
ABSTRACT
Cholangiocarcinoma (CCA) is a variety of biliary epithelial tumors involving intrahepatic, perihilar and distal bile duct. It is the most common malignant bile duct tumor in the liver and the second most common primary liver cancer, whose molecular mechanism not fully understood. Specifically, the relationship between CCA and chondroitin polymerizing factor (CHPF) is still not clear. In this study, detection of clinical specimens was performed to preliminarily study the role of CHPF in CCA. CCA cells with CHPF knockdown were constructed for in vitro study, which was also used in the construction of mice xenograft model for investigating the role of CHPF in the development of CCA. The results demonstrated that CHPF was significantly upregulated in CCA tissues compared with normal tissues. High expression of CHPF was correlated with more advanced tumor grade. Moreover, knockdown of CHPF significantly inhibited cell proliferation, cell migration, promoted cell apoptosis and arrest cell cycle in G2 phase in vitro, as well as suppressed tumor growth in vivo. In conclusion, CHPF was identified as a tumor promotor in the development and metastasis of CCA, which may provide a novel therapeutic target for the targeted therapy against CCA.
Introduction
Cholangiocarcinoma (CCA) is a variety of biliary epithelial tumors involving intrahepatic, perihilar and distal bile duct. It is the most common malignant bile duct tumor in the liver and the second most common primary liver cancer, accounting for 10–20% of primary liver cancer [Citation1,Citation2]. In the past decades, the morbidity and mortality of CCA are both increasing rapidly worldwide [Citation3]. Moreover, CCA is usually diagnosed in advanced stage because of its “silent” clinical features [Citation4]. Nowadays, surgery is still the most effective treatment for CCA, but only 30% of patients are diagnosed with resectable tumor [Citation4,Citation5]. Even treated with the most aggressive surgery, the recurrence rate of patients with CCA after resection is still high, which is usually between 67% and 75%, and the 5-year survival rate is still lower than 5% [Citation6]. For patients with advanced or unresectable CCA, they could only benefit from chemotherapy [Citation7]. However, due to the drawback of drug resistance, the long-term therapeutic effect is still very poor [Citation7,Citation8]. Therefore, deepening the understanding of molecular mechanism of CCA is in urgent need for development more effective treatment strategy against CCA [Citation8].
Chondroitin sulfate (CS) is a sulfated glycosaminoglycan that is widely distributed in the connective tissues of humans and other animals [Citation9,Citation10]. It is noteworthy that there is a correlation between CS and the regulation of various types of diseases such as osteoarthritis [Citation9,Citation11], cardiovascular and cerebrovascular diseases [Citation12], central nervous system related diseases [Citation13] and malignant tumors [Citation14]. Further analysis suggested that CS may exert anticancer effects through immunomodulatory and angiogenic inhibition [Citation15,Citation16]. The biosynthesis of CS is extremely complex, which requires the participation of a variety of glycosyltransferases. chondroitin polymerizing factor (CHPF) is an essential cofactor in the synthesis of double disaccharide units in CS mediated by human chondroitin synthase [Citation10,Citation17]. Except for its role in biosynthesis of CS, CHPF has also been reported to be associated with several types of human cancers [Citation18,Citation19]. For example, Cao et al. supported that the expression of CHPF in lung cancer can regulate cell proliferation and invasion [Citation20]. Xu et al. elucidated that CHPF is involved in the malignant proliferation and migration of hepatocellular carcinoma [Citation21]. Sun et al. put forward that CHPF could promote the progression of malignant melanoma by regulating CDK1 [Citation22]. However, the role in CCA played by CHPF has not been reported and remains unclear.
In this study, we identified CHPF as a tumor promotor in the development and progression of CCA and a potential therapeutic target for CCA treatment. CHPF was found to be upregulated in CCA tumor tissues, whose high expression was associated with more advanced malignant grade. The in vitro experiments clarified that knockdown of CHPF could inhibit CCA development through regulating cell growth, apoptosis and migration, while CHPF overexpression induced reversed effects. The inhibition of CCA by CHPF knockdown was also confirmed by in vivo experiments through constructing mice xenograft models.
Material and methods
Cell culture, transduction and antibodies
Human CCA cell lines QBC939, HCCC-9810 and HUCCT1 cells were purchased from BeNa Technology (Hangzhou, Zhejiang, China). QBC939 cells were cultured in 90% DMEM-H medium with 10% FBS containing glutamine and sodium pyruvate. HCCC-9810 and HUCCT1 cells were grown in 90% RPMI-1640 medium with 10% FBS additives containing glutamine. All culture medium was changed every 3 days and cells were humid maintained in a 37°C 5% CO2 incubator.
QBC939 and HCCC-9810 cells (2 × 105 cells/mL) were infected with 400 μL lentiviral vectors (1 × 107 TU/mL) additive with ENI.S and polybrene (10 µg/mL, Sigma-Aldrich, St Louis, MO, USA) at a MOI of 20 in 6-well plates. After cultured at 37°C with a 5% CO2 for 72 h, the fluorescence was observed under microscope with magnification of ×100 and ×200.
Antibodies used in our study were detailed as follows: CHPF (1:100, # ab224495 Abcam) for IHC and CHPF (1:1000), E-cadherin (1:1000, #3195, CST), N-cadherin (1:1000, # ab18203, Abcam), Vimentin (1:1000, # ab92547, Abcam) for WB. Inner control antibody GAPDH (1:3000, # AP0063, Bioworld), and horseradish peroxidase (HRP) Goat antirabbit IgG (1:3000, #A0208, Beyotime). Ki-67 (1:200, # Ab16667, Abcam) and HRP Goat antirabbit IgG (1:400, # ab6721, Abcam) for Ki-67 assay.
Immunohistochemistry (IHC)
Extrahepatic and intrahepatic bile duct adenocarcinoma tissue microarray chip was obtained from Xian Alenabio Co., Ltd (Xian, Shanxi, China), including 74 cases of bile duct adenocarcinoma tissue and 5 cases of bile duct normal tissue. Before the IHC experiment, the tissue microarray chip was baked at 60°C for 30 min in an oven. Next, the chip was dehydrated in xylene and rehydrated in graded alcohol (100%, 95%, 90%, and 70%). Antigen of the chip was recovered by boiling citric acid buffer for 30 min. After blocked with 3% H2O2 and rabbit serum, the chip was incubated with the primary polyclonal antibody of rabbit to CHPF at 4°C overnight. Subsequently, the chip was washed with PBS three times, and the second antibody was added and incubated for 2 h at room temperature. Finally, the chip was stained with DAB solution for 10 min and counterstained with hematoxylin for 5 min. All tissues in the chip were pictured with microscopic and all slides were viewed with ImageScope and CaseViewer. IHC scores were determined by staining percentage scores and staining intensity scores. Staining percentage scores were classified as: 1 (1–24%), 2 (25–49%), 3 (50–74%), 4 (75–100%) and staining intensity was scored as 0 (Signalless color), 1 (brown), 2 (light yellow), 3 (dark brown). CHPF expression outcomes in CCA tissues and para-normal tissues revealed here were analyzed.
Lentiviral vector construction and package
Short hairpin RNA and overexpression construction of CHPF was designed by Shanghai Biosciences, Co., Ltd (Shanghai, China) and the sequence was 5’-AGCTGGCCATGCTACTCTTTG-3’. The following primer sequences were used for amplifying: Forward, 5’-CCTATTTCCCATGATTCCTTCATA-3’ and reverse, 5’-GTAATACGGTTATCCACGCG-3’. 20 µL PCR volume contained 0.2 µL DNA, 0.4 µL forward primer and 0.4 µL reverse primer and the PCR cycling condition is 94°C 3 min, 94°C 30 s, 55°C 30 s, 72°C 30 s, 22 cycles, a final extension for 72°C for 5 min. The PCR products were verified by DNA sequencing and then target sequence was cloned into BR-V-108 lentiviral vector (Shanghai Biosciences, China). EndoFree Maxi Plasmid Kit (Tiangen Biotech, Beijing, China) was used for plasmid extraction. Qualified plasmid was used for packaging.
qRT-PCR
Transfected QBC939 and HCCC-9810 cells (LV-shCtrl and LV-shCHPF) were fully lysed and total RNA was extracted using TRIzol reagent (Sigma, St. Louis, MO, USA). Nanodrop 2000/2000 C spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) was used to evaluate the RNA quality according to the manufacturer’s instructions. Promega M-MLV kit (Heidelberg, Germany) was used to reverse transcribed RNA (2.0 μg) to cDNA. Two steps qRT-PCR was performed with SYBR Green mastermixs Kit (Vazyme, Nanjing, Jiangsu, China) and melting curve was drawn, the relative quantitative analysis in gene expression data was analyzed by the 2−ΔΔCt method. GAPDH act as the inner control, and the upstream and downstream primer sequences of human CHPF for the PCR reaction were 5’-GGAACGCACGTACCAGGAG-3’ and 5’-CGGGATGGTGCTGGAATACC-3’, respectively. The upstream and downstream primer sequences of GAPDH were 5’-TGACTTCAACAGCGACACCCA-3’ and 5’-CACCCTGTTGCTGTAGCCAAA-3’.
Western blotting
Lentivirus transfected QBC939 and HCCC-9810 cells (LV-shCtrl and LV-shCHPF) were fully lysed in ice-cold RIPA buffer (Millipore, Temecula, CA, USA) and total protein was collected. The protein concentration was detected by BCA Protein Assay Kit (HyClone-Pierce, Logan, UT, USA). A total of 20 μg per lane was separated by 10% SDS-PAGE (Invitrogen, Carlsbad, CA, USA) and then transferred onto PVDF membranes at 4°C. The PVDF membranes were blocked with TBST solution containing 5% degreased milk at room temperature for 1 h. Then the PVDF membranes were incubated at 4°C overnight with primary antibodies. After washing with TBST, the membranes were further incubated with second antibody HRP-conjugated Goat antirabbit IgG for 2 h at room temperature. The blots were visualized by enhanced chemiluminescence (ECL, Amersham, Chicago, IL, USA) and the density of the proteins band was analyzed by ImageJ software.
MTT assay
2500 lentivirus transfected QBC939 and HCCC-9810 cells were seeded into a 96-well plate with 100 mL culture medium. The detection time points were 24 h, 48 h, 72 h, 96 h, 120 h. Four hours before each detection time point, 20 μL 5 mg/mL MTT solution (GenView, El Monte, CA, USA) was added for coloring. After formazan was dissolved by DMSO solution, the absorbance values at 490 nm were measured by microplate reader (Tecan, Männedorf, Zürich, Switzerland) with a reference wavelength of 570 nm. The cell viability ratio was calculated.
Cell apoptosis and cycle assay
Lentivirus transfected QBC939 and HCCC-9810 cells were inoculated in a 6-well plate until cell density reached 85%. Cells were harvested and washed with 4°C ice-cold PBS. After centrifugation (1200 × g), cells were resuspended with 100 μL binding buffer.
For cell apoptosis, 10 μL Annexin V-APC (eBioscience, San Diego, CA, USA) was added and incubated at room temperature without light. After 15 min later, 300 μL binding buffer was added and apoptosis analysis was measured using FACSCalibur (BD Biosciences, San Jose, CA, USA).
For cell cycle, cells were stained by staining solution containing 40 × PI (2 mg/ml), 100 × RNase (10 mg/ml) and 1× PBS. FACSCalibur (BD Biosciences, San Jose, CA, USA) was used to detect cell cycle distribution at 200–300 Cell/s. The percentage of cells in G1, S, and G2-M phases were analyzed.
Wound-healing assay
Lentivirus transfected QBC939 and HCCC-9810 cells (5 × 104 cells/well) were plated into a 96-well dish in triplicate for culturing. After confluence of cells reached 90%, the medium was exchanged to medium with 0.5% FBS. Cell scratches across the cell layer were accomplished by a 96-wounding replicator (VP scientific, San Diego, CA, USA). Then the cell layers were gently washed with PBS. 24 and 48 h post scratching, photographs were taken by a fluorescence microscope and cell migration rates were calculated.
Human Apoptosis Antibody Array
In lentivirus transfected HCCC-9810 cells, the related genes in human apoptosis signal pathway were detected with Human Apoptosis Antibody Array (# ab134001, Abcam, Cambridge, MA, USA) following the manufacturer’s instructions. Briefly, lentivirus transfected HCCC-9810 cells at 90% confluence were collected, washed, lysed in ice-cold RIPA buffer (Millipore, Temecula, CA, USA) and then protein concentration was detected by BCA Protein Assay Kit (HyClone-Pierce, Logan, UT, USA). Protein were incubated with blocked array antibody membrane overnight at 4°C. After washing, 1:100 Detection Antibody Cocktail was added incubating for 1 h, followed by incubated with HRP linked streptavidin conjugate for 1 h. All spots were visualized by enhanced ECL (Amersham, Chicago, IL, USA) and the signal densities were analyzed with ImageJ software (National Institute of Health, Bethesda, MD, USA).
Xenograft mouse model experiments
Female BALB/c nude mice (aged 4 weeks, weighted 17–20 g) obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) were used in our study and the animal experiments were approved by Ethics committee of Hunan Provincial People’s Hospital. 4 × 106 LV-shCtrl or LV-shCHPF transfected HUCCT1 cells were subcutaneously injected into the 10 mice which were randomly divided into shCtrl and shCHPF groups. The mice were housed in a conditional environment with a 12-h dark/light cycle. The tumor size and weight of each mouse was monitored 2 times per week. For in vivo bioluminescence imaging, all mice were anesthetized by intraperitoneal injection of 0.7% pentobarbital sodium (10 uL/g) 9 weeks post-cell injection, and anesthetized mice were placed under the Berthold Technologies living imaging system and imaging was collected. Then anesthetized mice were sacrificed by cervical dislocation and the tumor tissues were harvested for Ki-67 staining assay.
Ki-67 staining assay
Mice tumor tissues were fixed in 10% formalin for 24 h and then were paraffin-embedded. 5 μm slides were cut and immersed in xylene and ethanol for deparaffinization and rehydration. Tissue slides were blocked with 3% PBS-H2O2 and were incubated with primary antibody Ki-67 at 4°C overnight. Then slides were incubated with HRP goat anti-rabbit IgG at room temperature for 2 h. Finally, all slides were stained by Hematoxylin (# BA4041, Baso) for 10 min and Eosin for 5 min (# BA4022, Baso, Zhuhai, Guangdong, China). Stained slides were examined at ×100 and ×200 objective lens microscopic.
Statistical analyses
All cell experiments were performed in triplicate and data were shown as mean ± SD. The significance of the differences between shCHPF and shCtrl group in cells experiment was determined using the two-tailed Student’s t-test. Mann–Whitney U analysis and Spearman rank correlation analysis were used while explaining the relationship between CHPF expression and tumor characteristics in patients with CCA. Statistical significance (P value) was calculated by SPSS 22.0 (IBM, SPSS, Chicago, IL, USA) with P value < 0.05 as statistically significant. Graphs were made using GraphPad Prism 6.01 (Graphpad Software, La Jolla, CA, USA).
Results
CHPF was highly expressed in CCA
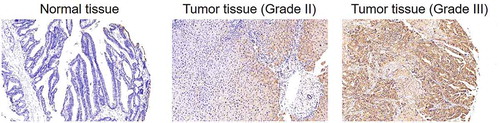
For exploring the role played by CHPF in the development of CCA, clinical specimens (including 74 CCA tissues and 5 normal tissues) were collected and subjected to IHC analysis of CHPF expression. As shown in and , CHPF was found to be expressed in both tissues, showing obviously higher expression in CCA tissues. Simultaneously, it was demonstrated that tumor tissues with higher grade were accompanied with higher expression of CHPF, indicating the potential linkage of them (). Consistently, the correlation analysis between CHPF expression and tumor characteristics of CCA patients showed significant association between CHPF expression and malignant grade (). Altogether, the upregulation of CHPF in CCA was illustrated, which indicated its potential role in the development of CCA.
Table 1. Expression patterns of CHPF in cholangiocarcinoma tissues and normal tissues revealed in immunohistochemistry analysis
Table 2. Relationship between CHPF expression and tumor characteristics in patients with cholangiocarcinoma
Table 3. Relationship between CHPF expression and tumor characteristics in patients with cholangiocarcinoma analyzed by Spearman rank correlation analysis
Figure 1. CHPF was upregulated in cholangiocarcinoma
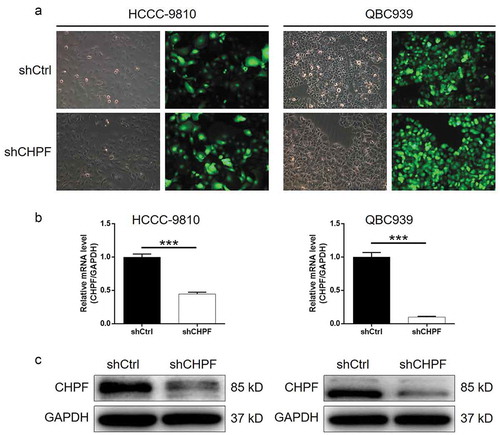
Construction of CHPF knockdown CCA cell lines
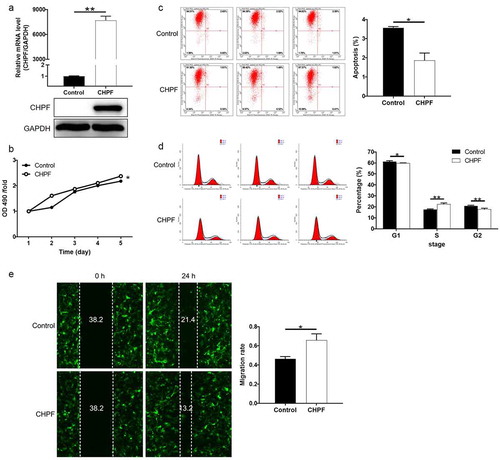
Considering the potential facilitation role of CHPF in CCA, we subsequently silenced CHPF in HCCC-9810 and QBC939 cells to investigate its functions in development of CCA in vitro. ShRNA targeting CHPF (shCHPF), and shCtrl as negative control, were designed, prepared, packaged into lentivirus vector, which was finally used for cell transfection. The fluorescence of green fluorescent protein on lentivirus vector was used as a representation of transfection efficiency, which indicated >80%, as well as successful transfection (). Moreover, results of qPCR showed that the mRNA level of CHPF was decreased by 55% and 90% in HCCC-9810 and QBC939 cells, respectively (P < 0.001, ). The successful knockdown of CHPF in HCCC-9810 and QBC939 cells was also verified by the detection of CHPF protein level by western blotting (). Collectively, these results suggested that the lentivirus expressing shCHPF could significantly downregulate the expression of CHPF in CCA cells.
Figure 2. Construction of cholangiocarcinoma cell models with CHPF knockdown
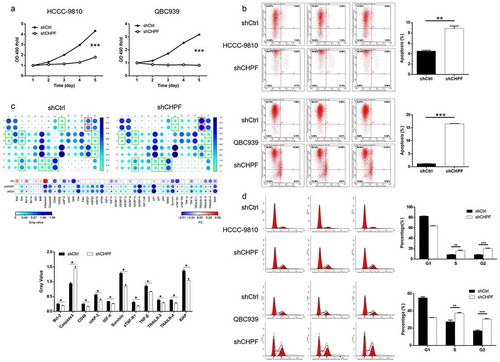
Knockdown of CHPF inhibited cell proliferation and induced cell apoptosis and cell cycle arrest
Next, cellular functions were detected in HCCC-9810 and QBC939 cells transfected with shCtrl or shCHPF to reveal the role of CHPF in development of CCA. It was demonstrated that the viability of CCA cells was significantly lower in shCHPF group during cell culture, indicating the inhibition of cell proliferation (P < 0.001, ). The detection of cell apoptosis by flow cytometry revealed the significantly increased apoptotic cell percentage in shCHPF group (P < 0.01, ). Moreover, a Human Apoptosis Antibody Array was performed to explore how knockdown of CHPF affects cell apoptosis. The comparison of the expression of apoptosis-related proteins in HCCC-9810 cells with or without CHPF knockdown identify a variety of significantly downregulated proteins, including Bcl-2, CD40, clAP-2, IGF-II, Survivin, sTNF-R1, TNF-β, TRAILR-3, TRAILR-4 and XIAP, and the upregulated Caspase3 (P < 0.05, ). Furthermore, the effects of CHPF on cell cycle distribution were also evaluated for interpreting its ability to regulate cell apoptosis. Similar results, that CHPF knockdown induced the arrest of cell cycle in S and G2 phase, were obtained in HCCC-9810 and QBC939 cells (P < 0.05, ).
Figure 3. CHPF knockdown inhibited cell proliferation and promoted cell apoptosis and cell cycle arrest in cholangiocarcinoma cells
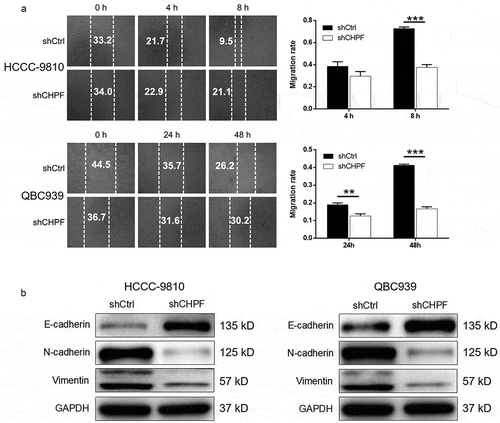
Knockdown of CHPF suppressed cell motility and downregulate EMT-related proteins
In order to explore whether CHPF depletion could regulate tumor metastasis of CCA, cell migration of HCCC-9810 and QBC939 cells transfected with corresponding lentivirus. The outcomes of wound-healing assay showed that knockdown of CHPF could significantly decrease the cell migration ability of HCCC-9810 and QBC939 cells (P < 0.001, ). Moreover, as well-known participations in tumor metastasis, epithelial-mesenchymal transition (EMT) related proteins including N-cadherin, Vimentin and Snail were detected in HCCC-9810 and QBC939 cells. As shown in , the expression levels of N-cadherin and Vimentin showed significant downregulation, while that of E-cadherin exhibited significant upregulation, upon silencing of CHPF, verifying the suppression of EMT as well as tumor metastasis by CHPF knockdown.
Figure 4. CHPF knockdown inhibited cell migration and expression of EMT-related proteins
Overexpression of CHPF promoted development of CCA in vitro
Although the inhibition of CCA by CHPF knockdown has been clearly elucidated by the above results, a gain-of-function assay was further performed to deepen the role of CHPF in CCA development. As shown in , upon the transfection of CHPF-overexpressing lentivirus, the expression of CHPF in HCCC-9810 cells was significantly enhanced. Subsequently, the detection of cell phenotypes showed that cells with overexpressed CHPF showed higher proliferative activity, lower apoptotic cell rate, higher cell percentage in S phase and stronger motility (. Notably, all the regulatory effects induced by CHPF overexpression were just the opposite of that induced by CHPF knockdown.
Figure 5. CHPF overexpression promoted cholangiocarcinoma development in vitro.
CHPF knockdown inhibited tumor growth of CCA in vivo
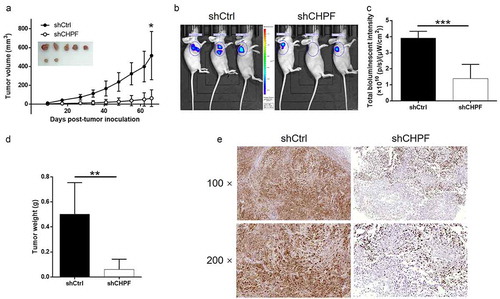
For the sake of verifying the role of CHPF in CCA in vivo, HUCCT1 cells with or without CHPF knockdown were subcutaneously injected into mice for constructing mice xenograft models. Throughout the culture of animal models, the growth of tumors was observed and the volumes were calculated based on the tumor size. We found that CHPF knockdown could obviously slowdown the rate of tumor growth in vivo (P < 0.01, ). Consistently, the suppression of tumor growth by CHPF knockdown was also visualized by fluorescence imaging which was facilitated by injection of D-Luciferin (P < 0.05, . After sacrificing the animals, the weights of tumors were measured, indicating smaller tumors in shCHPF group (P < 0.01, ). Besides, Ki-67, which was considered as representation of tumor growth, was also detected in the removed tumors and exhibited apparently downregulation in shCHPF group (). Altogether, the results suggested that knockdown of CHPF could restrain tumor growth in vivo.
Figure 6. CHPF knockdown inhibited tumor growth of cholangiocarcinoma in vivo.
Discussion
CS can be detected on the surface of extracellular matrix and cell membrane of a variety of tissues [Citation10], which is closely related to the biological processes such as the development of brain neural network, inflammation, infection, cell division and tissue morphology [Citation23]. At the same time, it imparts some physiological functions such as inhibition of axonal regeneration [Citation24], prevention of abnormal myocardial remodeling after spinal cord injury [Citation25] and so on. Moreover, previous studies suggested that CS has a certain regulatory capacity for malignant tumors. For example, the content of CS in glioblastoma is related to the proliferative activity of tumor cells and the prognosis of the disease [Citation26]. In addition, CS modified nanoparticles brusatol has an inhibitory effect on the proliferation, migration and invasion of cancer cells [Citation27]. Furthermore, as one of the six necessary glycosyltransferases in the biosynthesis of CS, CHPF is essential in the process of repeated disaccharide unit synthesis in CS [Citation10]. In view of the physiological functions of CS, CHPF was predicted to be involved in the regulation of cell division and differentiation, thus regulating the development of the body and the occurrence of diseases. Moreover, the role of CHPF in multiple types of malignant tumors has been extensively studied, such as lung adenocarcinoma [Citation28], non-small cell lung cancer [Citation29], esophageal squamous cell carcinoma [Citation30]. Recently, the expression of CHPF can regulate the proliferation and invasion of lung cancer cells [Citation20]. Additionally, it was revealed that CHPF could promote the progression of malignant melanoma by regulating CDK1 [Citation22]. However, to the best of our knowledge, the functional of CHPF in development of CCA has not been elucidated.
In this study, the difference in the expression of CHPF between CCA and normal tissues was detected by IHC, showing that the expression of CHPF was significantly upregulated in CCA. Further analysis of the expression of CHPF in CCA with different characteristics showed that the high level of CHPF was positively correlated with the advanced malignant grade, suggesting that CHPF has cancer-like characteristics in CCA. Subsequently, in vitro experiments demonstrated that knockdown of CHPF could inhibit CCA progression by slowing down cell growth, increasing the percentage of apoptosis, blocking cell cycle and inhibiting cell progression in S/G2 phase. Moreover, the promotion of cell apoptosis by CHPF knockdown was contributed to the regulation of various apoptosis-related proteins. On the contrary, CHPF overexpression could induce promotion effects on the development of CCA. In addition, in vivo experiments in mice further confirmed that knockdown of CHPF inhibited tumor formation. Altogether, the in vitro and in vivo studies on the role of CHPF in development of CCA identified it as a potential tumor promotor.
In addition to malignant proliferation, metastasis is also a major feature of tumor, which seriously restricts the therapeutic effect of tumor and reduces the survival period of patients [Citation31]. EMT is a process that plays an important role in tumor invasion and metastasis [Citation32–34]. During the process of invasion and metastasis, tumor cells change from epithelial phenotype to mesenchymal phenotype, and thus moving from primary focus to metastasis pathway [Citation35]. E-cadherin and N-cadherin are calcium containing transmembrane glycoprotein, which plays important roles in cell adhesion of epithelial cells. Studies have shown that the progression of EMT is accompanied by simultaneous downregulation of E-cadherin and upregulation of N-cadherin, which can lead to increased invasion and metastasis during tumor progression [Citation36]. For example, the inhibited expression of E-cadherin was identified to mediate the promotion effects of LINC00978 on migration and invasion of hepatocellular carcinoma cells [Citation37]. Consistently, the inhibition of cell migration induced by CHPF deficiency in our study could also be rationalized by the alteration of E-cadherin and N-cadherin. Moreover, Vimentin is an intermediate fibroin expressed by mesenchymal cells, and it is also one of the important markers of EMT in epithelial cells. Zhu et al. presented that CircNHSL1/miR-1306-3p/SIX1 axis could exert its regulatory role in the development and progression of gastric cancer through regulating Vimentin and EMT [Citation38]. Herein, we also found that Vimentin expression was significantly suppressed in cells with CHPF depletion.
Although there are still some limitations in our study, such as limited number of clinical samples and still unclear downstream regulatory mechanism. The results allow us to conclude that CHPF may act as a tumor promotor in the development and progression of CCA, which may be used as a novel therapeutic target for treating CCA in the future.
Author contributions
X.M. designed this program. X.D., J.Y. and B.J. operated the cell and animal experiments. X.D., W.D. and R.W. conducted the data collection and analysis. X.D. and H.Z. produced the manuscript which was checked by X.M. All the authors have confirmed the submission of this manuscript.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34.
- Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
- Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383(9935):2168–2179.
- Rizvi S, Khan SA, Hallemeier CL, et al. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol. 2017;15(2):95–111.
- Doherty B, Nambudiri VE, Palmer WC. Update on the diagnosis and treatment of cholangiocarcinoma. Curr Gastroenterol Rep. 2017;19(1):2.
- Esnaola NF, Meyer JE, Karachristos A, et al. Evaluation and management of intrahepatic and extrahepatic cholangiocarcinoma. Cancer-Am Cancer Soc. 2016;122:1349–1369.
- Rizvi S, Gores GJ. Pathogenesis, diagnosis, and management of Cholangiocarcinoma. Gastroenterology. 2013;145(6):1215–1229.
- Moeini A, Sia D, Bardeesy N, et al. Molecular pathogenesis and targeted therapies for intrahepatic Cholangiocarcinoma. Clin Cancer Res. 2016;22(2):291.
- Volpi N, Maccari F, Mantovani V. Chondroitin Sulfate and Glucosamine as disease modifying anti- osteoarthritis drugs (DMOADs). Curr Med Chem. 2016;23(11):1139–1151.
- Mikami T, Kitagawa H. Biosynthesis and function of chondroitin sulfate. Biochim Biophys Acta Gen Subj. 2013;1830(10):4719–4733.
- Bishnoi M, Jain A, Hurkat P, et al. Chondroitin sulphate: a focus on osteoarthritis. Glycoconjugate J. 2016;33(5):1–13.
- Maksimenko AV, Golubykh VL, Tischenko EG. Catalase and chondroitin sulfate derivatives against thrombotic effect induced by reactive oxygen species in a rat artery. Metab Eng. 2003;5(3):177–182.
- Rolls A, Shechter R, London A, et al. Two faces of chondroitin sulfate proteoglycan in spinal cord repair: a role in microglia/macrophage activation. PLoS Med. 2008;5(8):e171.
- Nadanaka S, Kinouchi H, Kitagawa H. Chondroitin sulfates-mediated N-cadherin/β-catenin signaling is associated with basal-like breast cancer cell invasion. J Biol Chem. 2017;293(2):444–465.
- Pantazaka E, Papadimitriou E. Chondroitin sulfate-cell membrane effectors as regulators of growth factor-mediated vascular and cancer cell migration. Biochim Biophys Acta. 2014;1840(8):2643–2650.
- Asimakopoulou AP, Theocharis AD, Tzanakakis GN, et al. The biological role of chondroitin sulfate in cancer and chondroitin-based anticancer agents. In Vivo. 2008;22(3):385–389.
- Kitagawa H, Izumikawa T, Uyama T, et al. Molecular cloning of a chondroitin polymerizing factor that cooperates with chondroitin synthase for chondroitin polymerization. J Biol Chem. 2003;278(26):23666–23671.
- Kalathas D, Triantaphyllidou IE, Mastronikolis NS, et al. The chondroitin/dermatan sulfate synthesizing and modifying enzymes in laryngeal cancer: expressional and epigenetic studies. Head Neck Oncol. 2010;2(1):27.
- Hwang S, Mahadevan S, Qadir F, et al. Identification of FOXM1-induced epigenetic markers for head and neck squamous cell carcinomas. Cancer-Am Cancer Soc. 2013;119:4249–4258.
- Cao C, Liu Y, Wang Q, et al. Expression of CHPF modulates cell proliferation and invasion in lung cancer. Braz J Med Biol Res = Rev Bras Pesqui Med Biol. 2020;53(5):e9021.
- Xu Q, Lin W, Tao C, et al. Chondroitin polymerizing factor (CHPF) contributes to malignant proliferation and migration of hepatocellular carcinoma cells. Biochem Cell Biol. 2020;98(3):362–369.
- Sun W, Zhao F, Xu Y, et al. Chondroitin polymerizing factor (CHPF) promotes development of malignant melanoma through regulation of CDK1. Cell Death Dis. 2020;11(7):496.
- Izumikawa T, Koike T, Shiozawa S, et al. Identification of chondroitin sulfate glucuronyltransferase as chondroitin synthase-3 involved in chondroitin polymerization. J Biol Chem. 2008;283(17):11396–11406.
- Takeda A, Okada S, Funakoshi K. Chondroitin sulfates do not impede axonal regeneration in goldfish spinal cord. Brain Res. 2017;16:23–29.
- Zhao R, Ackers-Johnson M, Stenzig J, et al. Targeting chondroitin sulfate glycosaminoglycans to treat cardiac fibrosis in pathological remodeling. Circulation. 2018;137(23):2497–2513.
- Tsidulko AY, Kazanskaya GM, Volkov AM, et al. Chondroitin sulfate content and decorin expression in glioblastoma are associated with proliferative activity of glioma cells and disease prognosis. Cell Tissue Res. 2020;379(1):147–155.
- Chen X, Yin T, Zhang B, et al. Inhibitory effects of brusatol delivered using glycosaminoglycan‑placental chondroitin sulfate A‑modified nanoparticles on the proliferation, migration and invasion of cancer cells. Int J Mol Med. 2020;46(2):817–827.
- Hou X, Zhang T, Da Z, et al. CHPF promotes lung adenocarcinoma proliferation and anti-apoptosis via the MAPK pathway. Pathol Res Pract. 2019;215(5):988–994.
- Hou X, Baloch Z, Zheng Z, et al. Knockdown of CHPF suppresses cell progression of non-small-cell lung cancer. Cancer Manag Res. 2019;11:3275–3283.
- Ye W, Zhu J, He D, et al. Increased CDKL3 expression predicts poor prognosis and enhances malignant phenotypes in esophageal squamous cell carcinoma. J Cell Biochem. 2019;120(5):7174–7184.
- Yeung KT, Yang J. Epithelial-mesenchymal transition in tumor metastasis. Mol Oncol. 2017;11(1):28–39.
- Diepenbruck M, Christofori G. Epithelial-mesenchymal transition (EMT) and metastasis: yes, no, maybe? Curr Opin Cell Biol. 2016;43:7–13.
- Liao TT, Yang MH. Revisiting epithelial-mesenchymal transition in cancer metastasis: the connection between epithelial plasticity and stemness. Mol Oncol. 2017;11(7):792–804.
- Marcucci F. G Stassi and RD Maria, Epithelial|[ndash]|mesenchymal transition: a new target in anticancer drug discovery. Nat Rev Drug Discov. 2016;15(5):311–325.
- Samy L. X Jian and D Rik, Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Bio. 2014;15:178–196.
- Loh C, Tang W, Sethi S, et al. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: signaling, Therapeutic Implications, and Challenges. Cells-Basel. 2019;8(10):1118.
- Xu X, Gu J, Ding X, et al., LINC00978 promotes the progression of hepatocellular carcinoma by regulating EZH2-mediated silencing of p21 and E-cadherin expression. Cell Death Dis. 2019;10(10):752.
- Zhu Z, Rong Z, Luo Z, et al. Circular RNA circNHSL1 promotes gastric cancer progression through the miR-1306-3p/SIX1/vimentin axis. Mol Cancer. 2019;18(1):126.