1 did not impair MRE11 foci-formation after CPT treatment. This finding is in agreement with the fact that inactivation of BRG1 did not impair the activation of the ATM kinase, which requires MRE11 recruitment (Supplementary Fig. S2H, I). We then tested whether CtIP recruitment to DSBs was affected by the inactivation of BRG1. Live cell laser micro irradiation experiments showed that cells lacking BRG1 were unable to recruit GFP-CtIP to laser-induced DSBs within 10 minutes, as observed for control cells (, b)). Moreover, foci-formation analysis of G2-synchronized cells also showed a reduced number of CtIP foci after CPT treatment in cells lacking BRG1, when compared to control cells (, d)). The defective recruitment of CtIP to DSBs in cells lacking BRG1 explains their defect in DNA end resection and HR.
Figure 4. Inactivation of BRG1 impairs the recruitment of CtIP to DSBs. (a) Live cell confocal microscopy was used to monitor the recruitment of GFP-CtIP to laser-induced DSBs. Control cells (Cas9) and cells lacking BRG1 (BRG1-KO) were transfected with GFP-CtIP and 24 h post-transfection cells were incubated with BrdU (10 μM for 24 h). Laser micro irradiation experiments were performed using a 405 nm laser. Images were collected before laser irradiation and every 10 sec after irradiation for 10 min. Representative images are shown. (b) Quantification of the recruitment of GFP-CtIP was performed using ImageJ. A minimum of 20 cells were measured per genotype. Graph represent averages ± SD. (c) Inactivation of BRG1 impairs CtIP foci formation after CPT. Control cells (Cas9) and cells lacking BRG1 (BRG1-KO) were synchronized at late S/G2 phase of the cell cycle by double thymidine block. Cells were released from the second thymidine block and allowed cell cycle progression for 8 h (late G2). Cells were then treated with CPT (1 μM for 1 h). Cells were fixed and immuno-labeled with an antibody against CtIP, and nuclei were stained with DAPI. Images were acquired and CtIP nuclear foci were counted using ImageJ and approximately 50 cells were counted by time point, per experiment. (d) Representative confocal microscopy images of CtIP foci are shown at the indicated times. (e) Control cells (Cas9) and cells lacking BRG1 (BRG1-KO) were transduced with a retrovirus expressing ER*-HA-I-PpoI. These cells were treated for 12 h with tamoxifen (2 μM, + I-PpoI) or not (- I-PpoI), crosslinked and nuclear extracts were prepared. Chromatin immunoprecipitation was performed for histone H3. Quantitative PCR was performed to determine the amount of histone H3 present at the specific locus (489 bp 3ʹ to the I-PpoI cut site in the rDNA region). Fold enrichment was calculated by dividing the percentage (%) of input of the + I-PpoI by the – I-PpoI. The % of input refers to the amount of DNA obtained from the immunoprecipitation of the given factor divided by the total amount of DNA (input). All experiments were done in triplicate and graphs represent averages of three independent experiments ± SD (* p < 0.05, ** p < 0.01, *** p < 0.001 by student t test)
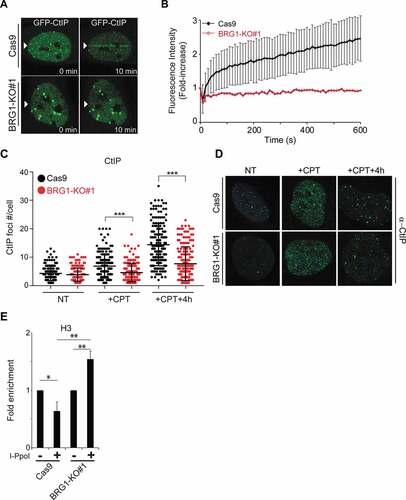
We next wanted to interrogate the function of BRG1 at DSBs in terms of chromatin structure. SWI/SNF complexes are known to push and slide nucleosomes in order to modulate access to certain genomic loci [Citation21]. Using the I-PpoI system we monitored the nucleosome density at DSBs by measuring histone H3 occupancy at these sites. We observed that while in control cells there was a reduction in nucleosome density at DSBs, cells lacking BRG1 showed an increase in nucleosome density at the break site ()). It is important to note that the basal levels of histone H3 were very similar at this site prior to the induction of the break and that the levels of damage at the site is comparable between cell lines, as indicated by the amount of γH2AX induced at this site (Supplementary Fig. S4C-D). This data is in agreement with our previous work showing that blocking the recruitment of BRG1 to DSBs by inactivating RB resulted in an increase in nucleosome density at the break sites [Citation24]. These findings suggest that BRG1 actively reduces nucleosome density at DSBs. It is likely that this change in chromatin structure at the break site helps in the recruitment or the retention of the CtIP nuclease at the break site, thus stimulating DNA end resection and HR.
BRG1 inactivation sensitizes cells to DNA damage
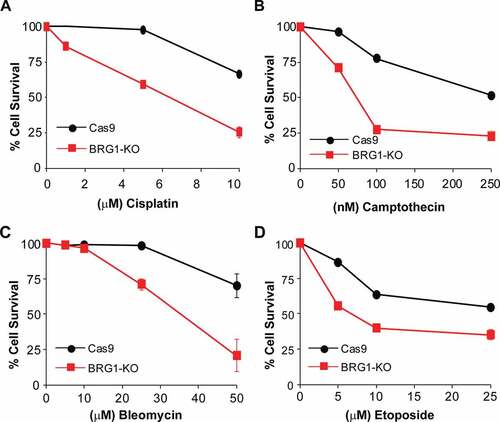
SWI/SNF chromatin remodeling complexes are characterized by the presence of one ATPase subunit (either BRG1 or BRM) [Citation20]. Downregulation of BRM however, seems to play a less pronounced role in HR ()) [Citation24]. As we have shown that BRG1 plays an important role in the repair of DSBs and HR, and HR-deficient cells often show sensitivity to DNA damage, we tested whether the inactivation of BRG1 resulted in sensitivity to chemotherapeutic agents that induce DSBs. As shown in , inactivation of BRG1 sensitized U2OS cells to bleomycin, camptothecin, cisplatin, and etoposide. The sensitivity of cells lacking BRG1 to these chemotherapeutic agents is in agreement with BRG1 playing a role in HR. Indeed, inactivation of CtIP has also been shown to result in sensitivity to these chemotherapeutic agents [Citation9]. We did not, however, observe that BRG1-KO cells were sensitive to the PARPi Olaparib by using the MTT-like assay. We then assessed colony formation ability of these cells in increasing concentrations of Olaparib and also observed no difference, when compared to control cells (Supplementary Fig. S5A). Next, we treated cells with CPT for 72 h or with Olaparib for 72 h and tested the activation of caspase 3/7 as a marker of cell death. We observed, first that a larger proportion of BRG1-KO cells initiated caspase 3/7 activation pathways upon CPT treatment, in agreement with their sensitivity to CPT. Second, we observe that there was no difference in caspase 3/7 activation between BRG1-KO cells and control cells upon olaparib treatment (Supplementary Fig. S5B). Taken together these findings show that inactivation of BRG1 results in cells that are sensitive to DSB-inducing agents, but not to the PARPi olaparib.
Figure 5. Inactivation of BRG1 renders cells sensitive to chemotherapeutic agents. Control cells (Cas9) and cells lacking BRG1 (BRG1-KO) were treated with increasing concentrations of (a) bleomycin, (b) camptothecin, (c) cisplatin, or (d) etoposide for 4 days and cell viability was measured by the MTT assay. All experiments were done in triplicate and each point represents averages of three independent experiments ± SD (* p < 0.05, ** p < 0.01, *** p < 0.001 by student t test)
Discussion
HR is a critical DNA repair pathway for maintaining genome integrity [Citation2,Citation43,Citation44]. Moreover, studies show that cancers deficient in HR are sensitive to a number of DNA damaging anti-cancer drugs and specifically to PARPi [Citation4,Citation12–14]. It is therefore of critical importance to identify new players in the HR pathway in order to test their feasibility as potential targets in the treatment of cancers. SWI/SNF complexes have been identified as a novel family of tumor suppressors [Citation23]. These complexes contain 10–15 subunits and multiple subunits within these complexes are mutated at very high frequencies in a variety of cancers, but the function of the majority of these subunits and their mechanism of tumor suppression is still unclear [Citation20,Citation21,Citation23]. Identifying subunits within the SWI/SNF complexes as important for HR, would also unveil DNA repair vulnerabilities in cancers bearing mutations in these subunits.
SWI/SNF chromatin remodelers have been primarily thought of as transcriptional regulators, but more recent studies have placed these complexes at the center of genome integrity [Citation24,Citation26,Citation35,Citation45–50]. Indeed, BRG1 itself is mutated at high frequencies in lung and skin cancers and has been shown to be important for the repair of UV-induced DNA damage [Citation25,Citation45]. In this study we investigated the role of the BRG1 ATPase in the repair of DSBs through HR and showed that BRG1 stimulates HR, while BRM seems to play a lesser role ()). Downregulation of both ATPases simultaneously did not further reduce HR efficiency, but it did cause cell cycle arrest and cell death. These findings are in agreement with our previous work, which showed that a BRG1-containing complex is important for HR [Citation24]. Moreover, we showed that the inactivation of RB and thus the destabilization the TopBP1-E2F1-RB complex responsible for the recruitment of BRG1 to DSBs, results in a DSB repair defect [Citation24]. We also recently showed that this TopBP1-E2F1-RB complex is also important for histone acetylation at DSBs and for the preservation of genome integrity [Citation51]. The function of BRG1 in HR that we demonstrate in this study is further supported by studies where testes-specific BRG1 inactivation in mice results in male-sterility, which suggests that BRG1 plays a role in meiotic recombination, which is analogous to HR [Citation52,Citation53]. Moreover, our observation of the minor role of BRM in HR is also supported by the fact that inactivation of BRM in mice results in normal mice (albeit somewhat heavier) that are fertile [Citation54]. The specific role BRM and the mechanism by which it affects HR must be the subject of a future study.
We show that the inactivation of BRG1 impairs the repair of DSBs and the activation of the ATR kinase (). The attenuated activation of the ATR kinase is likely due to the defect in DNA end resection displayed by cells lacking BRG1. This attenuated activation of the ATR kinase was previously reported in cells lacking both ATPases simultaneously, BRG1 and BRM [Citation26]. A previous study also reported that BRG1 stimulates the activation of the ATM kinase, but we observed ATM activation in multiple ways including γH2AX, pATM and pChk2 in cells lacking BRG1 either by shRNA or Cas9-mediated inactivation () and Supplementary Fig. S2H,I) [Citation27,Citation28]. In this study we show that BRG1 stimulates DNA end resection by promoting the recruitment of the CtIP nuclease (), while the recruitment of MRE11 was unaffected in the absence of BRG1. We still do not know exactly how does BRG1 stimulate the recruitment of CtIP to DSBs, as we did not detect a direct or indirect interaction between BRG1 and CtIP by co-immuno precipitation experiments. We propose that the removal of nucleosomes at DSBs is enough to stimulate or stabilize the recruitment of the CtIP nuclease (), as multiple studies have shown that nucleosomes block nucleases at DNA ends [Citation18,Citation19]. We do not know at this point whether BRM plays a role in the recruitment of CtIP or the reduction of nucleosome density at DSBs, but these repair defects that we report here occur in cells that express BRM. Future studies should aim to describe the specific function of BRM in HR.
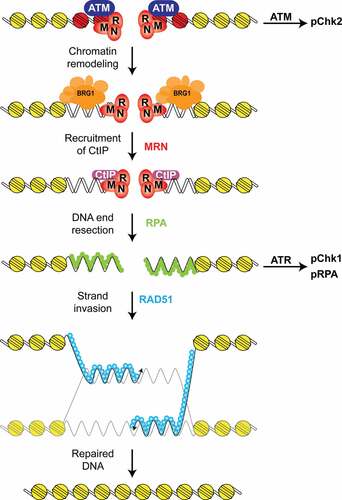
Figure 6. Proposed model for BRG1 function in DNA end resection and HR. First, the DSB is recognized by the MRN complex and the ATM kinase. This step also activates the ATM kinase, which phosphorylates the Chk2 kinase and histone H2AX (red). This step is followed by the recruitment of a SWI/SNF chromatin remodeling complex containing BRG1 (blue) that reduces nucleosome density at the DSB. After this chromatin remodeling step, the CtIP nuclease is recruited to the DSB and DNA end resection occurs. DNA end resection generates ssDNA, which is coated by RPA (green) and this structure activates the ATR kinase, which phosphorylates the Chk1 kinase. These ssDNA regions are later coated by RAD51 (light blue), which mediates the homology search and strand invasion steps of HR. Inactivation of BRG1 impairs the recruitment of the CtIP nuclease, and thus DNA end resection, ATR activation, RAD51 foci-formation, and HR
Multiple subunits of the SWI/SNF complexes have been implicated in DNA repair. Qi et al. found that BRG1 is important for HR, and proposed that BRG1 is responsible for the replacement of RPA by RAD51 [Citation49]. Although we did not observe sustained levels of RPA in the absence of BRG1 as they did, it is possible that the sustained RPA signal in their studies could also represent slower repair in the absence of BRG1. A BRG1-mediated reduction in nucleosome density at DSBs has been shown by us, and others including studies in yeast [Citation24,Citation35,Citation55]. This type of chromatin remodeling was also the proposed function for an ARID1A-containing SWI/SNF complex at DSBs [Citation46,Citation47]. The BAF200/ARID2 subunit of the SWI/SNF chromatin remodeler has also been shown to play a role in DSB repair [Citation48]. In this study de Castro et al. showed that inactivation of BAF200/ARID2 results in the impairment of RAD51 recruitment and defective HR. This is particularly interesting, in light of the fact that BAF200/ARID2 has been shown to be critical for the assembly of the canonical PBAF complex [Citation56]. Another study also showed that BRG1 and BAF180 were important for a different aspect of DSB repair, the silencing of transcription during DSB repair in the context of NHEJ [Citation50]. Future studies will aim to reconcile these differences and identify which complex (BAF vs. PBAF) and which subunits specifically within these complexes are important for the repair function of SWI/SNF complexes.
The requirement for chromatin remodeling of DNA ends for the process of DNA end resection has been widely proposed and multiple ATPases (INO80, p400, CHD4) have been identified as important for this function [Citation18,Citation19,Citation57,Citation58,Citation59,Citation60,Citation61]. It is still unclear why the cell would need several different chromatin remodelers to perform what is, in essence, the same function of remodeling chromatin at DNA breaks, but this seems to be the case. It is possible that there is a “chromatin code” that determines which chromatin remodeling complex acts at a specific genomic locus. If chromatin remodelers work in non-overlapping breaks or have non-overlapping functions, inactivation of multiple remodelers may display a much stronger defect in HR. It is important to note that while chromatin remodelers seem to play an important role in HR, they seem to play a less important role in the repair of DSBs mediated by NHEJ [Citation48,Citation55].
The inactivation of BRG1 also rendered cells sensitive to various chemotherapeutic agents that induce DSBs (). We did not find BRG1-KO cells to be hypersensitive to PARPi alone (Supplementary Fig. S5A). The lack of hypersensitivity to PARPi is puzzling, but can be explained in multiple ways; first, BRG1 may be required for the recruitment of CtIP of a particular set of DSBs and not all breaks. While we observe barely any recruitment of CtIP to DSBs within 10 minutes in our laser microirradiation experiments, there is lower levels of recruitment at much later time points. This delayed recruitment could explain why we still observe CtIP and RAD51 foci in BRG1-KO cells, albeit at much lower number than in control cells. This finding also suggests that BRG1 may not be an integral part of the HR pathway, but rather an ancillary factor that can be employed under certain chromatin conditions that would require it. Second, it is also possible that BRG1 may play two different roles in the repair process; one role in the recruitment of CtIP for a subset of breaks, and another role related to DDR signaling that may have a significant impact in cell viability after DNA damage. Third, and related to DDR signaling, BRG1 and other SWI/SNF subunits are known to play a role in the transcriptional response to DNA damage through p53 and controlling cell death and senescence pathways [Citation62,Citation63]. It is possible that the inactivation of BRG1 may impair the ability of cells to trigger cell death pathways upon PARPi treatment. All these possibilities could explain the lack of sensitivity of BRG1-KO cells to PARPi and will need to be further explored in the future.
The identification of BRG1 as a player in HR constitutes an important finding when considering that multiple components of the SWI/SNF complexes have been identified as a novel family of tumor suppressors [Citation20,Citation23]. The mechanism of tumor suppression of these genes is still unknown and it is possible that the defect in DNA repair that we demonstrate in this study contributes to the tumor suppressor capacity of this ATPase. Moreover, the fact that BRG1 is highly mutated in multiple cancers suggests that these cancers have a DNA repair vulnerability that can be exploited by radiotherapy or chemotherapy. Finally, this study will now allow us to identify which other SWI/SNF subunits are important for this function in HR, and which complex (BAF or PBAF) mediates this repair function. These future studies will advance our knowledge of the biology of these complexes and their functions as it relates to HR and genome stability.
Supplemental Material
Download Zip (7.9 MB)Acknowledgments
We thank Brian Zanotti and Dr. Ellen Kohlmeir for their technical assistance with the flow cytometry studies and the Midwestern University Core Facility, Downers Grove, Illinois. We also thank Karyn DiNovo (MWU) for her assistance with the confocal microscope. We thank Dr. Maria Jasin (MSKCC) for providing us with the DR-U2OS cells, Dr. Kei-ichi Takata (MDACC) for the pCAB-ISceI plasmid, Dr. Tanya Paull (UT Austin) for the pCDNA5-eGFP-CtIP plasmid, and Dr. Kyle Miller (UT Austin) for the pCDNA3-RFP-53BP1 plasmid and assistance with the micro irradiation experiments. We thank Dr. David G. Johnson (MDACC) for his comments and feedback. These studies were supported by Midwestern University startup funds (RVC) and the Midwestern University Kenneth A. Suarez Research Fellowship (EN and LV).
Disclosure statement
The authors declare no conflict of interests.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Piazza A, Heyer W-D. Homologous recombination and the formation of complex genomic rearrangements. Trends Cell Biol. 2019 Feb;29(2):135–149.
- Ranjha L, Howard SM, Cejka P. Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes. Chromosoma. 2018 Jun;127(2):187–214.
- Nickoloff JA, Jones D, Lee S-H, et al. Drugging the cancers addicted to DNA repair. JNCI. 2017 May 18;109(11):1–13.
- Talens F, Jalving M, Gietema JA, et al. Therapeutic targeting and patient selection for cancers with homologous recombination defects. Expert Opin Drug Discov. 2017 Jun;12(6):565–581.
- van Gent DC, Kanaar R. Exploiting DNA repair defects for novel cancer therapies. Mol Biol Cell. 2016 Jul 15;27(14):2145–2148.
- Davis AJ, Chen DJ. DNA double strand break repair via non-homologous end-joining. Transl Cancer Res. 2013 Jun;2(3):130–143.
- Stracker TH, Petrini JHJ. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol. 2011 Feb;12(2):90–103.
- Lee J-H, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005 Apr 22;308(5721):551–554.
- Sartori AA, Lukas C, Coates J, et al. Human CtIP promotes DNA end resection. Nature. 2007 Nov 22;450(7169):509–514.
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003 Jun 6;300(5625):1542–1548.
- Duursma AM, Driscoll R, Elias JE, et al. A role for the MRN complex in ATR activation via TOPBP1 recruitment. Mol Cell. 2013 Apr 11;50(1):116–122.
- O’Kane GM, Connor AA, Gallinger S. Characterization, detection, and treatment approaches for homologous recombination deficiency in cancer. Trends Mol Med. 2017 Dec;23(12):1121–1137.
- Carden CP, Yap TA, Kaye SB. PARP inhibition: targeting the Achilles’ heel of DNA repair to treat germline and sporadic ovarian cancers. Curr Opin Oncol. 2010 Sep;22(5):473–480.
- Dréan A, Lord CJ, Ashworth A. PARP inhibitor combination therapy. Crit Rev Oncol Hematol. 2016 Dec;108:73–85.
- Venkitaraman AR. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science. 2014 Mar 28;343(6178):1470–1475.
- Clouaire T, Legube GA. Snapshot on the Cis chromatin response to DNA double-strand breaks. Trends Genet. 2019 May;35(5):330–345.
- Xu Y, Price BD. Chromatin dynamics and the repair of DNA double strand breaks. Cell Cycle. 2011 Jan 15;10(2):261–267.
- Mimitou EP, Yamada S, Keeney S. A global view of meiotic double-strand break end resection. Science. 2017 Jan 6;355(6320):40–45.
- Adkins NL, Niu H, Sung P, et al. Nucleosome dynamics regulates DNA processing. Nat Struct Mol Biol. 2013 Jul;20(7):836–842.
- Pulice JL, Kadoch C. Composition and function of mammalian SWI/SNF chromatin remodeling complexes in human disease. Cold Spring Harb Symp Quant Biol. 2016;81:53–60.
- Wilson BG, Roberts CWM. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011 Jun 9;11(7):481–492. Nature Publishing Group.
- Clapier CR, Iwasa J, Cairns BR, et al. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol. 2017 Jul;18(7):407–422.
- Kadoch C, Hargreaves DC, Hodges C, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013 May 5;45(6):592–601.
- Velez-Cruz R, Manickavinayaham S, Biswas AK, et al. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. 2016 Nov 15;30(22):2500–2512.
- Zhang L, Chen H, Gong M, et al. The chromatin remodeling protein BRG1 modulates BRCA1 response to UV irradiation by regulating ATR/ATM activation. Front Oncol. 2013;3:7.
- Smith-Roe SL, Nakamura J, Holley D, et al. SWI/SNF complexes are required for full activation of the DNA-damage response. Oncotarget. 2015 Jan 20;6(2):732–745.
- Lee H-S, Park J-H, Kim SJ, et al. A cooperative activation loop among SWI/SNF, gamma-H2AX and H3 acetylation for DNA double-strand break repair. Embo J. 2010 Apr 21;29(8):1434–1445. EMBO Press.
- Kwon S-J, Park J-H, Park E-J, et al. ATM-mediated phosphorylation of the chromatin remodeling enzyme BRG1 modulates DNA double-strand break repair. Oncogene. 2015 Jan 15;34(3):303–313.
- Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013 Nov;8(11):2281–2308.
- Berkovich E, Monnat RJ, Kastan MB. Assessment of protein dynamics and DNA repair following generation of DNA double-strand breaks at defined genomic sites. Nat Protoc. 2008;3(5):915–922.
- Carey MF, Peterson CL, Smale ST. Chromatin immunoprecipitation (ChIP). Cold Spring Harb Protoc. 2009 Sep;2009(9):pdb.prot5279.
- Mukherjee B, Tomimatsu N, Burma S. Immunofluorescence-based methods to monitor DNA end resection. Methods Mol Biol. 2015;1292:67–75.
- Forment JV, Walker RV, Jackson SP. A high-throughput, flow cytometry-based method to quantify DNA-end resection in mammalian cells. Cytometry A. 2012 Oct;81(10):922–928. John Wiley & Sons, Ltd.
- Gong F, Chiu L-Y, Cox B, et al. Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev. 2015 Jan 15;29(2):197–211.
- Wiest NE, Houghtaling S, Sanchez JC, et al. The SWI/SNF ATP-dependent nucleosome remodeler promotes resection initiation at a DNA double-strand break in yeast. Nucleic Acids Res. 2017 Apr 8;45(10):5887–5900.
- Nakanishi K, Cavallo F, Brunet E, et al. Homologous recombination assay for interstrand cross-link repair. Methods Mol Biol. 2011;745:283–291.
- Majtnerová P, Roušar T. An overview of apoptosis assays detecting DNA fragmentation. Mol Biol Rep. 2018 Jul 18;45(5):1469–1478.
- Löbrich M, Shibata A, Beucher A, et al. gammaH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle. 2010 Feb 15;9(4):662–669.
- Szumiel I. Analysis of DNA double-strand breaks by means of γ-H2AX foci. Methods: chromosomal alterations. Springer, Heidelberg. p. 1–16. 2007 May 31.
- Bonner WM, Redon CE, Dickey JS, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008 Dec;8(12):957–967.
- Dunaief JL, Strober BE, Guha S, et al. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell. 1994 Oct 7;79(1):119–130.
- Takeda S, Nakamura K, Taniguchi Y, et al. Ctp1/CtIP and the MRN complex collaborate in the initial steps of homologous recombination. Mol Cell. 2007 Nov;28(3):351–352.
- Kass EM, Moynahan ME, Jasin M. When genome maintenance goes badly awry. Mol Cell. 2016 Jun 2;62(5):777–787.
- Jasin M, Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol. 2013 Nov;5(11):a012740.
- Hodges C, Kirkland JG, Crabtree GR. The many roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb Perspect Med. 2016 Aug 1;6(8):1–24.
- Shen J, Peng Y, Wei L, et al. ARID1A deficiency Impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 2015 Jul;5(7):752–767.
- Brownlee PM, Chambers AL, Cloney R, et al. BAF180 promotes cohesion and prevents genome instability and aneuploidy. Cell Rep. 2014 Mar 27;6(6):973–981.
- de Castro RO, Previato L, Goitea V, et al. The chromatin-remodeling subunit Baf200 promotes homology-directed DNA repair and regulates distinct chromatin-remodeling complexes. J Biol Chem. 2017 May 19;292(20):8459–8471.
- Qi W, Wang R, Chen H, et al. BRG1 promotes the repair of DNA double-strand breaks by facilitating the replacement of RPA with RAD51. J Cell Sci. 2015 Jan 14;128(2):317–330.
- Kakarougkas A, Ismail A, Chambers AL, et al. Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol Cell. 2014 Sep 4;55(5):723–732. The Authors.
- Manickavinayaham S, Velez-Cruz R, Biswas AK, et al. E2F1 acetylation directs p300/CBP-mediated histone acetylation at DNA double-strand breaks to facilitate repair. Nat Commun. 2019 Oct 30;10(1):4951.
- Kim Y, Fedoriw AM, Magnuson T. An essential role for a mammalian SWI/SNF chromatin-remodeling complex during male meiosis. Development. 2012 Mar;139(6):1133–1140.
- Wang J, Gu H, Lin H, et al. Essential roles of the chromatin remodeling factor BRG1 in spermatogenesis in mice. Biol Reprod. 2012 Jun;86(6):186.
- Reyes JC, Barra J, Muchardt C, et al. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2alpha). Embo J. 1998 Dec 1;17(23):6979–6991.
- Chen Y, Zhang H, Xu Z, et al. A PARP1-BRG1-SIRT1 axis promotes HR repair by reducing nucleosome density at DNA damage sites. Nucleic Acids Res. 2019 Sep 19;47(16):8563–8580.
- Mashtalir N, D’Avino AR, Michel BC, et al. Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell. 2018 Nov 15;175(5):1272–1288.e20.
- Chen X, Cui D, Papusha A, et al. The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature. 2012 Sep 27;489(7417):576–580.
- Costelloe T, Louge R, Tomimatsu N, et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature. 2012 Sep 27;489(7417):581–584.
- Chen H, Symington LS. Overcoming the chromatin barrier to end resection. Cell Res. 2013 Mar;23(3):317–319.
- Gospodinov A, Vaissiere T, Krastev DB, et al. Mammalian Ino80 mediates double-strand break repair through its role in DNA end strand resection. Mol Cell Biol. 2011 Dec 1;31(23):4735–4745.
- Xu Y, Sun Y, Jiang X, et al. The p400 ATPase regulates nucleosome stability and chromatin ubiquitination during DNA repair. J Cell Biol. 2010 Oct 4;191(1):31–43.
- Liu K, Luo Y, Lin F-T, et al. TopBP1 recruits Brg1/Brm to repress E2F1-induced apoptosis, a novel pRb-independent and E2F1-specific control for cell survival. Genes Dev. 2004 Mar 15;18(6):673–686.
- Burrows AE, Smogorzewska A, Elledge SJ. Polybromo-associated BRG1-associated factor components BRD7 and BAF180 are critical regulators of p53 required for induction of replicative senescence. Proc Natl Acad Sci U S A. 2010 Aug 10;107(32):14280–14285.