
ABSTRACT
The centrosome is a unique membraneless organelle that plays a pivotal role in the orderly progression of the cell cycle in animal cells. It has been shown that two pericentriolar scaffold proteins, Cep63 and Cep152, generate a heterotetrameric complex to self-assemble into a higher-order cylindrical architecture around a centriole. However, the mechanisms underlying how they reach their threshold concentrations in the vast intracellular space and generate a self-assembled architecture remain mysterious. Here we demonstrate that, like liquid-like assemblies, Cep63 and Cep152 cooperatively generate amorphous aggregates capable of undergoing dynamic turnover and inter-aggregate fusion in vivo and a significant level of internal rearrangemefnt within a condensate in vitro. Consistently, 1,6-hexanediol, a liquid–liquid phase separation disruptor, greatly diminished the ability of endogenous Cep63 and Cep152 to localize to centrosomes. Interestingly, a purified Cep63•Cep152 complex generated either a cylindrical structure or a vesicle-like hollow sphere in a spatially controlled manner. It also formed condensate-like solid spheres in the presence of a macromolecular crowder. At the molecular level, two hydrophobic motifs, one each from Cep63 and Cep152, were required for generating phase-separating condensates and a high molecular–weight assembly. Thus, we propose that the self-assembly of the Cep63•Cep152 complex is triggered by an intrinsic property of the complex undergoing density transition through the hydrophobic-motif-mediated phase separation.
Abbreviations: PCM, pericentriolar material; LLPS, liquid–liquid phase separation; MW, molecular-weight; CLEM, correlative light and electron microscopy; WT, wild-type; CMV, cytomegalovirus; FRAP, fluorescence recovery after photobleaching; FITC, fluorescein isothiocyanate; PCR, polymerase chain reaction; 3D-SIM, three-dimensional structured illumination microscopy; DMEM, Dulbecco’s Modified Eagle Medium; PEI Max, Polyethylenimine Max; PBS, phosphate-buffered saline; RT, room temperature; DAPI, 4', 6-diamidino-2-phenylindole; AOTF, acousto-optic tunable filter; LB, Luria broth; OD, optical density; IPTG, isopropyl β-D-1-thiogalactopyranoside; SDS-PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis
KEYWORDS:
Introduction
The centrosome, the main microtubule organizing center in animal cells, is a membraneless organelle composed of two microtubule-derived structures, called centrioles, and an amorphous mass of pericentriolar material (PCM) [Citation1,Citation2]. Information regarding how PCM proteins are organized is beginning to emerge. Super-resolution microscopic analyses of several pericentriolar scaffold proteins revealed that they are concentrically localized around a centriole in a highly organized manner [Citation3–6]. Subsequent studies revealed that one of the pericentriolar scaffold proteins, Cep152, displays an open-ended cylinder-like localization pattern around a centriole [Citation7] and forms a complex with another scaffold protein, Cep63, to self-assemble into a cylindrical architecture [Citation8]. Combined, these observations suggest that proper assembly and organization of pericentriolar scaffold proteins are likely critical for normal positioning of their client proteins and promoting distinct cellular processes in a spatially regulated manner. Not surprisingly, aberrations in the function of these scaffolds are associated with the development of many human diseases, including cancer, microcephaly, ciliopathy, and dwarfism [Citation9,Citation10].
Although the subcellular localization patterns of various pericentriolar proteins have been revealed recently, the molecular mechanisms by which these proteins are recruited to the PCM region and arranged around a centriole in an ordered manner remain undetermined. One of the attractive views is that they are effectively recruited to the region through liquid–liquid phase separation (LLPS) [Citation11,Citation12], a fundamental physicochemical process in which the clustering among specific component(s) drives the demixing of these components from their surrounding intracellular environment [Citation13–15]. This process, which is often driven by multivalent macromolecular interactions, is thought to be critical for increasing the local concentration of constituent components and promoting the formation of assemblies or membraneless organelles. Indeed, LLPS appears to be an underlying mechanism for generating various biomolecular condensates, such as PML bodies, P granules, stress granules, etc. (reviewed in [Citation13]). Aside from these classical phase-separating examples, Spd-5, a Caenorhabditis elegans centrosomal scaffold that shows micrometer-sized porous networks in vitro [Citation16], exhibits a degree of condensate-like properties in the presence of a macromolecular crowder [Citation17], suggesting that PCM assembly can be promoted through LLPS [Citation11]. Drosophila Cnn, a potential functional homologue of C. elegans Spd-5, also appears to form a large solid-like network structure but apparently through specific coiled-coil interactions [Citation18]. Whether the Cnn network exhibits condensate-like properties remains to be further investigated.
Here we demonstrate that, by combining both in vivo and in vitro studies, assemblies made of two pericentriolar scaffold proteins, Cep63 and Cep152, exhibit a significant degree of LLPS, including dynamic turnover of their components with those in the surroundings, fusion with other assemblies, and internal rearrangement within a condensate. Furthermore, we demonstrate that the Cep63•Cep152 complex is structurally versatile and capable of adopting multiple architectural arrangements. At the molecular level, a small hydrophobic residue-bearing region from both Cep63 and Cep152, lacking sequence signatures commonly associated with LLPS, plays a key role in phase-separating and architecting a higher-order self-assembly, suggesting that the Cep63•Cep152 assembly is driven by a small region with a unique physicochemical character.
Results
Ectopically expressed Cep63 and Cep152 generate high molecular-weight aggregates in a C-terminal hydrophobic-motif-dependent manner
Since both Cep63 and Cep152 possess multiple coiled-coil domains known to assemble into diverse subcellular structures [Citation19], we investigated the physicochemical characteristics of these proteins in vivo by ectopically expressing them in U2OS cells. To eliminate potential variations in the proteins’ assembling activity during mitosis, our analyses were limited to interphase cells, in which both proteins cooperatively generate a highly organized structure around a centriole [Citation3,Citation4]. For the convenience of tracking, a monomeric mCherry-fused Cep63 and a monomeric GFP (mGFP containing the A206K mutation) [Citation20]-fused Cep152 were used for analysis. Three-dimensional structured illumination microscopy (3D-SIM) showed that, although overexpression of either mCherry-Cep63 or mGFP-Cep152 alone yielded somewhat diffusive aggregates, co-expression of both proteins generated robustly aggregated assemblies around a centrosome () and Fig. S1A) and less frequently in other parts of cytosol (), 4th panel). Multiple clustered aggregates appeared to generate a larger ring-like assembly, up to approximately 1–2 μm in diameter. These observations suggest that Cep63 and Cep152 cooperatively self-organize into high molecular–weight (MW) assemblies in cytosol. These proteins efficiently localized to pericentriolar regions in a clear Cep63-in and Cep152-out fashion (), 3rd panel, arrows), indicating that these proteins are functional in vivo.
Figure 1. A hydrophobic-motif-dependent aggregative activity of Cep63 and Cep152. (a), 3D-SIM analysis for immunostained HEK293 cells co-infected with adenoviruses expressing mCherry-Cep63 and mGFP-Cep152. The location of endogenous centrioles is marked by anti-Cep192 signals. DAPI, chromosomal DNA stained by 4',6-diamidino-2-phenylindole. Arrowheads, a pair of endogenous centrioles. Asterisk, a ring-like aggregate found away from the two centrioles indicated by arrowheads. Note the cooperative generation of pericentrosomal aggregates by mCherry-Cep63 and mGFP-Cep152. Boxes, areas of enlargement. Scale bar, 2 µm. (b), CLEM analysis for HEK293 cells expressing mCherry-Cep63 and mGFP-Cep152. The endogenous centriole is marked by anti-Cep192 signal. Electron micrographs show an amorphous matrix-like aggregate around a centriole (asterisk). Box, area of enlargement. (c–e), Confocal microscopy, quantification, and immunoblotting analyses of HEK293 cells infected with adenoviruses expressing either WT Cep63 and Cep152 or their respective 4A and 5A hydrophobic mutants. Boxes in (c), areas of enlargement. Quantification in (d) was carried out by determining the total signal intensities (relative) of 12 randomly chosen fields (224.92 μm x 224.92 μm/field) from each sample using 2.5D intensity plots generated by the ZEN black software. Each microscopic field contains >50 cells. The data in (d) are shown in mean ± s. d. Exemplary intensity profiles for 2.5D plots are shown in Fig. S1B. ****, P < 0.0001 (unpaired two-tailed t-test). Immunoblots in (e) show the levels of WT and mutant forms of Cep63 and Cep152 expressed in the transfected cells. CBB, Coomassie Brilliant Blue–stained membrane. (f–h), 3D-SIM, quantification, and immunoblotting analyses of cells expressing endogenous promoter (Pendo)–controlled mCherry-Cep63-sil and mGFP-Cep152-sil and depleted of endogenous Cep63 and Cep152 by RNAi. Arrowheads in (f) indicate active, dot-state Plk4 signals. The data in (g) are shown in mean ± s. d. *, P < 0.05, ****, P < 0.0001 (unpaired two-tailed t-test). CBB, Coomassie Brilliant Blue–stained membrane
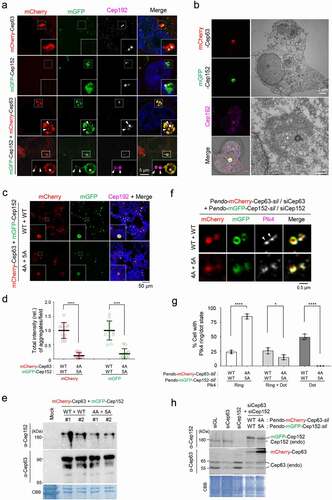
Given that the PCM is densely packed with electron-dense materials [Citation21], it would be difficult to determine whether and how Cep63 and Cep152 scaffolds generate distinguishable structures at their endogenous level. Therefore, we examined the architectural nature of ectopically expressed mCherry-Cep63•mGFP-Cep152 assemblies by carrying out correlative light and electron microscopy (CLEM). The results indicated that, although electron-dense signals correlating with the mChery and mGFP fluorescence were apparent around a centriole, they did not show any morphologically distinguishable structures ()). This suggests that Cep63 and Cep152 form amorphous aggregates through undefined weak interactions. Notably, these aggregates were capable of recruiting downstream components, such as Plk4, a key regulator of centriole duplication [Citation22,Citation23], and Sas6, a key element of the centriolar cartwheel structure [Citation24] (Fig. S1A).
A previous report showed that the interaction between Cep63 and Cep152 is mediated through their short C-terminal fragments (residues 490–541 for Cep63 and residues 1205–1272 for Cep152) containing a hydrophobic motif and a short alpha-helical fragment capable of forming a heterotetrameric complex between the two proteins [Citation8] (see ) below). Since the hydrophobic motifs (one each from Cep63 and Cep152) appear to be important for the proteins’ mutual interactions [Citation8], we examined whether they are also critical for generating Cep63•Cep152 aggregates in vivo. As expected, whereas the coexpression of mCherry-Cep63 and mGFP-Cep152 efficiently generated cytosolic assemblies, that of their respective 4A (L497A, I500A, F504A, L505A) and 5A (L1260A, I1261A, L1263A, I1266A, L1267A) hydrophobic mutants produced aggregates at a greatly diminished level ( and d) and Fig. S1B). The 4A and 5A mutants were expressed at levels similar to those of their respective wild-type (WT) Cep63 and Cep152 ()). Thus, the formation of the mCherry-Cep63•mGFP-Cep152 aggregates requires the hydrophobic-motif-mediated interactions.
Figure 2. Phase-separating activity of Cep63 and Cep152 in vivo. (a), FRAP analysis of adenovirus-expressed mCherry-Cep63 and mGFP-Cep152 aggregates generated in HEK293 cells. Images were acquired for 600 seconds at 2-second intervals. Representative confocal images acquired at the indicated time points are provided. Quantified relative signal intensities are shown in mean ± s.d. (n = 8 independent aggregates). (b), Time-lapse imaging showing the fusion of adenovirus-expressed mCherry-Cep63 and mGFP-Cep152 aggregates formed in U2OS cells. Images were acquired at 40-second intervals. The image at each time point was generated by projecting all z-stacks (greater than 21 stacks, 300-nm intervals). White arrowheads indicate two small condensate-like aggregates, which fuse into the bigger condensate-like aggregate (the middle sphere). Representative images are shown. Six out of 6 independent time-lapse experiments showed more than one fusion event during the course of experiment. (c), Quantification of confocal images for immunostained U2OS cells treated with either culture medium (mock) or 6% 1,6-hexanediol for the indicated lengths of time. Representative images are provided in Fig S2A. Relative fluorescence intensities for endogenous Cep63, Cep152, γ-tubulin, and glutamylated tubulin signals are shown in mean ± s.d. (n ≥ 50 cells/sample obtained from two independent experiments). ****, P < 0.0001 (one-way ANOVA). (d), 3D-SIM analyses for immunostained U2OS cells stably expressing endogenous promoter (Pendo)–controlled mCherry-Cep63-sil (WT or 4A) and mGFP-Cep152-sil (WT or 5A) and treated with centrinone and siRNAs for Cep63 and Cep152 (see the schematic diagram). Cep192, a centriole marker. N, nucleus. Arrowheads, nanoscale mCherry-Cep63•mGFP-Cep152 aggregates detected in cytosol after centrinone treatment. Note a greatly reduced number of aggregates in the 4A + 5A cells. (e), The cells in (d) were lysed in PBS and incubated in the presence of 5% glycerol. The resulting samples were subjected to confocal imaging and quantification. The total number of condensates (weak condensates in the 4A + 5A sample were marked by arrowheads) observed from seven randomly chosen fields for each group is provided, and the data are shown in mean ± s. d. (f), Confocal imaging and quantification of samples prepared from U2OS cell lysates transfected (to express in multiple copies) with Pendo-mCherry-Cep63-sil and Pendo-mGFP-Cep152-sil under Cep63 and Cep152 RNAi conditions. The lysates were incubated in PBS or PBS + 5% glycerol. The total number of condensates (arrowheads) observed from twelve randomly chosen fields for each sample is provided and the data are shown in mean ± s. d. Arrowhead, a small condensate observed in the 4A + 5A mutant sample
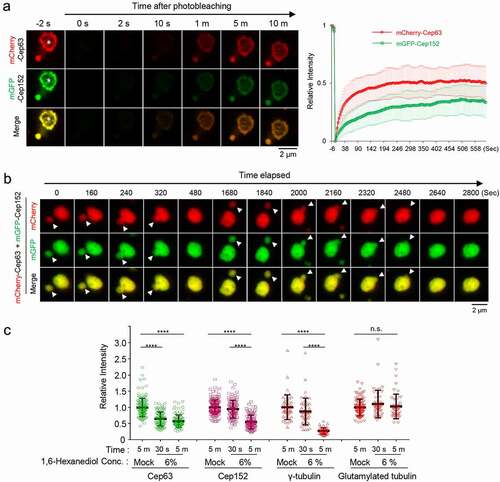
Figure 2. (Continued)
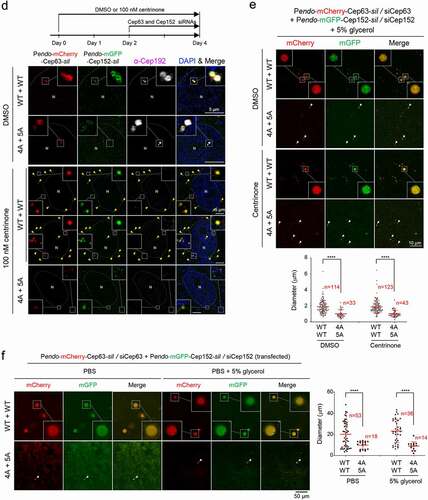
Figure 3. Formation of two morphologically distinct Cep63•Cep152 self-assemblies in vitro. (a), Schematic diagram showing the Cep63 (424–541)•Cep152 (1205–1295) complex and its corresponding 4A•5A hydrophobic mutant and purified proteins stained with Coomassie Brilliant Blue (CBB). (b and c), The proteins (125 μM) in (a) were incubated in an assembly buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5 mM TCEP) + 5% glycerol at 4 °C overnight and subjected to bright-field microscopy (b). The self-assemblies were stained with an anti-His antibody to confirm that spherical condensates are made of the His-Cep63 (424–541)•Cep152 (1205–1295) complex (c). A series of z-stack images in (c) revealing a spherical morphology is shown. The diameters of spherical assemblies were quantified and the results are shown in mean ± s.d. (n = 321 condensates obtained from two independent experiments). (d), 3D-SIM analysis of FITC-conjugated His-Cep63 (424–541)•Cep152 (1205–1295) self-assemblies generated either in a PCR tube (i.e., 3D space) or on a poly-L-lysine-coated slide glass (i.e., 2D surface) for 14 hours. Right panels, a series of z-stack images showing either a spherical (top) or cylindrical (bottom) morphology. Note of the different diameters for different z-stacks of the spherical assembly (top). 3D surface-rendered movies are provided in Movie S3. (e), 3D-SIM analysis of His-Cep63 (424–541)•Cep152 (1205–1295) self-assemblies generated in a 3D space in the presence of the indicated 5% polyethylene glycols (PEGs) for 14 hours
![Figure 3. Formation of two morphologically distinct Cep63•Cep152 self-assemblies in vitro. (a), Schematic diagram showing the Cep63 (424–541)•Cep152 (1205–1295) complex and its corresponding 4A•5A hydrophobic mutant and purified proteins stained with Coomassie Brilliant Blue (CBB). (b and c), The proteins (125 μM) in (a) were incubated in an assembly buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5 mM TCEP) + 5% glycerol at 4 °C overnight and subjected to bright-field microscopy (b). The self-assemblies were stained with an anti-His antibody to confirm that spherical condensates are made of the His-Cep63 (424–541)•Cep152 (1205–1295) complex (c). A series of z-stack images in (c) revealing a spherical morphology is shown. The diameters of spherical assemblies were quantified and the results are shown in mean ± s.d. (n = 321 condensates obtained from two independent experiments). (d), 3D-SIM analysis of FITC-conjugated His-Cep63 (424–541)•Cep152 (1205–1295) self-assemblies generated either in a PCR tube (i.e., 3D space) or on a poly-L-lysine-coated slide glass (i.e., 2D surface) for 14 hours. Right panels, a series of z-stack images showing either a spherical (top) or cylindrical (bottom) morphology. Note of the different diameters for different z-stacks of the spherical assembly (top). 3D surface-rendered movies are provided in Movie S3. (e), 3D-SIM analysis of His-Cep63 (424–541)•Cep152 (1205–1295) self-assemblies generated in a 3D space in the presence of the indicated 5% polyethylene glycols (PEGs) for 14 hours](/cms/asset/057759f5-723c-485d-87c2-cbbcc78a2f67/kccy_a_1843777_f0003_oc.jpg)
To investigate the physiological significance of forming Cep63•Cep152 aggregates in vivo, we examined whether a defect in this event influences Plk4’s capacity to carry out its ring-to-dot localization conversion [Citation25–27], an event required for triggering centriole biogenesis [Citation28,Citation29]. To this end, we generated U2OS cells stably expressing RNAi-insensitive mCherry-Cep63-sil and mGFP-Cep152-sil under their respective endogenous CEP63 and CEP152 promoters (i.e., Pendo-mCherry-Cep63-sil and Pendo-mGFP-Cep152-sil) or their respective clustering-impaired 4A and 5A mutants. The cells were depleted of endogenous Cep63 and Cep152 by RNAi and then subjected to immunostaining analyses. As expected, both endogenous promoter–controlled mCherry-Cep63 and mGFP-Cep152 properly localize to the pericentriolar region in a Cep63-in and Cep152-out fashion (Fig. S1C). Under these conditions, cells expressing WT Cep63 and Cep152 exhibited an active, dot-state Plk4 in greater than 76% of the population (26% one-dot state [mother centriole] and 50% two-dot state [both mother and daughter centrioles]) (). In contrast, the Cep63 (4A) – and Cep152 (5A)–expressing cells displayed inactive two-ring-state signals in greater than 85% of the population. Immunoblotting analyses showed that both exogenous proteins were expressed at levels largely comparable to those of their respective endogenous proteins ()).
In a second experiment, we also generated U2OS cells stably expressing cytomegalovirus (CMV) promoter–controlled Flag-Cep63 WT or 4A or untagged Cep152 WT or 5A using a lentivirus-based expression system. (This system offers a modest level of expression even under the CMV promoter control.) Under Cep63 and Cep152 RNAi conditions, we observed that either the 4A or 5A mutation alone was sufficient to disrupt Plk4’s ring-to-dot conversion (Fig. S1D–F). These data are consistent with the previous observation that Cep63 (4A) or Cep152 (5A) mutations greatly cripple the recruitment of Sas6 to centrosomes [Citation8]. Taken together, these findings suggest that the hydrophobic motif–mediated clustering of Cep63 and Cep152 is crucial for Plk4’s LLPS-mediated ring-to-dot conversion and Plk4-dependent centriole biogenesis.
Cep63 and Cep152 exhibit characteristics of LLPS in vivo
The condensate-like Cep63•Cep152 aggregates ()), together with their amorphous electron-dense signals ()), suggested that Cep63 and Cep152 may possess unusual physicochemical properties that enable them to generate highly associative pericentriolar deposits. Therefore, we examined whether the Cep63•Cep152 aggregates exhibit dynamic molecular nature, as previously observed in LLPS [Citation13–15]. Fluorescence recovery after photobleaching (FRAP) showed that both mCherry-Cep63 and mGFP-Cep152 in the aggregates recovered fluorescence within minutes () and Movie S1), suggesting that Cep63 and Cep152 in the aggregates undergo a dynamic turnover with those in the surroundings. Notably, the degrees of fluorescence recovery between mCherry and mGFP were somewhat different. Since equal rates of fluorescence recovery are expected for each component of a stable heterotetrameric complex, this observation suggests that the Cep63•Cep152 aggregates possess a heterogeneous nature.
We then examined whether the mCherry-Cep63•mGFP-Cep152 aggregates can fuse with one another to generate a larger aggregate in vivo, one of the characteristics that distinguishes liquid-like condensates from solid-like precipitable aggregates [Citation15]. Time-lapse microscopic analysis showed that the mCherry-Cep63•mGFP-Cep152 aggregates exhibited a stochastic movement in cytosol and underwent fusion efficiently (i.e., six out of six independent experiments) over a period of several minutes () and Movie S2). This finding along with the dynamic turnover of the assembly shown in ), strongly suggests that the mCherry-Cep63•mGFP-Cep152 aggregates exhibit the physiochemical properties of LLPS-induced biomolecular condensates.
To determine whether endogenous Cep63 and Cep152 exhibit any properties of LLPS-induced assemblies at their native location, we examined whether the assembly of the Cep63-Cep152 structure around a centriole is sensitive to the presence of 6% 1,6-hexanediol, a well-characterized disruptor of the weak hydrophobic protein–protein interactions required for generating dynamic liquid-like assemblies [Citation30,Citation31]. 1,6-hexanediol has been shown to effectively disassemble several assemblies with liquid-like properties, such as nuclear pore complex [Citation32,Citation33] and stress granules [Citation30,Citation34,Citation35]). To this end, U2OS cells treated with 1,6-hexanediol for a short period (30 seconds or five minutes) were fixed immediately and immunostained for further analyses. Quantification of centrosome-localized signal intensities revealed that, although the Cep63 signal diminished rapidly (approximately 40% reduction during the first 30-second treatment), Cep152 showed a somewhat delayed response () and Fig. S2A). Five minutes after treatment, the levels of both Cep63 and Cep152 were reduced to approximately 55–58% of the signals treated with control medium () and Fig. S2A). Notably, the signal intensity of γ-tubulin present in the PCM was also greatly diminished, whereas that of glutamylated tubulin found in the centriolar microtubule triplets [Citation36] was not () and Fig. S2A).
Knowing that Cep63 and Cep152 cooperate to generate a cylindrical architecture around a centriole [Citation8], the 1,6-hexanediol-sensitive endogenous Cep63 and Cep152 signals in ) may represent pericentriolar cylindrical architecture, the formation of which is driven in part by LLPS. To corroborate this finding, we used U2OS cells stably expressing endogenous promoter (Pendo)–controlled mCherry-Cep63-sil and mGFP-Cep152-sil to directly image mCherry and mGFP fluorescence signals because this approach allows us to avoid potential artificial signals that could be introduced by immunostaining procedures. Notably, cells expressing mCherry-Cep63 and mGFP-Cep152, but not their respective clustering-impaired 4A and 5A mutants, under Cep63 and Cep152 RNAi conditions occasionally exhibited a low level of flare-like fluorescent signals in addition to the strong toroidal signal around a centriole (Fig. S2B). In line with these observations, treatment with centrinone, which is known to deplete centrioles [Citation37], helped reveal small but distinguishable mCherry-Cep63•mGFP-Cep152 aggregates scattered in cytosol () and Fig. S2C). Under the same conditions, the hydrophobic 4A and 5A mutants yielded visibly less-distinct aggregates barely detectable by direct imaging (), bottom panel).
We then prepared cell lysates with the cells in ) in phosphate-buffered saline (PBS) and mounted them on a coverslip for further examination. The results showed that the number and diameter of the condensate-like aggregates were significantly greater in the mCherry-Cep63•mGFP-Cep152 sample than those in the 4A•5A mutant sample ()). The presence of 5% glycerol as a macromolecular crowding agent was required to detect the Cep63•Cep152 aggregates. The glycerol effect observed here is somewhat similar to the previous finding that the crowding agent can induce the formation of a phase-separated spherical condensate from a network-like structure [Citation17].
To further examine whether the formation of the condensate-like Cep63•Cep152 aggregates is concentration-dependent, we transfected the Pendo-controlled mCherry-Cep63-sil and mGFP-Cep152-sil to increase their expression levels, then examined the Cep63•Cep152 condensates after preparing cell lysates in the absence or presence of 5% glycerol ()). As expected, both the number and diameter of the Cep63•Cep152 condensates were significantly (>10-fold in diameter) increased under these conditions, and the 4A and 5A mutations greatly crippled this event. Provision of 5% glycerol appeared to enhance the degree of condensation (judging from a clearly demixed peripheral morphology) but did not alter the overall degree of condensate formation (), right).
To examine whether LLPS could play a role in driving the cylindrical Cep63•Cep152 self-assemblies in vitro, we examined the effect of 1,6-hexanediol in the process. The self-assemblies were generated using either a more soluble, minimized mCherry-Cep63 (424–541)•mGFP-Cep152 (1205–1295) complex or individually purified mCherry-Cep63 (full-length) and mGFP-Cep152 (924–1295; the longest fragment that we could purify), as described previously [Citation8] (Fig. S3 A and B). The results showed that, as expected, if LLPS drives the Cep63•Cep152 self-assembly, the provision of 1,6-hexanediol eliminated the ability of these proteins to generate a cylindrical architecture on a 2D surface (Fig. S3B). Notably, however, preassembled cylinders were somewhat resistant to 1,6-hexanediol, even after three hours of incubation (Fig. S3B). This is likely because disassembling a preassembled architecture generated through a cumulative free-energy-minimizing process may not be energetically favorable. Since the nature of the two self-assemblies shown in Fig. S3B was similar, the minimized mCherry-Cep63 (424–541)•mGFP-Cep152 (1205–1295) complex was subjected to further characterization.
The Cep63•Cep152 complex: a versatile structural module capable of forming morphologically distinct assemblies
To understand the underlying mechanism of how the Cep63•Cep152 complex generates a self-assembly, we investigated the complex’s intrinsic capacity to form a higher-order structure. Ectopic expression of the mCherry-Cep63 (424–541) and mGFP-Cep152 (1205–1295) complex, but not the proteins’ respective assembly-defective 4A and 5A hydrophobic mutants (see –e)), led to the formation of hollow spherical assemblies in the Escherichia coli cytosol (Fig. S4A). Co-expression of non-fused Cep63 (424–541) and Cep152 (1205–1295) also showed anti-His-antibody-stainable spherical condensates (Fig. S4B) that can be purified through density gradient centrifugation (Fig. S4C). These findings suggest that, in addition to the previously reported cylindrical self-assemblies [Citation8], the Cep63•Cep152 complex is capable of generating a morphologically distinct spherical assembly.
To further characterize the Cep63 (424–541)•Cep152 (1205–1295) assemblies, the His-Cep63 (424–541)•Cep152 (1205–1295) complex and its 4A•5A mutant [Citation8] were purified from E. coli. The proteins were then reconstituted in a physiological buffer (20 mM Tris-HCl, [pH 7.5], 150 mM NaCl, 0.5 mM TCEP + 5% glycerol) and placed in a dish (with poly-L-lysine-untreated surface) for overnight incubation (16 hours) at 4 °C ()). Strikingly, the His-Cep63 (424–541)•Cep152 (1205–1295) complex, but not the 4A•5A mutant, generated spherical condensates that can be easily discernable under a bright-field microscope ()). Subsequent immunostaining with anti-His antibody confirmed that the spherical condensates are indeed made of the His-Cep63 (424–541)•Cep152 (1205–1295) complex ()). The size of the condensates varied, with most condensates approximately 1 μm in diameter ()). These findings suggest that, unlike the previously characterized mCherry-Cep63 (424–541)•mGFP-Cep152 (1205–1295) complex that generates cylindrical self-assemblies on a poly-L-lysine-treated glass surface (i.e., 2D surface) [Citation8], the His-Cep63 (424–541)•Cep152 (1205–1295) complex generated spherical assemblies in a dish (i.e., 3D space).
The hollow spherical signals observed with anti-His staining could be the consequence of an indirect immunostaining that decorates only the surface protein. Therefore, to directly examine the effect of 2D or 3D space on the assembly of the His-Cep63 (424–541)•Cep152 (1205–1295) complex, a fluorescein isothiocyanate (FITC)-conjugated complex was generated and incubated either in a polymerase chain reaction (PCR) tube (i.e., 3D space) or on a slide glass surface (i.e., 2D surface) at 4 C. 3D-SIM analysis of the assemblies showed that the FITC-conjugated complex generated self-assembled hollow spheres in a 3D space (), top, and Movie S3, left), suggesting that the His-Cep63 (424–541)•Cep152 (1205–1295) complex bears a capacity to organize into a higher-order vesicle-like architecture. In sharp contrast, however, the same complex generated cylindrical self-assemblies on a 2D surface (), bottom, and Movie S3, right). Thus, the His-Cep63 (424–541)•Cep152 (1205–1295) complex can self-assemble into a morphologically distinct higher-order structure depending on its spatial cues. Similar results were obtained with the mCherry-Cep63 (424–541)•mGFP-Cep152 (1205–1295) or its longer mCherry-Cep63•mGFP-Cep152 (924–1295) complex (Fig. S4 D and E and Movie S4).
Although condensate-forming efficiencies vary depending on the molecular weight (MW) of polyethylene glycols (PEGs), 9% [w/v] PEG 3350 (MW, 3,350 Da) has been used to effectively mimic the high density of intracellular components and induce the condensation of Spd-5, a key PCM scaffold protein in C. elegans [Citation17]. In analyses with the FITC-conjugated His-Cep63 (424–541)•Cep152 (1205–1295) complex, we observed that provision of 5% PEG 3350 was sufficient to effectively induce the formation of solid spherical condensates (hereinafter referred to as “condensates” for simplicity) in a 3D space ()). Provision of PEGs with a larger MW (PEG 6000 or 8000) or at a higher concentration induced a threadlike twisted morphology with an increased signal intensity, suggestive of over-condensation. The mCherry-Cep63 (424–541)•mGFP-Cep152 (1205–1295) complex exhibited morphologies similar to those of FITC-conjugated complex (Fig. S4F). A low level of hollow spheres was detectable under the conditions that contain PEG 3350, but not PEG 6000 or PEG 8000.
Cep63-Cep152 self-assemblies are highly dynamic, exhibiting LLPS properties in vitro
The flexible nature of the Cep63-Cep152 self-assemblies and PEG-induced condensates in ) suggests that the components of these higher-order structures are likely dynamic. To determine the dynamic nature of these structures, the FITC-conjugated His-Cep63 (424–541)•Cep152 (1205–1295) complex bearing the capacity to form higher-order self-assemblies ( and Fig. S4) was used to generate either hollow spherical self-assemblies ()) or PEG 3350-induced, inside-filled condensates ()) for less than one hour (to avoid potential over-condensation). Because their cylindrical assemblies were extensively characterized previously [Citation8], they were not included in this study. The resulting samples were subjected to FRAP analyses either before (to examine a turnover rate) or after (to examine an internal rearrangement capacity) washing out unincorporated components in solution. In a turnover experiment with unwashed hollow spheres, the fluorescence of the bleached region recovered rapidly over a timescale of several minutes after the photobleaching (0 second) (), left). Thus, the vesicle-like self-assembly undergoes a dynamic exchange of its components with those in the surroundings. In a second experiment with washed spheres, however, the level of internal fluorescence recovery was very low or almost undetectable (), right), suggesting that the spherical self-assemblies are well organized with limited mobility within the spherical self-assembly.
The rounded morphology of the His-Cep63 (424–541)•Cep152 (1205–1295) condensates formed in the presence of 5% PEG 3350 ()) suggests that the condensates’ components are not likely arranged in an organized fashion. FRAP analyses revealed that, although the condensates showed a somewhat slower turnover rate than the hollow spheres in ) (), left), they exhibited a noticeably increased capacity for internal fluorescence recovery than the latter (), right). It is possible that the condensates’ decreased surface area may have slowed their turnover rate, while the increase in the shared interface between the bleached and nonbleached regions may have enhanced the rate of their internal rearrangement. These findings, together with the data shown in ), suggest that although the assemblies generated by the His-Cep63 (424–541)•Cep152 (1205–1295) complex exhibit dynamic turnover of their components, they exhibit a somewhat limited capacity in rearranging their components within hollow spheres and inside-filled condensates. Experiments carried out with condensates made of His-Cep63 (full-length) and a longer Cep152 (924–1295) also exhibited low but significant levels of turnover and internal rearrangement (Fig. S5). Notably, however, their rates of fluorescence recovery for both turnover and internal rearrangement appeared to be slower than those of the shorter Cep63 (424–541)•Cep152 (1205–1295) complex. This is to be expected, if a longer form engages in additional inter-heterotetrameric interactions that may further stabilize the assemblies.
Discussion
Phase separation of Cep63 and Cep152 triggers the formation of a higher-order structure
A large body of evidence suggests that Cep63 and Cep152 are mutually required to localize at centrosomes [Citation25,Citation38,Citation39]. However, the mechanism underlying how Cep63 and Cep152 are recruited to the PCM region and self-assembled into a higher-order architecture around a centriole remains largely elusive. A quantitative mass spectrometry analysis reveals that approximately 540 copies of Cep152 are present per centrosome, while approximately 20-fold more copies are present in the cytosol of a cell [Citation40]. Given that the average volume of human cells (3,000 μm3 for HeLa cells [Citation41]) is approximately 100,000-fold larger than that of the Cep63-Cep152 cylindrical architecture (approximately 0.03 μm3 for a Cep152 cylinder of approximately 0.35 μm [D] and 0.3 μm [H]), a mechanism may exist to efficiently amass Cep63 and Cep152 from a vast intracellular space above the threshold concentration required for inducing self-assembly.
Here we provide several lines of evidence that Cep63 and Cep152 exhibit a significant level of capacity to undergo LLPS and induce morphologically distinct self-assemblies (). First, ectopically expressed Cep63 and Cep152 cooperatively generated spherical aggregates that show a densely deposited matrix-like morphology with no distinguishable structures (). Second, in vivo Cep63•Cep152 assemblies exhibited dynamic turnover and underwent fusion among the assemblies () and Movies S1 and S2), thus demonstrating their liquid-like property. Third, in vitro-generated spherical Cep63•Cep152 assemblies also showed dynamic turnover and a significant level of internal rearrangement within a condensate ( and Fig. S5), further demonstrating a mobile nature of its components. Fourth, both centrosome-localized endogenous Cep63 and Cep152 signals and in vitro-assembled Cep63•Cep152 cylindrical self-assembly were sensitive to 1,6-hexanediol () and Fig. S3B), a well-characterized LLPS disruptor proposed to distinguish between liquid-like and solid-like assemblies [Citation31]. Consistent with this finding, Cep63 and Cep152 expressed under their respective endogenous promoters yielded a weakly detectable level of condensates, which became easily discernable after centrinone treatment ( and Fig. S2B). However, because of the small diameter of the in vivo Cep63•Cep152 cylindrical assembly (~250 nm for Cep63 and ~350 nm for Cep152 [Citation8]) at the endogenous location, we were not able to directly examine the LLPS properties of the native assembly.
Figure 4. Dynamic nature of spherical assemblies or condensates generated by the Cep63 (424–541)•Cep152 (1205–1295) complex in vitro. A and B, The fluorescence recovery after photobleaching (FRAP) analysis for His-Cep63 (424–541)•Cep152 (1205–1295)-generated hollowed spherical assemblies (a) or inside-filled spherical condensates formed in the presence of 5% PEG 3350 (b). Both the turnover of its constituents with those in the surroundings (left) and their internal rearrangement within a condensate (right) were determined. Images were taken every three seconds for 360 seconds and representative images at indicated time points were provided on top. To calculate the efficiency of internal rearrangement, relative signal intensities were determined by dividing the intensities in photobleached area (“b”) by the intensities in their corresponding unbleached area (“a”). Graphs are shown in mean ± s.d. (n = 23 for turnover and n = 15 for internal rearrangement for [a]. n = 14 for turnover and n = 13 for internal rearrangement for [b]). The red dotted lines indicate the level of the fluorescence intensity immediately after photobleaching
![Figure 4. Dynamic nature of spherical assemblies or condensates generated by the Cep63 (424–541)•Cep152 (1205–1295) complex in vitro. A and B, The fluorescence recovery after photobleaching (FRAP) analysis for His-Cep63 (424–541)•Cep152 (1205–1295)-generated hollowed spherical assemblies (a) or inside-filled spherical condensates formed in the presence of 5% PEG 3350 (b). Both the turnover of its constituents with those in the surroundings (left) and their internal rearrangement within a condensate (right) were determined. Images were taken every three seconds for 360 seconds and representative images at indicated time points were provided on top. To calculate the efficiency of internal rearrangement, relative signal intensities were determined by dividing the intensities in photobleached area (“b”) by the intensities in their corresponding unbleached area (“a”). Graphs are shown in mean ± s.d. (n = 23 for turnover and n = 15 for internal rearrangement for [a]. n = 14 for turnover and n = 13 for internal rearrangement for [b]). The red dotted lines indicate the level of the fluorescence intensity immediately after photobleaching](/cms/asset/fd7a686a-f5b6-41e7-83c7-0fe6e92c2014/kccy_a_1843777_f0004_oc.jpg)
Figure 5. Model illustrating the versatile capacity of Cep63 and Cep152 to phase-separate into morphologically distinct higher-order self-assemblies. Cep63 and Cep152 interact with each other and generate a heterotetrameric complex through the Cep63 (424–541) and Cep152 (1205–1272) regions [Citation8]. In the presence of the intact hydrophobic motifs (one each from Cep63 and Cep152), the resulting complex can phase-separate into a condensate or an organized assembly, as evidenced by its ability to generate a densely deposited matrix-like aggregate that exhibits dynamic turnover and fusion and undergoes an internal rearrangement of its constituents. Our data demonstrate that the complex possesses a versatile capacity to generate inside-filled condensates (in the presence of a macromolecular crowder), hollow spherical assemblies (in a 3D space), or cylindrical assemblies (in a 2D surface) under different physicochemical and spatial cues. We propose that an unknown element(s) may dictate the Cep63•Cep152 complex to assume a cylindrical localization pattern around a centriole in vivo. A arrow denotes that a spherical condensate may convert to a vesicle-like, hollow spherical assembly, although its transition to a solid-like state cannot be ruled out
![Figure 5. Model illustrating the versatile capacity of Cep63 and Cep152 to phase-separate into morphologically distinct higher-order self-assemblies. Cep63 and Cep152 interact with each other and generate a heterotetrameric complex through the Cep63 (424–541) and Cep152 (1205–1272) regions [Citation8]. In the presence of the intact hydrophobic motifs (one each from Cep63 and Cep152), the resulting complex can phase-separate into a condensate or an organized assembly, as evidenced by its ability to generate a densely deposited matrix-like aggregate that exhibits dynamic turnover and fusion and undergoes an internal rearrangement of its constituents. Our data demonstrate that the complex possesses a versatile capacity to generate inside-filled condensates (in the presence of a macromolecular crowder), hollow spherical assemblies (in a 3D space), or cylindrical assemblies (in a 2D surface) under different physicochemical and spatial cues. We propose that an unknown element(s) may dictate the Cep63•Cep152 complex to assume a cylindrical localization pattern around a centriole in vivo. A arrow denotes that a spherical condensate may convert to a vesicle-like, hollow spherical assembly, although its transition to a solid-like state cannot be ruled out](/cms/asset/a045090a-f77e-4151-8f66-a6cbb407c7b4/kccy_a_1843777_f0005_oc.jpg)
The capacity of Cep63•Cep152 condensates to carry out a substantial level of internal rearrangement ( and Fig. S5) is striking, considering that the best-characterized PCM scaffold, C. elegans Spd5, exhibits a liquid drop-like character only at the very early stage (less than 2 minutes) of its aggregation [Citation17]. However, the low level of internal rearrangement for the self-organized hollow spheres suggests that, once assembled, higher-order Cep63•Cep152 architectures could be gel- or solid-like, thus making them distinct from the classical LLPS condensates found in RNA or stress granules [Citation30,Citation34,Citation35,Citation42]. In fact, Cep63 and Cep152 are made of largely coiled-coil domains with no apparent intrinsically disordered domains [Citation13,Citation43] commonly found in multivalent proteins that undergo LLPS. In addition, they ultimately engage in a specific 2:2 α-helical interactions (as opposed to less-specific transient interactions observed in LLPS) to provide a structural stability to their self-assembly [Citation8]. A low degree of liquid-phase behavior would be desirable, if Cep63 and Cep152 were to form a higher-order architecture stable enough to serve as a platform for various centrosome-mediated cellular processes. A somewhat limited capacity of centrosomal scaffold proteins in forming condensates has been discussed in recent reviews [Citation12,Citation44].
Hydrophobic motif-derived clustering: An underlying event for promoting the condensation and self-assembly of the Cep63•Cep152 complex
We previously observed that the hydrophobic motifs present in the Cep63 (424–541)•Cep152 (1205–1295) complex play an important role in forming a higher-order complex and promoting Cep152-dependent Plk4 function in centriole biogenesis [Citation8]. Here we demonstrated that the Cep63 (4A) and Cep152 (5A) mutants bearing Ala mutations at their hydrophobic motifs were severely impaired in generating condensed assemblies both in vivo and in vitro () and Fig. S4A–C). These data strongly suggest that the hydrophobic motifs from Cep63 and Cep152 either exhibit or promote the Cep63•Cep152 complex’s LLPS properties that underlie its capacity to form a higher-order self-assembly (). Hydrophobic-interaction-based LLPS has been described previously [Citation45–47]. Further investigation into the structure and physicochemical properties of these hydrophobic residue-enriched regions will be important for better understanding the molecular mechanism underlying the self-assembly of the Cep63•Cep152 complex.
Our results showed that the assemblies generated by the Cep63 (424–541)•Cep152 (1205–1295) complex were significantly more dynamic than those of a larger Cep63 (full-length)•Cep152 (924–1295) complex (compare data in with those in Fig. S5). This is not surprising because additional interactions along the other parts of the complex may likely confer an extra solidity on the higher-order self-assembly. A physically stable Cep63•Cep152 architecture will be important to ensure that various intracellular processes can occur securely in a distinct subcentrosomal space.
Versatile nature of the Cep63•Cep152 complex in generating morphologically distinct higher-order structures
Like ectopically expressed Cep63 and Cep152 that self-assemble into a spherical morphology in bacterial cells (Fig. S4A–C), recombinant Cep63 and Cep152 self-organize into a hollow sphere in a 3D space (–d) and Fig. S4 D and E). On the other hand, these proteins form a cylindrical architecture when they are placed on a 2D surface, such as slide glass [Citation8] () and Fig. S4 D and E). Interestingly, provision of macromolecular crowders, such as PEG 6000 or PEG 8000, which could help mimic the high concentration (200–300 mg/ml) of macromolecules inside the cell [Citation48], effectively elicited the formation of inside-filled amorphous condensates often connected to one another () and Fig. S4F). The amorphous condensates could represent less-organized assembly products, which may have either partially or improperly engaged protein–protein interactions. These findings demonstrate the versatile nature of the Cep63•Cep152 complex capable of generating multiple morphologically distinct assemblies (). This is somewhat similar to the self-assembly of lipid amphiphiles that can assume a variety of structures as a result of the building block molecule’s altered packing properties under different conditions [Citation49,Citation50]. Thus, we propose that the Cep63•Cep152 complex is structurally flexible with a capacity to absorb different constraints imposed by distinct higher-order structures. Furthermore, its localization around a barrel-shaped centriole and interaction with the centriole-associated Cep57 [Citation51] may have determined the overall geometric shape of the Cep63•Cep152 self-assembly in the native pericentriolar environment.
Multiple morphologies of the Cep63•Cep152 assemblies predict the plastic feature of the Cep63•Cep152 self-assembly. This argument is consistent with the ability of the self-assembly to undergo dynamic turnover and internal rearrangement ( and Fig. S5). Given that diverse mechanochemical processes, such as microtubule assembly/disassembly and motor-driven microtubule sliding, constantly occur throughout the cell cycle, the plasticity of the Cep63•Cep152-assembled pericentriolar architecture could be very important for offering resilience to protect the architecture from mechanical stresses. Notably, multiple mutations in Cep63 or Cep152 are associated with the development of various human diseases [Citation52–55] and are found in cancer patients (https://portal.gdc.cancer.gov/). Therefore, it will be interesting to further investigate whether these mutations alter the structure, dynamics, and mechanical properties of the Cep63•Cep152 architecture as well as how these alterations influence the Cep63•Cep152 complex’s function in promoting proper centriole duplication and the maintenance of genomic stability.
Materials and methods
Plasmid construction
pShuttle-CMV vector [Citation56]-based constructs expressing mCherry-Cep63 (pKM6953), mCherry-Cep63 (4A) (pKM6954), mGFP-Cep152 (pKM6957), and mGFP-Cep152 (5A) (pKM6958) were generated by inserting corresponding BglII-NotI fragments into the pShuttle-CMV vector digested with the same enzymes. The resulting pShuttle-CMV constructs were used to generate pAdEasy-1-based adenoviral constructs (pKM6952, pKM7109, pKM7106, and pKM7110) according to the procedure published previously [Citation56].
pCI-neo-mCherry-Cep63 (pKM4916), pEGFP (A206K)-C1-Cep152 (pKM6005), pCI-neo-Flag-Plk4 (pKM3445), pCI-neo-HA-STIL (pKM4483), and pCI-neo-HA-Sas6 (pKM4381) were reported previously [Citation8].
To generate Pendo-mCherry-Cep63-sil WT and 4A (pKM7441 and pKM7442, respectively) and Pendo-mGFP-Cep152-sil WT and 5A (pKM7446 and pKM7447, respectively) constructs, the promoter regions of CEP63 (nt −481 to −1) and CEP152 (nt −461 to −1) were synthesized (GenScript) and inserted upstream into the mCherry-Cep63-sil and mGFP-Cep152-sil genes, respectively. The BglII-NotI fragment of Pendo-mCherry-Cep63-sil or the BglII-EcoRV fragment of Pendo-mGFP-Cep152-sil were cloned into the pCI-neo vector digested with BglII-NotI or BglII-SmaI, respectively.
Lentiviral pHR'.J-CMV-SV-puro-based constructs expressing FLAG3-Cep63-sil WT or 4A (pKM5659 or pKM5660) or untagged Cep152-sil WT or 5A (pKM5767 or pKM5768) were reported previously [Citation8]. Cep63-sil and Cep152-sil contain RNAi-insensitive silent mutations.
To generate a bacterial expression construct expressing both Cep63 (full-length) and Cep152 (924–1295) (pKM6370), a BamHI-NotI fragment of Cep63 and a NdeI-XhoI fragment of Cep152 (924–1295) were cloned into the respective sites in pETDuet-1 vector (Novagen). Other bacterial expression constructs–pETDuet1-His-mCherry-Cep63 (pKM6066), pRSFDuet1-His-mGFP-Cep152 (924–1295) (pKM6368), pETDuet1-His-mGFP-Cep152 (1205–1295)•mCherry-Cep63 P1(424–541) (pKM6714) and its respective 4A•5A mutant (pKM6715), and pETDuet1-His-Cep63 (424–541)•Cep152 (1205–1295) (pKM5615) and its respective 4A•5A mutant (pKM5616) – were described previously [Citation8].
All the constructs used for this study are summarized in Table S1.
Cell culture, adenovirus generation, transfection, stable cell line generation, and centrinone treatment
HEK293 cells (American Type Culture Collection; ATCC) were maintained in a high-glucose Dulbecco’s Modified Eagle Medium (DMEM) medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (HyClone Laboratories), 2 mM L-glutamine (Thermo Fisher Scientific), Minimum Essential Medium non-essential amino acid solution (Thermo Fisher Scientific), 12.3 mM HEPES (Thermo Fisher Scientific), 100 unit/ml penicillin, and 100 µg/ml streptomycin (Thermo Fisher Scientific). U2OS cells (ATCC) were maintained in McCoy’s 5A medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum, 100 unit/ml penicillin, and 100 µg/ml streptomycin.
AdEasy-1-based adenoviruses were amplified and used for infection, as previously described [Citation56]. Where indicated, transfection was carried out using Polyethylenimine Max (PEI Max; Polysciences). For the experiment shown in Fig. S1A, HEK293 cells were co-transfected with Flag-Plk4, HA-STIL, and HA-SAS6 using the PEI Max reagent. Six hours after transfection, cells were washed with PBS and coinfected with adenoviruses expressing mCherry-Cep63 and mGFP-Cep152 for eight hours. The resulting cells were then washed with PBS and treated with 200 nM okadaic acid (Cell Signaling) to activate Plk4 [Citation27] for two hours before subjecting them to immunostaining.
To generate a stable cell line expressing Pendo-mCherry-Cep63-sil and Pendo-mGFP-Cep152-sil, U2OS cells were transfected with linearized pKM7441 and pKM7446 by digesting them with AclI and BspHI, respectively. The cells were then treated with 500 µg/ml of neomycin (G-418 sulfate; USB Corporation) and 300 µg/ml hygromycin B (Roche) 3 days post-transfection and selected for at least 14 days. A stable cell line expressing Pendo-mCherry-Cep63-sil (4A) and Pendo-mGFP-Cep152-sil (5A) mutants was generated as described above with linearized pKM7442 and pKM7447 constructs. For the data shown in ) and Fig. S2C, cells were treated with 100 nM of centrinone [Citation37] for 4 days to inhibit centriole duplication (judging from the loss of Cep192 signal). Two days before fixing the cells for immunostaining, cells were silenced for endogenous Cep63 and Cep152 by treating them with their respective siRNAs. siRNA sequences are listed in Table S2.
Lentivirus production, infection, selection of stable cells, and subsequent analyses were performed as described previously [Citation8].
Immunostaining and immunoblotting analyses
For immunostaining, cells cultured on poly-L-lysine (Sigma-Aldrich)-coated No. 1.5 coverslips were fixed with 4% paraformaldehyde for 30 minutes at 4 °C and permeabilized with 0.1% Triton X-100 for five minutes at room temperature (RT). The resulting cells were then placed in a blocking buffer (5% bovine serum albumin in PBS) for one hour, washed, and reacted with indicated primary antibodies in the blocking buffer for two hours, at RT. For immunostaining with a TRITC-conjugated γ-tubulin antibody (Santa Cruz Biotechnology), cells were fixed with cold 100% methanol (–20 °C) for one minute, reacted with the primary antibody in the blocking buffer for two hours, at RT, washed two times with a wash buffer (PBS + 0.1% of Tween-20 [Sigma-Aldrich]), and labeled with an appropriate Alexa fluorophore–conjugated secondary antibody in the blocking buffer for one hour, at RT. After washing three times, samples were then treated with 4', 6-diamidino-2-phenylindole (DAPI) to visualize DNA and mounted with the ProLong Gold antifade (Thermo Fisher Scientific) for imaging.
Immunoblotting analysis was performed according to standard procedures by using an enhanced chemiluminescence detection system (Thermo Fisher Scientific), and immunoblotted signals were captured using a chemiluminescence imager (ChemiDoc™ Imaging Systems, BioRad Laboratories).
All the antibodies used for this study are listed in Table S3.
Confocal microscopy, 3D-SIM, and image analysis, and surface rendering
Confocal images (for )) were acquired using a Zeiss LSM780 equipped with a plan-apochromat 63x (N.A. 1.4) oil-immersion objective lens, 34-channel GaAsP spectral detector (Carl Zeiss Microscopy, LLC.), and 8-bit, 0.5-μm z-steps. To quantify fluorescence signal intensities, images were acquired under the same settings and the images obtained after the maximum-intensity projection of z-stacks were analyzed using the Zeiss ZEN v2.1 software (Carl Zeiss Microscopy, LLC).
3D-SIM images were acquired using Zeiss ELYRA S1 super-resolution microscope (Zeiss) equipped with Alpha Plan-Apo 63x/1.46 oil objective, 405 nm/488 nm/562 nm/640 nm laser illumination, and standard excitation and emission filter sets. Acquired images were processed by ZEN black software (Zeiss) for further analyses. The Zeiss ZEN software enabled an inter-signal distance of up to 33 nm/pixel. The z-stack interval was at 110 nm/stack. 3D surface-rendering for 3D-SIM images was carried out using Imaris software version 7.7.0 (Bitplane). The 2.5D intensity plots for mCherry-Cep63 and mGFP-Cep152 (Fig. S1B) were generated by using the ZEN black software.
Correlative and light electron microscopy
For CLEM in ), HEK293 cells transfected with constructs expressing mCherry-Cep63 and mGFP-Cep152 were grown on a 35-mm No. 1.5 glass-bottom gridded dish (MatTek Corporation) for 14 hours and fixed with 4% paraformaldehyde for 30 minutes at 4 °C. The resulting cells were permeabilized with 0.1% Triton X-100 for five minutes at RT, immunostained with anti-Cep192 and Alexa 647 fluorophore-conjugated secondary antibody, and then imaged using a Zeiss ELYRA 3D-SIM system. After fixing the cells with 2% glutaraldehyde in the 0.1 M sodium cacodylate buffer (pH 7.4), the resulting samples were processed and embedded essentially as described above. The area containing the cells identified by 3D-SIM was sectioned and then prepared for thin-section transmission electron microscopy. Images were acquired using the Hitachi H-7650 transmission electron microscope.
Fluorescence recovery after photobleaching
For the data shown in ) and Movie S1, HEK293 cells were cultured on the Lab-Tek II chambered coverglass No. 1.5 borosilicate glass bottom (Thermo Fisher Scientific) at 37 °C and infect with adenoviruses expressing mCherry-Cep63 and mGFP-Cep152 for eight hours. The cells were washed twice with cold PBS and subjected to time-lapse imaging using a Zeiss LSM880 Airyscan microscope. Photobleaching in the specific region of interest was performed using 100% acousto-optic tunable filter (AOTF)-modulated transmission power of the 488-nm laser, 10 iterations, 90% 561-nm laser, 10 iterations, and 4.10-µs pixel dwell time. Fluorescence recovery was monitored every two seconds over a 600-second time period.
The hollow spherical assemblies for the FITC-conjugated His-Cep63 (424–541)•Cep152 (1205–1295) complex in ) were generated by incubating the complex (6 µM) in the assembly buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5 mM TCEP) in a PCR tube (i.e., 3D space) at 4 °C for 14 hours. The inside-filled condensates shown in ) were generated under the same conditions except that the protein was incubated in the assembly buffer + 5% PEG 3350 for one hour. To quantify the turnover rate, samples obtained above (spherical assemblies or condensates) were transferred onto the Lab-Tek II chambered coverglass and subjected to FRAP analysis. To observe the capacity of internal rearrangement, the samples (spherical assemblies and condensates) were centrifuged at 18,000 × g (Beckman Coulter Microfuge 22 R, F241.5P, 14,000 rpm) at 4 °C for 15 minutes to eliminate free molecules, then resuspended in the assembly buffer in the absence of PEG 3350. FRAP analyses were performed essentially as for ) except that 405-nm laser was used for 20 iterations with 2.62-µs pixel dwell time. Fluorescence recovery was monitored by collecting images every three seconds over a 360-second time period. For the data shown in Fig. S5, FITC-conjugated His-Cep63•Cep152 (924–1295) condensates were generated by incubating the complex (5 µM) in the assembly buffer + 5% PEG 3350 in a PCR tube at 4 °C for 30 minutes. The procedures taken for turnover and internal rearrangement were the same as described for ).
All the Airyscan images were processed using the ZEN 2.3 SP1 image processing software.
Live-cell time-lapse microscopy
For the data shown in ) and Movie S2, U2OS cells were cultured on a Lab-Tek II chambered coverglass with a No. 1.5 borosilicate glass bottom (Thermo Fisher Scientific), infected with adenoviruses expressing mCherry-Cep63 and mGFP-Cep152, and subjected to live-cell imaging using a using a Zeiss LSM880 Airyscan microscope with a 32-channel GaAsP Airyscan detector, a plan-apochromat 63x (N.A. 1.4) oil-immersion objective lens, and a PeCon heated stage incubator (temperature, humidity, and 5% CO2). Images were acquired every 40 seconds up to 4,000 seconds. The processed images were analyzed using ZEN v2.1 software (Zeiss).
OptiPrep (Iodixanol) density gradient ultracentrifugation
To prepare the discontinuous iodixanol gradient, 50% (w/v), 40% (w/v), 30%(w/v), and 20% (w/v) iodixanol solutions were made by diluting 60% (w/v) OptiPrep density gradient medium (Sigma-Aldrich; D1556) with the assembly buffer. To prepare cell lysates, 20 ml of E. coli Rosetta cells expressing the gene of interest were cultured in Luria broth (LB) medium at 37 °C until their optical density (OD) reached to 0.8, then cultured in the presence of 0.5 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) at 30 °C for four hours. Cells were lysed in an ice-cold TBSN buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.5% NP-40, 5 mM EGTA, 2 mM MgCl2, 1.5 mM EDTA, and protease inhibitor cocktail [Roche]) by ultrasonication. The gradient was formed by adding 1 ml each of 50%, 40%, 30%, and 20% iodixanol solutions into a 13-mm × 64-mm thickwall polycarbonate tube (Beckman Coulter Life Sciences). The cell lysates (1 ml) were overlaid onto the top of the gradient and ultracentrifugation was performed at 122,800 × g (Beckman Coulter SW 55 Ti, 36,000 rpm) at 4 °C for two hours. Ten individual 500 µl gradient fractions were collected using Gilson Minipuls 2 peristaltic pump (Gilson) from bottom to top. Collected fractions were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE).
Protein expression and purification
All proteins for structural study were expressed in E. coli Rosetta strain (Novagen). Cells were cultured in LB medium at 37 °C until their OD reached to 0.8, and then proteins were expressed overnight at 16 °C with 0.5 mM IPTG.
For purification of mCherry-Cep63 (424–541)•mGFP-Cep152 (1205–1295) (pKM6714), His-Cep63 (424–541)•Cep152 (1205–1295) (pKM5615), and His-Cep63 (424–541) (4A)•Cep152 (1205–1295) (5A) (pKM5616) complexes, cells were lysed in an ice-cold buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 5% [v/v] glycerol, and 0.5 mM TCEP) by ultrasonication and precleared by centrifugation at 49,000 × g (Sorvall RC-5 C Plus SS-34, 20,000 rpm) for 20 minutes. The supernatant was applied to a HisTrap HP column (GE Healthcare) and the bound protein was eluted with the lysis buffer containing 150 mM imidazole. The eluted protein samples were subjected to HiLoad 16/60 Superdex 200 (GE Healthcare) size-exclusion chromatography in the final buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, and 0.5 mM TCEP).
To purify His-mCherry-Cep63 (pKM6066) and mGFP-Cep152 (924–1295) (pKM6368), cells were lysed in an ice-cold buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 10% [v/v] glycerol, and 0.5 mM TCEP) by ultrasonication and precleared as above. The supernatant was applied to a HisTrap HP column (GE Healthcare), and the bound protein was eluted with the lysis buffer with increasing concentrations of imidazole (20–300 mM). Samples were dialyzed with a low-salt binding buffer (20 mM Tris-HCl [pH7.5], 20 mM NaCl, 10% [v/v] glycerol, 0.5 mM TCEP) for four hours and then subjected to HiTrap Q HP (GE Healthcare) anion-exchange chromatography. The bound target protein was eluted with increasing concentrations of NaCl (20–500 mM). The eluted protein sample was subjected to size-exclusion chromatography with HiLoad 16/60 Superdex 200 (GE Healthcare) in the final buffer.
To purify the Cep63•Cep152 (924–1295) complex (pKM6370), the cells that expressed both Cep63 and Cep152 (924–1295) were lysed by ultra-sonication in an ice-cold lysis buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 10% [v/v] glycerol and 0.5 mM TCEP). The resulting supernatant was applied to a HisTrap HP column (GE Healthcare), and the bound protein was eluted with the lysis buffer containing an additional 500 mM imidazole. The eluted protein was subjected to HiLoad 16/60 Superdex 200 (GE Healthcare) size-exclusion chromatography equilibrated with the final buffer + 5% (v/v) glycerol.
All purified proteins showed more than 95% purity as estimated by SDS-PAGE.
Visualizing Cep63•Cep152 condensates and assemblies with FITC isomer I
To visualize complexes of His-Cep63 (424–541)•Cep152 (1205–1295) and its 4A•5A mutant, and His-Cep63 (full length)•Cep152 (924–1295), 20 µM of each of the indicated protein complexes in the gel filtration final buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, and 0.5 mM TCEP) were reacted with 800 µM of FITC isomer I (Sigma) in 0.1 M carbonate-bicarbonate buffer (pH 9.0) in a PCR tube for three hours at 4 °C, to decorate surface Lys residues. After three hours, reacted proteins were dialyzed in the assembly buffer at 4 C for two hours in the dark to eliminate free FITC molecules.
Detection of cytosolic Cep63•Cep152 condensates in vitro
To detect Cep63•Cep152 condensates in vitro, U2OS cells stably expressing ()) or transfected with ()) Pendo-mCherry-Cep63-sil and Pendo-mGFP-Cep152-sil under the control of endogenous CEP63 and CEP152 promoters, respectively, were depleted of endogenous Cep63 and Cep152 by treating them with 20 nM of their respective siRNAs (Table S2). The cells were harvested and lysed in PBS by 5-second pulses (15 seconds total) at the 80% pulse mode using a 130 W ultrasonic processor (Cole-Parmer). The resulting lysates were clarified by centrifugation at 20,000 × g for 10 minutes, incubated in PBS ()) or PBS + 5% glycerol ()), placed on a 5 K poly-L-lysine-coated slide glass, and subjected to confocal microscopy.
In vitro self-assemblies
To generate the condensates shown in ) and Fig. S4F, the indicated complex (6 µM) were incubated in the assembly buffer + 5% PEG 3350 in a PCR tube (i.e., 3D space) for 14 hours at 4 °C.
In vitro hollow spherical assemblies in ) (upper panel) and Fig. S4 D and E (upper panels) were generated by incubating the indicated complex (6 µM) in the assembly buffer without PEG in a PCR tube for 14 hours at 4 °C. The resulting samples were mounted on the slide glass with SlowFade (Thermo Fisher Scientific) for 3D-SIM imaging. In vitro cylindrical structure self-assemblies shown in ) (lower panel), Fig. S3B, and Fig. S4 D and E (lower panels) were generated as described previously [Citation8]. To examine the effect of an LLPS disruptor, 1,6-hexanediol, shown in Fig. S3B, either cylindrical assemblies were formed in the presence/absence of 6% 1,6-hexanediol, or preassembled cylinders were treated with 6% 1,6-hexanediol for the indicated lengths of time. The resulting samples were embedded with SlowFade (Thermo Fisher Scientific) for 3D-SIM.
Statistical analysis
All experiments were performed independently at least two times. All values are given as mean ± s. d. P values were calculated by one-way ANOVA (for ) only) and unpaired t test from the mean data of each group in GraphPad Prism (* P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001).
Author contributions
J.I.A., L.M., L.Z., and T.S.K. designed and performed experiments and interpreted data; M.J.K., B.Y.K., J.E.P, and K.S.L. provided insightful comments and interpret the data; and K.S.L., J.I.A., and L.Z. wrote the manuscript.
Supplemental Material
Download Zip (21.9 MB)Acknowledgments
We are grateful to Jung Mi Lim and Eunhye Lee for helping with OptiPrep and condensate analyses, respectively. We also thank Langston Lim for assisting 3D-SIM imaging and Yanling Liu for generating 3D surface-rendered movies. This research was supported by the Intramural Research Program of National Cancer Institute, National Institutes of Health (K.S.L.), the R&D Convergence Program (CAP-16-03-KRIBB) of National Research Council of Science & Technology of South Korea (B.Y.K), and the KRIBB Research Initiative Program (No. KGS1001911), Korea Research Institute of Bioscience and Biotechnology, Republic of Korea (J.I.A).
Disclosure statement
The authors declare that they have no conflicts of interest with the contents of this article.
Data availability
All the data are in the manuscript.
Supplementary materials
Supplemental data for this article can be accessed here.
Related Research Data
References
- Conduit PT, Wainman A, Raff JW. Centrosome function and assembly in animal cells. Nat Rev Mol Cell Biol. 2015;16(10):611–624.
- Vertii A, Hehnly H, Doxsey S. The centrosome, a multitalented renaissance organelle. Cold Spring Harb Perspect Biol. 2016;8(12):a025049.
- Lawo S, Hasegan M, Gupta GD, et al. Subdiffraction imaging of centrosomes reveals higher-order organizational features of pericentriolar material. Nat Cell Bio. 2012;14(11):1148–1158.
- Mennella VK, Eszthelyi B, McDonald KL, et al. Subdiffraction-resolution fluorescence microscopy reveals a domain of the centrosome critical for pericentriolar material organization. Nat Cell Bio. 2012;14(11):1159–1168.
- Fu J, Glover DM. Structured illumination of the interface between centriole and peri-centriolar material. Open Biol. 2012;2(8):120104.
- Sonnen KF, Schermelleh L, Leonhardt H, et al. 3D-structured illumination microscopy provides novel insight into architecture of human centrosomes. Biol Open. 2012;1(10):965–976.
- Olivier N, Keller D, Gonczy P, et al. Resolution doubling in 3D-STORM imaging through improved buffers. PLoS One. 2013;8:e69004.
- Kim TS, Zhang L, Il Ahn J, et al. Molecular architecture of a cylindrical self-assembly at human centrosomes. Nat Commun. 2019;10(1):1151.
- Gonczy P. Centrosomes and cancer: revisiting a long-standing relationship. Nat Rev Cancer. 2015;15(11):639–652.
- Nigg EA, Holland AJ. Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat Rev Mol Cell Biol. 2018;19:297–312.
- Woodruff JB, Wueseke O, Hyman AA. Pericentriolar material structure and dynamics. Philos Trans R Soc Lond B Biol Sci. 2014;369(1650):20130459.
- Woodruff JB. Assembly of mitotic structures through phase separation. J Mol Biol. 2018;430(23):4762–4772.
- Banani SF, Lee HO, Hyman AA, et al. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18(5):285–298.
- Hyman AA, Weber CA, Julicher F. Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol. 2014;30(1):39–58.
- Alberti S, Gladfelter A, Mittag T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell. 2019;176(3):419–434.
- Woodruff JB, Wueseke O, Viscardi V, et al. Regulated assembly of a supramolecular centrosome scaffold in vitro. Science. 2015;348(6236):808–812.
- Woodruff JB, Ferreira Gomes B, Widlund PO, et al. The centrosome is a selective condensate that nucleates microtubules by concentrating tubulin. Cell. 2017;169(6):1066–1077 e1010.
- Feng Z, Caballe A, Wainman A, et al. Structural basis for mitotic centrosome assembly in flies. Cell. 2017;169(6):1078–1089.
- Lupas AN, Bassler J. Coiled Coils - A model system for the 21st century. Trends Biochem Sci. 2017;42(2):130–140.
- Shaner NC, Patterson GH, Davidson MW. Advances in fluorescent protein technology. J Cell Sci. 2007;120(24):4247–4260.
- Schnackenberg BJ, Khodjakov A, Rieder CL, et al. The disassembly and reassembly of functional centrosomes in vitro. Proc Natl Acad Sci USA. 1998;95(16):9295–9300.
- Arquint C, Nigg EA. The PLK4-STIL-SAS-6 module at the core of centriole duplication. Biochem Soc Trans. 2016;44(5):1253–1263.
- Zitouni S, Nabais C, Jana SC, et al. Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol. 2014;15(7):433–452.
- Kitagawa D, Vakonakis I, Olieric N, et al. Structural basis of the 9-fold symmetry of centrioles. Cell. 2011;144(3):364–375.
- Park SY, Park JE, Kim TS, et al. Molecular basis for unidirectional scaffold switching of human Plk4 in centriole biogenesis. Nat Struct Mol Biol. 2014;21(8):696–703.
- Kim T-S, Park J-E, Shukla A, et al. Hierarchical recruitment of Plk4 and regulation of centriole biogenesis by two centrosomal scaffolds, Cep192 and Cep152. Proc Natl Acad Sci USA. 2013;110(50):E4849–4857.
- Ohta M, Ashikawa T, Nozaki Y, et al. Direct interaction of Plk4 with STIL ensures formation of a single procentriole per parental centriole. Nat Commun. 2014;5(1):5267.
- Park JE, Zhang L, Bang JK, et al. Phase separation of Polo-like kinase 4 by autoactivation and clustering drives centriole biogenesis. Nat Commun. 2019;10(1):4959.
- Yamamoto S, Kitagawa D. Self-organization of Plk4 regulates symmetry breaking in centriole duplication. Nat Commun. 2019;10(1):1810.
- Kroschwald S, Maharana S, Mateju D, et al. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. Elife. 2015;4:e06807.
- Kroschwald S, Maharana S, Simon A. Hexanediol: a chemical probe to investigate the material properties of membrane-less compartments. Matters. 2017;1–8. DOI:10.19185/matters.201702000010
- Patel SS, Belmont BJ, Sante JM, et al. Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell. 2007;129(1):83–96.
- Ribbeck K, Gorlich D. The permeability barrier of nuclear pore complexes appears to operate via hydrophobic exclusion. Embo J. 2002;21(11):2664–2671.
- Wheeler JR, Matheny T, Jain S, et al. Distinct stages in stress granule assembly and disassembly. Elife. 2016;5:5. pii: e18413.
- Molliex A, Temirov J, Lee J, et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163(1):123–133.
- Kann ML, Soues S, Levilliers N, et al. Glutamylated tubulin: diversity of expression and distribution of isoforms. Cell Motil Cytoskeleton. 2003;55(1):14–25..
- Wong YL, Anzola JV, Davis RL, et al. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science. 2015;348(6239):1155–1160.
- Brown NJ, Marjanovic M, Luders J, et al. Cep63 and cep152 cooperate to ensure centriole duplication. PLoS One. 2013;8(7):e69986.
- Lukinavicius G, Lavogina D, Orpinell M, et al. Selective chemical crosslinking reveals a Cep57-Cep63-Cep152 centrosomal complex. Curr Biol. 2013;23(3):265–270.
- Bauer M, Cubizolles F, Schmidt A, et al. Quantitative analysis of human centrosome architecture by targeted proteomics and fluorescence imaging. Embo J. 2016;35(19):2152–2166.
- Milo R, Phillips R. Cell biology by the numbers. Garland Sci. 2015;44. Chapter 1.
- Kato M, Han TW, Xie S, et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149(4):753–767.
- Harmon TS, Holehouse AS, Rosen MK, et al. Intrinsically disordered linkers determine the interplay between phase separation and gelation in multivalent proteins. Elife. 2017;6. DOI:10.7554/eLife.30294.
- Raff JW. phase separation and the centrosome: a fait accompli? Trends Cell Biol. 2019;29(8):612–622.
- Jiang H, Wang S, Huang Y, et al. Phase transition of spindle-associated protein regulate spindle apparatus assembly. Cell. 2015;163(1):108–122.
- Quiroz FG, Chilkoti A. Sequence heuristics to encode phase behaviour in intrinsically disordered protein polymers. Nat Mater. 2015;14(11):1164–1171.
- Reichheld SE, Muiznieks LD, Keeley FW, et al. Direct observation of structure and dynamics during phase separation of an elastomeric protein. Proc Natl Acad Sci USA. 2017;114(22):E4408–E4415.
- Ellis RJ. Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci. 2001;26(10):597–604.
- Israelachvili JN. Intermolecular and surface forces. In: Part III. Self-assembling structures and biological systems. 3rd. Academic press (Elsevier); 2011:501.
- Mai Y, Eisenberg A. Self-assembly of block copolymers. Chem Soc Rev. 2012;41(18):5969–5985.
- Wei Z, Kim TS, Ahn JI, et al. Requirement of the Cep57-Cep63 interaction for proper Cep152 recruitment and centriole duplication. Mol Cell Biol. 2020;40(10):e00535–00519.
- Klingseisen A, Jackson AP. Mechanisms and pathways of growth failure in primordial dwarfism. Genes Dev. 2011;25(19):2011–2024.
- Sir JH, Barr AR, Nicholas AK, et al. A primary microcephaly protein complex forms a ring around parental centrioles. Nat Genet. 2011;43(11):1147–1153.
- Guernsey DL, Jiang H, Hussin J, et al. Mutations in centrosomal protein CEP152 in primary microcephaly families linked to MCPH4. Am J Hum Genet. 2010;87(1):40–51.
- Kalay E, Yigit G, Aslan Y, et al. CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat Genet. 2011;43(1):23–26.
- Luo J, Deng ZL, Luo X, et al. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc. 2007;2(5):1236–1247.