
ABSTRACT
The colorectal cancer (CRC) dictates a common malignancy with high recurrence rate. Long non-coding RNAs (lncRNAs) belong to a class of regulatory factors involved in multiple cancers. In current work, we have uncovered a novel lncRNA named TSLC8. TSLC8 was dramatically downregulated in CRC samples and cell lines. Reintroduction of TSLC8 inhibited tumor sphere formation and viability in CRC cells. In vivo experiments further confirmed the tumor suppressive function of TSLC8. Ectopic TSLC8 expression elevates puma abundance whereas this effect is mediated by TSLC8-puma binding and stabilization. FOXO1 can transcriptionally induce TSLC8 expression. Epigenetic investigation suggested that TSLC8 locus was hypermethylated in CRC leading to diminished TSLC8 expression. Our current work has identified a novel tumor suppressive function of TSLC8, whose reduced expression may facilitate malignant phenotypes during CRC progression.
KEYWORDS:
Introduction
The colorectal cancer (CRC) belongs to the one of the leading causes of tumor-related mortality worldwide [Citation1]. The incidence rate of colorectal cancer is relatively high with around 1.4 million cases each year [Citation1]. Notably, the proportion of CRC incidence among women is slightly lower than men and CRC patients are usually diagnosed at late stage [Citation2]. Most CRC cases may sporadically arise, while the other cases occur with concomitant inflammatory bowel diseases and genetic syndromes [Citation3]. Although therapeutic interventions have made great progress in recent years, the death rate for CRC patients remain unsatisfactory primarily owing to frequent recurrence and metastasis [Citation4].
The long non-coding RNAs (lncRNAs) are designated as a class of transcripts with minimal or no protein coding capacity and composed of over 200 nucleotides in length [Citation5]. The lncRNAs may be divided into following categories which are sense, antisense RNAs with proximal coding transcripts and enhancer RNAs [Citation6]. It is shown that lncRNAs play essential roles during cancer transformation probably by affecting multiple cellular processes such as cell cycle, apoptosis and differentiation [Citation7]. LncRNAs also show tissue specific expressions with diverse regulatory mechanisms such as modulation of nuclear architecture, influencing gene expression in cis/trans and regulating interacted proteins/RNAs [Citation8]. For example, lncRNA B4GALT1-AS1 may advance colon cancer stemness and other malignant phenotypes via relocating the well-known yes‐associated protein (YAP) into the nucleus and increasing its transcriptional activities [Citation9]. LncRNA CPS1-IT1 can repress epithelial-mesenchymal transition (EMT) and metastasis by deactivating hypoxia-inducible factor-1 alpha (HIF-1α) and decreasing the expression of autophagy related protein (LC3) in colorectal cancer [Citation10]. Although several lncRNAs have been significantly associated with CRC development, the function of lncRNAs remain largely unknown.
In current work, we reported a novel lncRNA ENSG00000272128.1, which we termed Tumor Suppressive LncRNA on Chromosome 8 (TSLC8) in CRC. By lncRNA sequencing for both CRC tissues and cell lines, we noted that TSLC8 was substantially downregulated in cancerous cells. Functional studies revealed that reintroducing TSLC8 in CRC cells could activate the expression of PUMA (p53 up-regulated modulator of apoptosis). TSLC8 could bind puma transcript and stabilize it. The transcription factor FOXO1 could induce TSLC8 expression. Furthermore, loss of TSLC8 expression is primarily mediated by epigenetic silencing in TSLC8 promoter locus, and removing DNA methylation could rehabilitate TSLC8 expression. Our current work identifies TSLC8 as an anti-tumor lncRNA, and methylation mediated suppression of TSLC8 locus may contribute significantly to colorectal cancer progression.
Materials and methods
Cell lines, primary cells and human specimens
The CRC cell lines and normal FHC cells were purchased from the Shanghai Cell Biology Institute. The preparation of primary colon cells was done as previous described [Citation11]. All transformed cells were verified for their identify prior to experiments. Cells were cultured under the situation with 5% CO2 and in Dulbecco’s modified Eagle’s medium (DMEM, Sigma) at 37 °C with 8% fetal bovine serum (FBS, Sigma) and 150 μg/ml streptomycin (Sigma). Human CRC cancerous and adjacent normal tissues (mucosa) were trimmed from surgical specimens from the colorectal cancer. The crypt isolation method was used to discriminate cancerous and normal epithelia [Citation12]. All patients did not receive chemotherapy before surgical resection. This experimental protocol was formally obtained and approved by Human Research Ethics Committee (HREC) at The First Affiliated Hospital of Jinzhou Medical University.
Whole genome bisulfite sequencing (WGBS) and RNA-seq
For cell lines and primary cells, 2 μg RNAs were subject to RiboZero (Illumina, Shanghai) and library preparation by TruSeq Kit (Illumina, Shanghai). Samples were multiplexed and underwent paired-end 60-bp sequencing. The Illumina HiSeq2000 system was used. The bases 1–10 of output reads were depleted for FASTQ data. Filtering was implemented to leave the reads with scores >25 for 95% of reading lengths. Then, the FASTQ data files were aligned to the transcriptome Lincipedia 2.1 and RefSeqv58 by Bowtie (v1.1). The transcripts with <10 reads were removed from analyses using EdgeR package. The WGBS was performed primarily according to a previous work [Citation13] and analyzed in BGI (Beijing).
In vivo tumorigenesis
1 × 106 tumor cells were injected subcutaneously into nude mice (4 ~ 5 weeks old, female, N = 6 for each group). All mice were housed at 21–22 °C at 12/12 light-dark cycle with free access to food and water. The ellipsoid formula was used to determine the tumor volume: 1/2*D*d2, where “D” is the longest diameter and “d” represents the smallest diameter. After 4 weeks, mice were sacrificed, tumors were collected and weighed. All animal studies were performed in accordance with an Institutional Animal Care and Use Committee (IACUC) of The First Affiliated Hospital of Jinzhou Medical University.
RNA-RNA in vitro interaction
20 μL Protein A/G Magnetic Beads (Sigma) were washed twice using RIP buffer (Millipore) and then incubated with BrU antibody for 1 h at room temperature. After conjugation, beads were further washed twice with RIP wash buffer and then resuspended in buffer containing 30 mM EDTA (Millipore) and RNase Inhibitor (Millipore). 20 pmol of BrU-labeled RNAs were incubated with beads in buffer for 2 h. During incubation, 3 pmol RNA fragments were appended into individual tubes and incubation was performed overnight at 4 °C. Beads were digested with proteinase K buffer to recover RNAs with RIP Wash Buffer, 1% SDS (Millipore) and 1.2 μg/μl proteinase K (Millipore) at 50 °C for 20 min. RNAs were extracted using miRNeasy kit (Qiagen), followed by reverse transcription with Superscript III (Invitrogen). The amount of recovered RNA fragments was evaluated by qPCR.
Reverse transcription quantitative-PCR (RT-qPCR)
Total RNAs were extracted using TRIzol RNA Purification Kit (Thermo Fisher Scientific, Shanghai) according to manufacturer’s guidelines. All primers were designed and obtained from Life Technologies (Shanghai). For details, see Table S1. QuantiTect Reverse Transcription Kit (Qiagen) was used for reverse transcription. With a final concentration of 20 ng/μL, the products were then subject to quantitative PCR. GAPDH was the internal control.
Viability assay
The Cell Counting Kit-8 toolkit (CCK-8, Dojindo) was used according to the manufacturer’s protocols. After culturing cells for 24 hours, cells were re-suspended and then loaded into a 12-well plate (~2 × 105 cells/well). Notably, 25 μl CCK-8 solutions were applied in current study. Optical density readings at 450 nm (O.D. 450) were recorded by Spectramax M5 microplate monitor (Molecular Devices, Shanghai).
Stable cell line generation
The Mycoplasma-negative HCT-116 cells were transfected with either TSLC8 short hairpin RNA (shRNA) construct or a scramble control (Origene, Shanghai). The shRNAs for puma were also designed and obtained from (Origene, Shanghai). Totally, ~1 × 106 cells were subject to puromycin selection (0.8 μg/mL). For overexpression, SW480 cells were transfected with TSLC8 full-length vector (Origene, Shanghai) or the control pWPXL lentiviral vector (Origene, Shanghai). The transfection conditions were optimized with G418 concentration of 600 μg/mL. For shRNA sequences, see Table S1.
Tumor sphere formation
Single-cell suspensions were first generation by digestion and then removed into a 12-well plate in serum-free MEM with 20 ng/ml EGF (Epidermal Growth Factor), 10 ng/ml bFGF (basic fibroblast growth factor), 100 ng/ml streptomycin and B27 (Sigma). After culturing for 5 days, the cells in non-adherent cultures were quantified by Multisizer 3 Coulter Counter (Beckman).
Statistics
The statistical analysis was determined using SPSS (version 16). ANOVA was used for comparison among multiple groups. The t test was used for cell line comparison.
Results
Identifying TSLC8 as a potential tumor suppressive lncRNA in CRC
To identify CRC-associated lncRNAs potentially implicated in tumor suppression, we performed lncRNA profiling assays using RNA sequencing. We implemented lncRNA profiling between CRC tissues and normal surrounding tissues. Totally, 6598 lncRNAs were screened for differential expression. The results suggested 218 markedly downregulated genes ()). Notably, three novel transcripts were uncovered and we selected the one with the highest fold reduction (ENSG00000272128.1, Table S2).
Figure 1. Identification of decreased TSLC8 in CRC. (a) The lncRNA sequencing for normal colon mucosa (normal) versus CRC tissues (tumor). Only statistically significance lncRNAs were shown. (b) Volcano plots for (a). Up: significantly upregulated genes; Down: significantly downregulated genes. (c) Relative TSLC8 expression in FHC and different CRC cell lines. N = 3 for each. (d) Relative TSLC8 expression in normal and tumorous tissues. N = 116. (e) The association of TSLC8 expression with TNM stages and metastatic status. (f) Nucleocytoplasmic separation assay to quantify the fraction of TSLC8. (g) The localization of TSLC8 was revealed by fluorescence in situ hybridization (FISH). **: P < 0.01
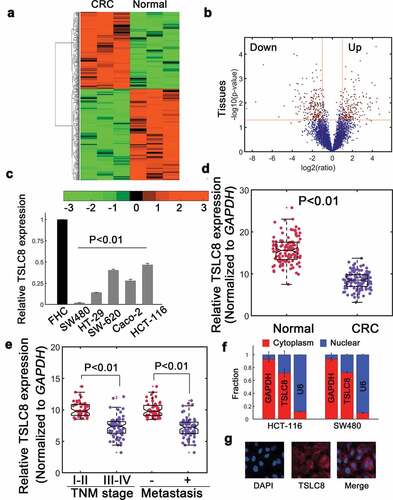
The lncRNA transcript ENSG00000272128.1 was located on chromosome 8 (GRCh38: CM000670.2, Ch8, 38,099,471–38,099,931 at forward strand). We termed it Tumor Suppressive LncRNA on Chromosome 8 (TSLC8) in current work. The full length 461-nt transcript had minimal protein coding potential (estimated coding probability 0.0098, Figure S1A and S1B). Northern blot further detected high endogenous TSLC8 expression in normal FHC cells and reduced expression in several CRC cell lines (Figure S1C). Consistently, RT-qPCR verification also suggested that TSLC8 was significantly downregulated in various CRC cell lines ()). Meanwhile, TSLC8 expression was repressed in CRC samples (n = 116, ), P < 0.01). TSLC8 expression was significantly correlated with TNM stages, metastatic status ()) and tumor size, whereas it was not associated with age, gender or location (Table S3). Nucleocytoplasmic fractionation assay suggested that TSLC8 was primarily located in the cytoplasm ()). The fluorescence in situ hybridization further revealed that a predominantly cytoplasmic distribution ()). These data identified that TSLC8 was significantly downregulated in CRC and may be correlated with CRC development.
Introducing TSLC8 inhibits malignancy and promotes apoptosis in CRC
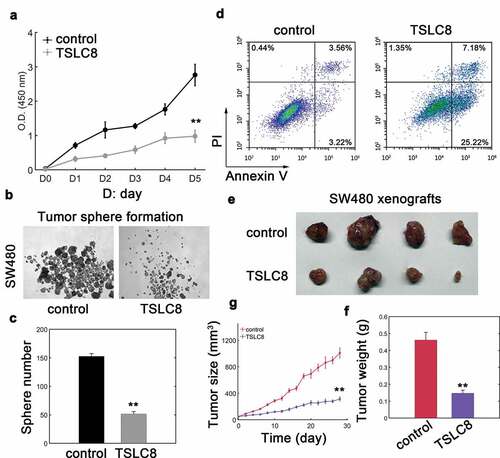
Since SW480 cells showed significantly lowered expression of TSLC8, we then chose SW480 cell for analysis. We found that SW480 cells transfected with pWPXL-TSLC8 vector markedly upregulated TSLC8 expression in SW480 cells (Figure S2A). TSLC8 overexpression then strongly inhibited CRC cell viability ()). Instead, the tumor sphere formation was also dramatically repressed with TSLC8 overexpression ()). As expected, introducing TSLC8 promoted apoptosis in SW480 cells ()). We next investigated whether TSLC8 had in vivo effect on CRC cell tumorigenesis. Results suggested that TLSC8 overexpression could significantly reduce xenograft tumor growth and tumor weight ()). The inhibitory effect on xenograft tumors was evident at very early stages with smaller tumor volumes ()). These results suggested that TSLC8 overexpression suppressed malignant phenotypes of CRC both in vitro and in vivo.
Figure 2. Ectopic expression of TSLC8 inhibits malignancy of CRC cells. (a) Cell viability of SW480 cells transfected with lentiviral control (control) or lentiviral vector containing TSLC8 (TSLC8). D stands for day. N = 3 for each time point. (b) Tumor sphere formation assay for SW480 cells with or without TSLC8 overexpression. (c) The quantification for (B). Triplicate results were shown. (d) Flow cytometry to detect apoptosis for SW480 cells transfected with a lentiviral control (control) or a vector with TSLC8 (TSLC8). (e) Representative tumor images for SW480 xenografts transfected with the lentiviral control or TSLC8 overexpressing vector. (f) Quantification results for (E). N = 6. (g) Dynamic tumor growth of SW480 xenografts with or without TSLC8 overexpression for 28 days. N = 6. **: P < 0.01
Silencing TSLC8 expression advances CRC development
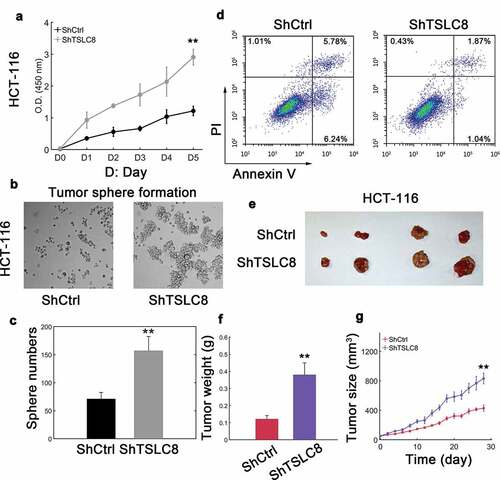
We noticed that some CRC cell lines had relatively higher TSLC8 expression compared with other cell lines ()). We then chose HCT-116 cells to knock down TSLC8 expression and evaluated associated effects. ShRNA mediated knockdown significantly lowered TSLC8 expression in HCT-116 cells (Figure S2B). Not surprisingly, reducing TSLC8 expression dramatically facilitated CRC cell viability () and S3A) as well as tumor sphere formation ()). However, if TSLC8 expression was restored, the viability and migratory ability of CRC cells would be markedly inhibited (Figure S3A and S3B). Meanwhile, the apoptotic rates were markedly lowered with TSLC8 silence ()). The in vivo effect was also evaluated and we found that TSLC8 knockdown substantially increased tumor weight and xenograft tumor growth ()). The tumor size was larger with TSLC8 silence during the transplantation experiments ()). These data again argued that knocking down TSLC8 expression may advance the tumorigenic effects of CRC cells.
Figure 3. Silencing TSLC8 advances malignant phenotypes in CRC cells. (a) Cell viability of HCT-116 cells transfected with a scramble control (ShCtrl) or shRNA#2 targeting TSLC8 (ShTSLC8). “D” is for day. N = 3 for each time point. (b) Tumor sphere formation assay for HCT-116 cells with or without TSLC8 knockdown by shRNA. (c) The quantification for (B), N = 3. (d) Flow cytometry to detect apoptosis for HCT-116 cells transfected with a scramble control (control) or a vector with TSLC8 (TSLC8). (e) Representative tumor images for SW480 xenografts transfected with the scramble control (control) or TSLC8 short hairpin RNA (ShTSLC8). (f) Quantification for (E). N = 6. (g) Tumor growth of HCT-116 xenografts transfected with scramble control or siRNA targeting TSLC8. N = 6. **: P < 0.01
Identification of puma as TSLC8 binding target
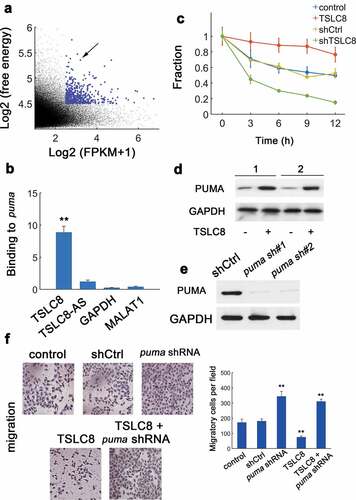
To determine putative TSLC8 targets, ntraRNA was utilized to implement an unbiased prediction [Citation14]. Results showed that puma mRNA was a putative target for TSLC8 binding ()). In vitro RNA-RNA assay confirmed the TSLC8-puma interaction (), GAPDH and lncRNA MALAT1 were negative controls). Furthermore, TSLC8 overexpression markedly increased the puma mRNA levels ()). In addition, TSLC8 silence significantly decreased the level of puma transcripts ()). Consistently, we found elevated PUMA protein expression in SW480 and HT-29 cells with TSLC8 overexpression ()). Two shRNAs for puma were constructed and efficiently downregulated puma expression (The shRNA puma #2 was used thereafter, )). We found that silencing puma alone could increase the cellular migration ()). Overexpression of TSLC8 consistently inhibited the migration ()). However, the tumor inhibitory role of TSLC8 overexpression was counteracted with concomitant puma silence ()). Therefore, TSLC8 may promote PUMA expression by stabilizing puma transcripts to inhibit CRC cells.
Figure 4. Identification of binding between TSLC8 and puma. (a) Prediction of TSLC8-binding partners. y: log2-absolute RNA binding energy between TSLC8 and putative targets; x: log2-average expression of targets. (b) In vitro RNA-RNA binding assay for TSLC8 and puma (antisense TSLC8 termed TSLC8-AS, GAPDH and non-target lncRNA MALAT1 as negative controls). N = 3. (c) The PCR for puma after RNA synthesis blockage with α-amanitin (30 μM) in SW480 cells transfected with shCtrl, shTSLC8, control or TSLC8 overexpressing vector. N = 3 for each time point. (d) Western blots for empty pWPXL vector transfected cells or cells overexpressing TSLC8. 1: SW480 cell; 2: HT-29 cell. (e) Western blot showing the efficacy of two different puma shRNA constructs (puma sh#1 and #2) in HCT-116 cells. The shRNA puma #2 was used. (f) Migration assay for HCT-116 cells transfected with indicated vectors (left). The quantification results were shown on the right. **: P < 0.01
FOXO1 facilitates TSLC8 expression
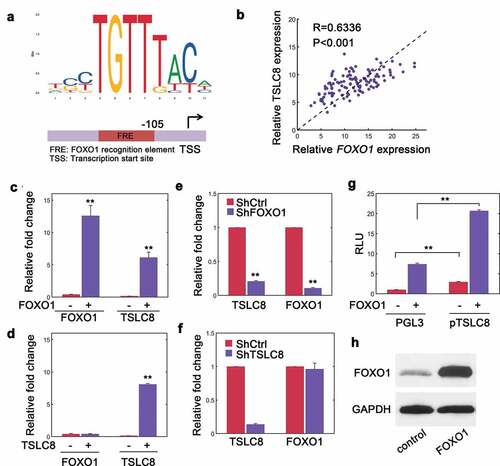
To search for the regulatory mechanisms of TSLC8 expression, we noted that there was a potential FOXO1 recognition element (FRE) upstream of TSLC8 transcription start site (TSS) ()). This transcription factor has critical roles during tumor suppression [Citation15]. To confirm whether FOXO1 regulated TSLC8 expression, we conducted a correlation study. We found that FOXO1 transcripts and TSLC8 were significantly correlated (R = 0.6336, P < 0.001, )). Ectopic expression of FOXO1 facilitated endogenous TSLC8 expression ()). However, when TSLC8 was overexpressed, concomitant elevation in FOXO1 levels was not observed implying that this regulation was unidirectional ()). When we knocked down FOXO1 expression, we observed a dramatic decrease in both FOXO1 and TSLC8 expression ()). As expected, depleting TSLC8 alone did not affect FOXO1 levels ()). In addition, overexpressing FOXO1 could stimulate the promotor activity of TSLC8 compared with empty control ()). The chromatin immunoprecipitation (ChIP) assays further revealed that FOXO1 directly bound to the TSLC8 promoter (Figure S4). As a verification, FOXO1 vector transfection significantly upregulated FOXO1 expression ()). There results suggested that FOXO1 might dictate TSLC8 expression in CRC cells.
Figure 5. FOXO1 regulates TSLC8 expression. (a) Consensus sequences for FOXO1 binding sequence obtained from JASPAR (jaspar.genereg.net). A FOXO1 recognition element (FRE) was shown upstream of TSLC8 transcription start site (TSS). (b) RT-qPCR verification of correlated expression between TSLC8 and FOXO1 in human samples. R = 0.6336, P < 0.001. (c) Relative FOXO1 and TSLC8 expression in SW480 cells transfected with pWPXL control or lentiviral pWPXL-FOXO1 vector (FOXO1). (d) Relative FOXO1 and TSLC8 expression in SW480 cells transfected with pWPXL control or lentiviral pWPXL-TSLC8 vector (TSLC8). (e) FOXO1 and TSLC8 expression in HCT-116 transfected with short hairpin RNA scrambled control (ShCtrl) or shRNA targeting FOXO1 (ShFOXO1#1 was used). (f) FOXO1 and TSLC8 expression in HCT-116 shRNA scrambled control (ShCtrl) or shRNA targeting TSLC8 (ShTSLC8). (g) Luciferase reporter assay in SW480 cells with TSLC8 promoter in the presence or absence of lentiviral FOXO1 transfection. The luciferase signals were normalized and adjusted for background PGL3 contents. RLU stands for relative light units. **: P < 0.01. (h) Verification of efficiency for FOXO1 overexpression at protein level in SW480 cells. Triplicates were shown unless otherwise specified
The TSLC8 locus was epigenetically regulated in CRC by hypermethylation
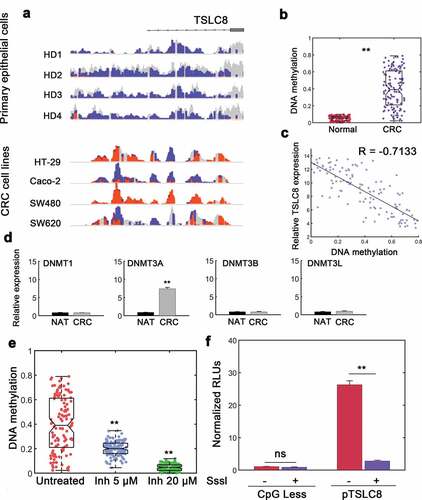
We observed significant loss of TSLC8 expression in CRC cells and samples, next we sought to determine the regulatory mechanism underlying TSLC8 silence. We conducted WGBS on primary colon cells from healthy donors and compared the results to profiling of CRC cell lines. We found that ~15 kB unmethylated regions in primary normal colon cells whereas it was hypermethylated in CRC cells ()). We also measured the methylation levels of CRC and surrounding normal tissues. The results also showed hypermethylation in CRC ()). The methylation status and TSLC8 expression was also negatively correlated ()). Quantification of de novo DNA methyltransferase (DNMT) family enzymes revealed that DNMT3A might be responsible for the observed hypermethylation ()). We then investigated the effect of reducing DNMT3A enzymatic activity using a previously synthesized DNMT3A inhibitor [Citation16]. We found markedly repressed methylation in the CpG island proximal to TSLC8 promoter region with DNMT3A inhibitor treatment ()). Furthermore, DNMT3A levels negatively correlated with TSLC8 expression (Figure S5). Notably, this effect was also dose-dependent ()). We also inserted the TSLC8 promoter construct into a CpGLess vector and subject this construct to SssI-induced methylation in vitro. We found a significant reduction in TSLC8 promoter activity when TSLC8 promoter was modified by hypermethylation ()). These data suggested that hypermethylation contributed to the decrease in TSLC8 expression leading to diminished downstream effects in CRC cells.
Figure 6. DNA hypermethylation deactivates the expression at TSLC8 promoter locus. (a) IGV (Integrative Genomics Viewer) tracks demonstrating WGBS for primary colon epithelial cells from 4 healthy donors (HD) and reduced representation bisulfite sequencing from CRC cell lines. Orange, methylated locus; blue, unmethylated locus; gray, non-CpG sequences. (b) Methylation of TSLC8 promoter (chr8:38,099,163, −442 from TSS) compared with normal adjacent colon tissues in Infinium human 450 K methylation array (Illumina). N = 116. (c) Correlation between methylation status and TSLC8 expression. N = 116. (d) Relative RNA expression of DNMT (DNA Methyltransferase) enzymes in normal adjacent tissues (NATs) and CRC tissues. (e) Methylation levels of SW480 cells left untreated or treated with increasing doses of DNMT3A inhibitor (5 μM and 20 μM). N = 116. (f) Luciferase reporter assays for TSLC8 promoter (pTSLC8) with or without CpG-methyltransferase (SssI) mediated methylation. Data were normalized to CpGLess empty vectors. N = 3. ns: not significant; **: P < 0.01
Discussion
In current work, we have first unraveled a novel lncRNA termed TSLC8 with potential tumor suppressive function in CRC. TSLC8 was frequently downregulated in various CRC cell lines and tumor specimens. We noted that reintroduction of TSLC8 in CRC cells inhibited viability, tumor sphere formation and promoted apoptosis. Functional silencing for TSLC8 in CRC cells profoundly advanced malignant phenotypes whereas apoptosis was consistently inhibited. Mechanistic study suggested that TSLC8 actively stabilizes puma transcripts. FOXO1 was responsible for the induction of TSLC8 and hypermethylation of TSLC8 locus may disrupt TSLC8 expression in CRC and increased expression of DNMT3A might contribute to the observed hypermethylation. It is still needed to further explore the enzymatic activity of DNMTs and its involvement in CRC progression.
We have designated TSLC8 as a potential tumor suppressive lncRNA at least in CRC owing to lowered expression of TSLC8 in CRC and inhibition of CRC malignancy by ectopic TSLC8 overexpression. However, we have not examined the effect of TSLC8 in other types of epithelial cancers such as renal cell carcinoma (RCC), lung squamous cell carcinoma (lung-SCC) and hepatocellular carcinoma (HCC) [Citation1] or the effect of TSLC8 is only restricted in colorectal cancers. This may point to several interesting area for research in future.
FOXO proteins belong to a superfamily of forkhead box containing transcription factor and FOXO1 (previously termed as FKHR, also known as FOXO1a) is one subfamily member [Citation15]. FOXOs have multiple tumor suppressive functions such as checkpoint control and apoptosis [Citation17,Citation18]. The anti-tumor function of FOXO1 is diverse, ranging from inhibiting Cyclin D1/2, cross-talking with tumor suppressor p53 to transcriptionally inducing p27cip/kip, Bim, Fas ligand and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression (reviewed in [Citation15]). The Akt signaling can inactivate FOXOs via phosphorylation whereas the protein phosphatase PPA2 can instead dephosphorylate FOXOs to facilitate apoptosis [Citation19]. In a recent report, Chae et al. showed that a histone methyltransferase G9a (EHMT2) was overexpressed in colon cancer [Citation20]. G9a mediated K273 methylation of FOXO1 increases the interaction between FOXO1 and an E3-ligase SKP2 to target FOXO1 for degradation [Citation20]. Therefore, FOXO1 is frequently downregulated in colon cancer implying a tumor suppressive role for FOXO1 [Citation20]. In current study, FOXO1 has been identified as the transcription factor responsible for TSLC8 induction. The tumor suppressive activity of FOXO1 can therefore be ascribed to its downstream target TSLC8. Collectively, our current work has uncovered a novel layer of complexity for the well-known transcription factor FOXO1 for its tumor suppressive function. The diverse and seemingly redundant roles of FOXO1 may constitute “heterogenous redundancy” and guarantee robust tumor suppression [Citation21].
Notably, many cancers show hypermethylation especially in CpG islands although these epigenetic alterations are thought as passive events and do not confer survival or proliferative advantages [Citation22]. Therefore, it is really interesting in the context of TSLC8 hypermethylation in current work. The hypermethylation within TSLC8 promoter may to some extent result in TSLC8 silence and establish a causal relationship between DNA hypermethylation at TSLC8 locus and colon cancer progression. However, whether FOXO1 locus is also subject to epigenetic silence via DNA hypermethylation remains elusive and requires further investigation. Furthermore, our experiments also support that using epigenetic drugs (e.g. 5-azacytidine) might provide fruitful insight into the treatment of colorectal cancer because reactivation of TSLC8 expression induce tumor suppression.
We have shown that puma expression was enhanced by TSLC8. TSLC8 could stabilize puma transcripts. A major challenge in CRC is the frequent mutations in tumor suppressor p53 gene and reduced sensitivity to agents [Citation23]. However, PUMA is a potent apoptosis inducer in colorectal cancer cells irrespective of p53 status [Citation24]. The stabilization of puma transcripts and upregulation of PUMA proteins may possibly bypass the need for functional p53 and exerts a tumor suppressive function especially in p53-negative or mutated CRC cells [Citation25]. Future experiments will be performed by qRT-PCR of puma transcripts in conditions by which TSCL8 is knocked-down and/or overexpressed at fixed time points without synthesis blockage.
In conclusion, we have uncovered a novel lncRNA termed TSLC8 which stabilizes puma mRNA. FOXO1 transcription factor is responsible for TSLC8 induction. Therefore, the FOXO1-TSLC8-puma axis has unraveled a novel aspect in tumorigenesis and provided a potential clue for therapeutic targeting of TSLC8 to treat colorectal cancer.
Author contributions
Z.A.D., T.Y. and G.L. conceived the study. Z.A.D., T.Y., M.N.S., Y.C., G.L. performed the experiments and analyzed the data. T.Y., Z.A.D. and G.L. drafted the manuscript; Z.A.D., T.Y., M.N.S., Y.C., G.L. contributed to data analysis. All authors approved the final version.
Supplemental Material
Download MS Word (1.1 MB)Disclosure statement
The authors declare no conflicts of interest
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34.
- Seo GH, Choe EK, Park KJ, et al. Incidence of clinically relevant incisional hernia after colon cancer surgery and its risk factors: a nationwide claims study. World J Surg. 2018;42:1192–1199.
- Sun YH, Li J, Shu HJ, et al. Serum immunoinflammation-related protein complexes discriminate between inflammatory bowel disease and colorectal cancer. Clin Transl Oncol. 2019;21:1680–1686.
- Smith JJ, Deane NG, Wu F, et al. Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology. 2010;138:958–968.
- Lin C-P, He L. Noncoding RNAs in cancer development. Ann Rev Cancer Biol. 2017;1:163–184.
- Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789.
- Yang X, Ikhwanuddin M, Li X, et al. Comparative transcriptome analysis provides insights into differentially expressed genes and long non-coding RNAs between ovary and testis of the mud crab (scylla paramamosain). Mar Biotechnol (NY). 2018;20:20–34.
- Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154(1):26–46.
- Zhang Y, Fang Z, Guo X, et al. lncRNA B4GALT1-AS1 promotes colon cancer cell stemness and migration by recruiting YAP to the nucleus and enhancing YAP transcriptional activity. J Cell Physiol. 2019;234:18524–18534.
- Zhang W, Yuan W, Song J, et al. LncRNA CPS1-IT1 suppresses EMT and metastasis of colorectal cancer by inhibiting hypoxia-induced autophagy through inactivation of HIF-1alpha. Biochimie. 2018;144:21–27.
- Wilhelm A, Jahns F, Bocker S, et al. Culturing explanted colon crypts highly improves viability of primary non-transformed human colon epithelial cells. Toxicol In Vitro. 2012;26:133–141.
- Habano W, Gamo T, Terashima J, et al. Involvement of promoter methylation in the regulation of Pregnane X receptor in colon cancer cells. BMC Cancer. 2011;11:81.
- Stueve TR, Li WQ, Shi J, et al. Epigenome-wide analysis of DNA methylation in lung tissue shows concordance with blood studies and identifies tobacco smoke-inducible enhancers. Hum Mol Genet. 2017;26:3014–3027.
- Mann M, Wright PR, Backofen R. IntaRNA 2.0: enhanced and customizable prediction of RNA-RNA interactions. Nucleic Acids Res. 2017;45:W435–W9.
- Ma J, Matkar S, He X, et al. FOXO family in regulating cancer and metabolism. Semin Cancer Biol. 2018;50:32–41.
- Erdmann A, Menon Y, Gros C, et al. Design and synthesis of new non nucleoside inhibitors of DNMT3A. Bioorg Med Chem. 2015;23:5946–5953.
- Ramaswamy S, Nakamura N, Sansal I, et al. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell. 2002;2:81–91.
- Lee SY, Lee GR, Woo DH, et al. Depletion of Aurora A leads to upregulation of FoxO1 to induce cell cycle arrest in hepatocellular carcinoma cells. Cell Cycle. 2013;12:67–75.
- Yan L, Lavin VA, Moser LR, et al. PP2A regulates the pro-apoptotic activity of FOXO1. J Biol Chem. 2008;283:7411–7420.
- Chae YC, Kim JY, Park JW, et al. FOXO1 degradation via G9a-mediated methylation promotes cell proliferation in colon cancer. Nucleic Acids Res. 2019;47:1692–1705.
- Kitano H. Towards a theory of biological robustness. Mol Syst Biol. 2007;3:137.
- Gutierrez-Arcelus M, Lappalainen T, Montgomery SB, et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. Elife. 2013;2:e00523.
- Chauvier D, Morjani H, Manfait M. Ceramide involvement in homocamptothecin- and camptothecin-induced cytotoxicity and apoptosis in colon HT29 cells. Int J Oncol. 2002;20:855–863.
- Yu J, Wang Z, Kinzler KW, et al. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci U S A. 2003;100:1931–1936.
- Li L, Lin L, Li M, et al. Gilteritinib induces PUMA-dependent apoptotic cell death via AKT/GSK-3beta/NF-kappaB pathway in colorectal cancer cells. J Cell Mol Med. 2020;24:2308–2318.