
ABSTRACT
The nuclear envelope (NE) is a critical barrier between the cytosol and nucleus that is key for compartmentalization within the cell and serves an essential role in organizing and protecting genomic DNA. Rupturing of the NE through loss of constitutive NE proteins and/or mechanical force applied to the nucleus results in the unregulated mixing of cytosolic and nuclear compartments, leading to DNA damage and genomic instability. Nuclear rupture has recently gained interest as a mechanism that may participate in various NE-associated diseases as well as cancer. Remarkably, these rupturing events are often transient, with cells being capable of rapidly repairing nuclear ruptures. Recently, we identified Barrier-to-Autointegration Factor (BAF), a DNA-binding protein involved in post-mitotic NE reformation and cytosolic viral regulation, as an essential protein for nuclear rupture repair. During interphase, the highly mobile cytosolic BAF is primed to monitor for a compromised NE by rapidly binding to newly exposed nuclear DNA and subsequently recruiting the factors necessary for NE repair. This review highlights the recent findings of BAF’s roles in rupture repair, and offers perspectives on how regulatory factors that control BAF activity may potentially alter the cellular response to nuclear ruptures and how BAF may participate in human disease.
Introduction to nuclear envelope rupture
The nuclear envelope (NE) is a dynamic, double-membraned bilayer derived from and contiguous with the endoplasmic reticulum (ER) that defines the boundary of the nucleus and establishes a nucleoplasm that is distinct from the cytosol. The NE plays an essential role in organizing and protecting genomic DNA, and establishes cellular compartmentalization that enables a whole host of fundamental cell signaling and regulatory processes. In addition to its role as a protective barrier, the NE hosts a diverse assembly of proteins with structural and functional roles. Underneath the NE lies the nuclear lamina, an intertwined network of intermediate filaments that mechanically stabilizes the NE and functions to regulate nuclear organization and gene expression [Citation1–4]. Nuclear pore complexes (NPCs) are large channels embedded within the NE that enable regulated trafficking of proteins, mRNA, and other large macromolecules between the cytosolic and nuclear compartments [Citation5,Citation6]. The NE is also enriched with numerous transmembrane proteins that predominantly reside within the inner nuclear membrane (INM), although a few are also enriched on the outer nuclear membrane (ONM) [Citation7–10]. The nucleus is mechanically coupled to the cytoskeleton via a structure called the Linker of Nucleoskeleton and Cytoskeleton (LINC)-complex that is fundamentally formed by an interaction between specific transmembrane proteins that span the INM and ONM [Citation9,Citation11,Citation12]. In cells that undergo open mitosis, including plant and animal cells, the NE is rapidly disassembled during mitosis, creating a transient loss of the nuclear-cytosolic barrier which is reestablished by reassembly of the NE during the final stages of mitosis [Citation13,Citation14]. This process of NE breakdown and reformation is carefully orchestrated during the cell cycle to prevent DNA damage and ensure that reforming nuclei incorporate a complete genome. Alternative mitotic mechanisms also exist in various eukaryotic species, with examples including semi-open mitosis in Schizosaccharomyces japonicas (S. japonicas) and Caenorhabditis elegans (C. elegans) [Citation15], as well as closed mitosis in Saccharomyces cerevisiae (S. cerevisiae) [Citation16].
Unlike the well-regulated NE breakdown that occurs during open mitosis, the physical breach of both the INM and ONM during interphase, a process called “nuclear rupture”, creates the potential for an uncontrolled intermixing of nuclear and cytosolic compartments. Nuclear ruptures result from loss of NE integrity that can occur subsequent to a variety of physiologically relevant and naturally occurring circumstances [Citation17–20]. Loss of NE integrity can arise from functional loss of NE protein constituents, caused by mutations in and/or downregulated expression of genes encoding key proteins of the NE and nuclear lamina [Citation21–25]. These disruptions to NE architecture can result in characteristically abnormal nuclei found in laminopathy and cancer cells, which are observed to undergo frequent nuclear rupture [Citation26,Citation27]. Chromosomal missegregation from mitotic errors can produce micronuclei, which exhibit structurally aberrant NEs that are susceptible to rupture [Citation28]. The degradation and/or disassembly of the NE is utilized as a nuclear intrusion mechanism by some viruses [Citation29,Citation30]. Mechanical strain placed on the NE from external compressive and/or tensile forces can also induce NE rupturing. Compressive forces on the nucleus can occur when cells migrate through constrained environments, such as cancer cell migration or cell extravasation, where the nucleus cannot deform as readily as the rest of the cell [Citation31–33]. It is also plausible that tensile forces participate in these migration-based ruptures, likely via LINC-mediated pulling of the NE by the cytoskeleton [Citation23,Citation34]. Similarly, mechanical trauma to tissue could potentially lead to nuclear rupturing from either compressive and/or tensile forces on the nucleus.
Loss of NE integrity has been implicated as a potential cellular mechanism promoting diseases such as cancer [Citation17,Citation22,Citation27], diseases resulting from mutations in the genes encoding lamins [Citation35,Citation36], and possibly autoimmune or inflammatory disorders potentially caused by leakage of innate immune triggering molecules from the nucleus into the cytoplasm [Citation37]. Currently, there is a growing body of evidence supporting the notion that cells with higher frequencies of NE rupture exhibit increased amounts of DNA damage and activation of DNA repair pathways [Citation22,Citation35,Citation38,Citation39]. Additionally, NE rupture has been associated with abnormal chromosome rearrangements [Citation40,Citation41], mislocalization of cytoplasmic and nuclear constituents [Citation26], auto-activation of innate immune signaling [Citation42,Citation43], and has also been implicated as a possible mechanism behind cell cycle arrest [Citation44] and modulating differentiation in cells migrating through constricted spaces [Citation45].
Strikingly, cells appear to possess a functional mechanism to reseal the NE following rupture and reestablish the barrier function of the NE, sometimes within minutes [Citation22,Citation27,Citation39,Citation46,Citation47]. Resealing membrane holes in the NE are performed by the endosomal sorting complex required for transport III (ESCRT-III), a dynamic protein complex that sculpts membranes for the energetically favorable fusion of gaps in the NE. Initiating membrane sealing is performed by the ESCRT-III adaptor Chmp7, which localizes to NE holes and transiently recruits the central core of the ESCRT-III subunits and the AAA ATPase VPS4, creating a spiral-like hub that drives the closure of the NE gap [for review, see Citation48–50]. ESCRT-III has been shown to functionally reseal the NE during mitotic exit [Citation51–53], during interphase after NE rupture [Citation22,Citation39,Citation54], upon NPC damage [Citation55], and on occurrences of micronuclei envelope rupture [Citation56, Citation57,Preprint]. The evolutionary emergence of ESCRT-III in NE repair would suggest that nuclear rupture is an anticipated and certainly recoverable event that does not necessarily condemn the cell to a catastrophic fate and perhaps even lead to adaptive alterations in the nuclear and cellular phenotypes.
While the causes of NE rupture and its associated cellular consequences have begun to be studied, the mechanisms behind the repair of nuclear ruptures have remained unclear. Perhaps unsurprisingly, some proteins with established roles in reforming and resealing the NE after mitosis are also found to localize to NE ruptures [Citation22,Citation39,Citation46,Citation47,Citation58], suggesting a cross-functionality in post-mitotic NE reformation and NE rupture repair. Recently we reported that the evolutionary conserved protein barrier-to-autointegration factor (BAF), which has well-characterized roles in post-mitotic NE reformation, is a crucial participant in enabling the repair of NE ruptures. BAF rapidly migrates and enriches to sites of rupture, where it subsequently organizes the necessary repair machinery for efficient NE resealing. Here, we review recent findings of BAF’s roles in NE repair and offer perspectives on how regulatory factors that control BAF activity may potentially alter the cellular response to NE ruptures. Additional insights on BAF’s role in diseases of the NE are also discussed.
The role of BAF at NE ruptures
BAF is a small 89-aa double-stranded DNA (dsDNA)-binding protein with considerable sequence conservation across all metazoans species [Citation59]. BAF forms obligate dimers, with each monomer binding to dsDNA (hereafter referred to simply as DNA) in a sequence-independent manner, allowing for the looping and condensation of DNA through intramolecular cross-bridging [Citation60]. Along with DNA, BAF also binds to the LAP2-Emerin-MAN1 (LEM) domain, a motif found in a family of proteins known collectively as the LEM-domain proteins of the INM, ER, and nucleoplasm. Other reported protein interactors of BAF include A-type lamins [Citation61], histones [Citation62], and various transcriptional regulators [Citation63,Citation64]. Additionally, BAF is a reported participant in oxidative DNA damage repair [Citation65] and a regulator of cytoplasmic DNA levels, which can activate innate immune responses [Citation66]. BAF is widely distributed in the cell, with populations residing in the cytoplasm, nucleus, and NE. The nuclear/NE population of BAF binds to genomic DNA, lamins, and LEM-domain proteins along the INM, whereas the cytosolic population has no clear-binding partners other than perhaps the resident LEM-domain proteins, Ankle1 and Ankle2, as well as other LEM-domain proteins that transit from the INM into the ER. BAF’s small size would typically permit passive diffusion through NPCs; however, photobleaching studies reveal minimal trafficking of GFP-BAF between the cytosolic and nuclear compartments during interphase despite BAF being extremely mobile in any cellular compartment [Citation67]. This profound compartmentalization appears to change during S-phase, when a shift of BAF into the nucleus has been observed [Citation68]. The mechanism for this compartmentalization of BAF during interphase is unclear, but could be due in part to formation of higher-order structures that exceed the NPC barrier limit. Compartmental differences in BAF behavior are potentially regulated by the vaccinia-related kinases (VRKs), a family of kinases with structural similarities to the vaccinia B1 kinase that phosphorylate BAF’s N-terminus and reduces its capacity for DNA binding [Citation69].
Functionally, BAF plays a necessary role in recruiting LEM-domain proteins, A-type lamins, and nascent NE membranes to the chromatin surface during mitotic exit, making it a critical component in post-mitotic NE reformation [Citation70–73]. Another study highlighted BAF’s role in coating and crosslinking decondensing chromosomes after mitosis to promote formation of a single intact nucleus and observed that membrane recruitment is surprisingly unaffected by BAF depletion or expression of a BAF mutant deficient in LEM-domain binding [Citation74]. However, it was recently reported that this same BAF mutant is unable to recruit LEM domain proteins to sites of nuclear rupture or enable efficient repair after rupture [Citation47]. These results appear to suggest fundamental differences in NE reformation mechanisms between rupture repair and post-mitotic NE reformation, and support the need for further studies on the role of BAF in these vital cellular processes. There is also a well-established role for BAF in binding to and regulating the activity and/or fate of viral DNA in the cytosol during infection. BAF has been shown to prevent the self-destruction of retroviral DNA by blocking auto-integration and promoting integration into the host genome [Citation75–78]. Conversely, BAF can also act as a host defense protein against cytosolic viral DNA by preventing its genomic replication [Citation79,Citation80] and transcription [Citation81,Citation82]. Recently, it was reported that BAF accumulates at NE ruptures during interphase [Citation22], and follow-up studies revealed BAF’s essential role in NE rupture repair [Citation46,Citation47]. The current model places BAF at the nexus between sensing NE ruptures and initiating their subsequent repair ( A-D).
Figure 1. A model for BAF-initiated repair of NE ruptures. (a) Before rupture, a nuclear pool of predominantly phosphorylated BAF and a cytosolic pool of predominantly non-phosphorylated BAF is separated by a functional NE barrier. (b) Upon loss of NE integrity, cytosolic BAF is recruited to the nuclear rupture, most likely via binding to nuclear DNA. Additional mechanisms for BAF recruitment and stabilization at rupture may exist, as well as a mechanism to prevent passive diffusion of larger nuclear and cytoplasmic constituents through the NE hole via BAF-mediated DNA condensation that acts as a “clot”. (c) BAF recruits LEM-domain proteins, ESCRT-III and membranes to the rupture site for repair. Other unknown cellular components may also depend on BAF for their recruitment, as well as an ESCRT-III independent mechanism for membrane resealing. Additionally, BAF phosphoregulation may play an important role in NE rupture repair. (d) Following restoration of the NE barrier following rupture there is a resultant compartmental redistribution of BAF from the cytoplasm into the nucleus, as well as a remnant enrichment of LEM-domain proteins at the site of NE repair
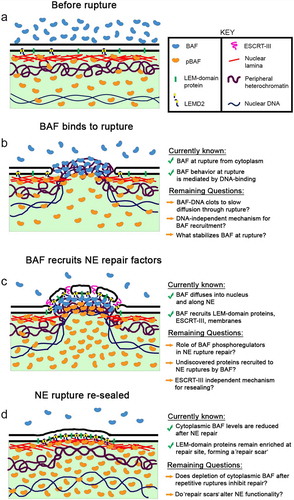
Upon loss of NE integrity, BAF rapidly localizes to and enriches at sites of NE rupture [Citation22,Citation46,Citation47]. BAF’s ability to enrich at NE ruptures appears to be largely driven by binding exposed nuclear DNA that emerges from rupture site, mirroring its function in recognizing and binding DNA that enters the cytoplasm during viral infection. Enrichment also coincides with the localization of cyclic GMP-AMP synthase (cGAS), a DNA-binding protein of the innate immune cGAS-STING pathway that is localized to the cytoplasm during interphase and is used as a reporter of NE rupture [Citation22,Citation39,Citation46]. Additionally, expression of fluorescent proteins tagged with a nuclear localization sequence (NLS), which are sequestered in the nucleus with an intact NE but leak into the cytoplasm upon rupture, also coincide with BAF enrichment [Citation47]. GFP-BAF is known to be highly mobile in both cytosolic and nuclear compartments, and BAF at ruptures appears to originate predominantly from the cytoplasm, with only a slight contribution from the nuclear BAF population [Citation46]. The comparatively high contribution from the cytoplasm is most likely due to a non-phosphorylated BAF pool in that compartment that is primed to bind to exogenous DNA, such as can occur during viral infections [Citation83]. Strikingly, photobleaching studies reveal that upon nuclear rupture BAF only moves from the cytoplasm into the nucleus with no leakage of the nuclear BAF into the cytoplasm [Citation46]. This is likely due in large part to a substantial pool of interactors in the nucleus such as DNA, LEM domain proteins and A-type lamins that serve to retain nuclear BAF until the nucleus can reseal. Following recruitment of BAF at a rupture via nuclear DNA, there is a gradual diffusion of BAF from the rupture site inward into the nucleus and along the NE (). This “wave” of BAF diffusion, driven largely by BAF’s interaction with DNA, disperses throughout the nucleus and NE, resulting in a cellular re-distribution of BAF into the nucleus following rupture. The mechanism likely to participate in establishing BAF’s ability to rapidly localize to nuclear ruptures and then gradually diffuse into the nucleoplasm is a population of predominantly phosphorylated BAF in the nucleus, likely established by nucleoplasmic VRK1 [Citation69,Citation84] and INM VRK2 [Citation85]. In agreement with this model is the observation that GFP-BAF variants possessing either a mutation in a key DNA-binding site (K6A) or mimicking phosphorylated N-terminal residues (MEEEQ) disperse into the nucleus faster, while a phosphorylation-null mutant (MAAAQ) remains highly retained on the NE after rupture with little to no diffusion [Citation46]. This compartmental variance in interphase BAF phosphorylation also serves to promote BAF’s ability to bind to exogenous DNA in the cytoplasm. Although DNA is likely the major driver behind BAF recruitment to ruptures, it is possible that other BAF interactors also play a role in retaining a subpopulation of BAF at the rupture site during and potentially even after repair.
Functionally, BAF appears to be a necessary component of rupture repair, as its cellular depletion results in failure to efficiently recruit LEM-domain proteins and Chmp7 to NE ruptures, as well as a substantial delay in reestablishing a functional NE barrier [Citation46,Citation47]. BAF’s recruitment of transmembrane LEM-domain proteins, including those of the INM (LEMD2, Emerin, Man1, Lap2β) and ER (Ankle2), are key to the repair of nuclear ruptures (), evidenced by an inability to efficiently repair nuclear ruptures upon simultaneous depletion of Emerin, LEMD2 and Ankle2 [Citation46] or by expressing a GFP-BAF variant lacking a LEM-domain binding domain [Citation47]. While it is possible that they play secondary roles, such as the stabilization of BAF and/or lamins at ruptures, it seems most likely that role of these transmembrane LEM-domain proteins in NE repair is to provide a mechanism to recruit and stably tether NE membranes to the rupture site in a BAF-dependent fashion (). Additionally, at least some of these transmembrane LEM-domain proteins may also enable an active membrane resealing process, as has been suggested for LEMD2 in post-mitotic nuclear envelope reformation [Citation86]. The recruitment of Chmp7 () to nuclear ruptures is dependent on LEMD2 [Citation46]. Interestingly, loss of ESCRT-III delays but does not abolish repair of ruptures created by constrained cell migration [Citation22,Citation39,Citation87], and has little effect on the repair of ruptures induced from laser-wounding the NE [Citation46]. The lack of a repair delay after loss of LEMD2 or Chmp7 observed in Halfmann et al. could be explained by differences in experimental approach, as the mild, transient ruptures induced from laser wounding might be more easily repaired than large ruptures that accompany prolonged constrained migration through a narrow channel. Regardless, these studies demonstrate that ESCRT-III is important for efficient NE repair, at least for larger ruptures, but appears dispensable for restoration of NE barrier function, and suggests a potential for redundancy in NE membrane repair mechanisms. It is possible that BAF-dependent recruitment of transmembrane LEM-domain proteins is sufficient to reseal the NE, as it may be energetically favorable for fusion of stabilized and closely apposed high curvature membranes. Alternatively, there may be other yet to be appreciated mechanisms that promote membrane re-sealing. The perinuclear space and nuclear interior contain elevated concentrations of calcium (Ca2+) relative to the cytosol, which when released from their respective compartments, may recruit calcium-binding proteins (e.g. Annexins) that aid in resealing NE ruptures, as suggested by others [Citation37]. Additionally, it has been suggested that locally controlled glycerophospholipid synthesis regulates ER sheet insertion into NE holes during NE reformation in C. elegans [Citation88], a phenomenon that might extend to repairing ruptures in the NE. Finally, it is possible that there is no membrane repair in the absence of ESCRT-III, but that the NE barrier function is nevertheless restored via unknown mechanisms, perhaps with leakage being prevented by BAF forming a functional clot or plug by binding to and tightly cross-linking DNA.
Regardless of the sealing mechanism, loss of BAF results in a systemic failure to recruit downstream repair proteins to NE ruptures and impairs restoration of the NE barrier [Citation46,Citation47]. Interestingly, BAF may not always be required for repair of all NE ruptures. In LaminB1-deficient cells, small spontaneous ruptures are able to repair even after BAF depletion but at a considerably slower rate, at least for spontaneous ruptures [Citation47]. In these same cells, laser induced ruptures lead to profound leakage of a rupture reporter, but appear to repair without delay [Citation47]. These observations suggest the existence of a repair mechanism independent of BAF, at least in cells deficient in lamin-B1, which perhaps depends on the severity of damage to underlying lamina and/or the physical size of the NE rupture. It seems that a small rupture may be able to mediate repair, albeit less efficiently, without BAF recruitment of LEM domain proteins. Alternatively, the observation of NE rupture repair during BAF depletion in Young et al. (represented by re-import of cytoplasmic RFP-NLS following NE rupture) may not actually represent a restored membrane at the rupture site, but nuclear import outpacing the rate of RFP-NLS diffusion from a small persistent NE rupture that is incapable of repair. Secondary methods to confirm a functionally restored NE may help clarify this possibility. In any event, BAF appears to function as a major lynchpin in the NE repair pathway, as it actively surveilles for breaches in the NE during interphase and once “activated” it spatially and temporally recruits repair machinery to the site of NE injury.
Following nuclear rupture, BAF is depleted from the cytoplasm [Citation46], most likely from its profound relocation into the nucleus where it is subsequently retained following NE repair (). It is unclear if this rapid redistribution of BAF during interphase carries any downstream consequences to cellular activity or fate. Since BAF has no substantial intercompartmental mobility during interphase [Citation67], multiple nuclear ruptures could deplete the reserve of cytosolic non-phosphorylated BAF, progressively impairing the repair of repeated rupture, as well as impeding other cellular mechanisms that require a sufficient pool of cytosolic BAF during interphase (e.g. viral DNA sensing). Additionally, the substantial accumulations of LEM-domain proteins at sites of nuclear rupture clearly reflect a necessary mechanism to recruit and retain those proteins that may require BAF to initiate, but likely require other proteins, such as the A-type lamins, to maintain since the LEM protein accumulations persist long after the BAF accumulation has dissipated. This observed “repair scar”, albeit having a poorly defined lifespan, represents a historic landmark of NE rupture repair that may lead to altered NE structural integrity or functionality ().
The possibility remains for yet-undiscovered roles for BAF at NE ruptures, outside of the reported membrane repair function. The ability of the BAF to cross-bridge nuclear DNA may generate a mechanically stiff chromatin-based scaffold that promotes efficient membrane docking to the site of rupture (), similar to BAF’s reported role in post-mitotic nuclear reformation [Citation74]. It is also conceivable that the presence of densely packed BAF-DNA complexes at the NE rupture may act as a temporary permeability barrier that slows the passive diffusion of large macromolecules through the rupture hole until the NE is resealed, as suggested by others [Citation49,Citation89]. NE rupturing has been implicated in the compartmental mislocalization of cyclins, transcription factors, promyelocytic leukemia nuclear bodies [Citation26], DNA damage repair proteins [Citation90], and may extend to yet unknown cellular factors. Given that mislocalization of these critical factors from their respective compartments may perturb normal cellular function over time, it is perhaps likely that the ability to slow or block their rapid diffusion through the rupture may be as critical as rupture repair itself. To push this concept further, chromatin compaction by BAF may even act to reestablish a cytoplasmic-nucleoplasmic barrier in small ruptures without complete membrane sealing. Future studies on NE repair may benefit from investigating the hypothetical “clotting” activity of BAF, which may be dependent solely on BAF-DNA ultra-structures for sufficient rupture blockage. Additionally, other BAF-binding molecules, such as lamins or histones, may also be involved in restricting diffusion through NE ruptures.
Potential impact of BAF regulation on NE rupture repair
Like most proteins, BAF’s function can be modulated through post-translational modification. BAF phosphorylation by the VRKs reduce DNA binding affinity, while BAF dephosphorylation by various phosphatases promote these interactions. Thus, BAF phosphoregulation offers the potential for tight regulatory control of this highly dynamic protein in a number of cellular processes. NE breakdown during mitotic entry is enabled, in part, by VRK1 phosphorylation of BAF, which disrupts the tethering of chromatin to NE transmembrane proteins to promote NE disassembly in mammalian cells [Citation69,Citation84], as well as in C. elegans [Citation70] and Drosophila melanogaster [Citation91]. Upon mitotic exit, BAF dephosphorylation by PP2A/PP4/Ankle2 restores linkages between chromatin and the NE and aides in NE reformation [Citation92,Citation93]. The cyclic phosphorylation of BAF during the cell cycle is observed to be critical for proper NE breakdown and reformation, as disruptions in this process are linked to mitotic errors [Citation94] and post-mitotic NE reformation abnormalities [Citation92]. Similar regulatory activity takes place during viral infection, as BAF phosphorylation by the viral kinase B1 can inhibit BAF-viral DNA interactions and promote viral replication in the cytosol [Citation80]. BAF phosphorylation, mobility, and localization are also shown to be influenced by environmental stresses in C. elegans [Citation95], although an exact mechanism remains elusive. These known mediators of BAF regulation raise the possibility that a similar mechanism may actively govern the sealing of NE ruptures (), perhaps by establishing the capacity of BAF to respond to NE rupture by enabling a population of DNA-binding competent BAF (pre-rupture regulation) and/or by modulating BAF-binding partner interactions during the NE repair process (post-rupture regulation). BAF phosphoregulation in the context of NE rupture repair remains poorly understand, yet one that may reveal fundamental and perhaps even pharmacologically regulatable processes in NE repair.
BAF phosphorylation is known to be regulated by the VRKs, a family of proteins with high homology to the B1 protein kinase encoded by the vaccinia virus [Citation96,Citation97]. In mammalian cells, the family consists of three major proteins. VRK1 is a soluble, nuclear protein with known roles in BAF phosphorylation during mitotic entry [Citation69,Citation70,Citation84,Citation91], as well as reported roles in regulation of the DNA damage response [Citation98,Citation99]. VRK2 is composed of two major splice isoforms, VRK2A and VRK2B. VRK2A localizes along the NE and ER due to an anchoring transmembrane domain and an interaction with A-type lamins, and is reported to phosphorylate BAF in vivo, whereas the less abundantly expressed VRK2B [Citation100] lacks a transmembrane domain and resides mainly in the nucleoplasm and cytoplasm [Citation85]. The predominantly nuclear VRK3 contains key amino acid differences in a conserved catalytic domain, making its ability to phosphorylate BAF unclear [Citation101,Citation102], although it has more recently reported to phosphorylate BAF on Ser-4 [Citation103]. Based on gene expression databases (BioGPS, Human Protein Atlas), VRK3 expression appears highest in the testis, perhaps indicating evolution toward a specific role in mammalian gametogenesis. VRKs have been shown to phosphorylate BAF on thr2, thr3, and ser4 residues on the extreme N-terminus of the protein, inhibiting BAF’s interactions with DNA and possibly to a lesser extent with LEM-domain proteins [Citation69]. These BAF modifications are reported to lower its affinity to DNA and the NE, thus promoting the transient “touch-and-go” nature of BAF’s interactions with its binding partners [Citation67]. Additionally, the observation that BAF’s rate of diffusion from the NE is enhanced in the absence of the NE-associated VRK2A suggests a role for VRK2A in regulating BAF-binding interactions and thus mobility at the NE [Citation85].
BAF’s phosphorylation state can also influence its subcellular distribution, as ectopic expression of an unphosphorylatable BAF variant (MAAAQ) was shown to localize almost exclusively to the nucleus [Citation83], likely due to an inability to efficiently detach from chromatin during mitosis and/or to excessively re-associate with nuclear DNA prior to NE reformation. In contrast, ectopic expression of phosphomimetic BAF variants containing either aspartic acid [Citation83] or glutamic acid [Citation46] phosphomimetic substitutions of its N-terminal phosphorylation residues exhibit a sub-cellular distribution similar to the WT. VRK1 overexpression is reported to diminish nuclear GFP-BAF, suggesting that VRK1-mediated phosphorylation of BAF inhibits its associations with binding partners in the nucleus and INM, thereby “driving” it into the cytoplasm [Citation69]. Given that BAF mobilization and subsequent recruitment of repair factors appears dependent on interactions with DNA and LEM-domain proteins, it is possible that BAF phosphorylation via VRK1 and/or VRK2 functions to regulate these interactions and the NE repair process. This could occur through the phosphorylation of BAF at the rupture site, at the membrane and/or in the nucleoplasm which could promote BAF release from the DNA and facilitate efficient rupture repair, as well as serve to help return the newly repaired NE to a state that is physiologically normal during interphase. Future investigations into VRK-BAF regulatory roles in NE repair may illuminate new insights into the NE repair process.
BAF’s role in surveilling for NE ruptures draws comparisons with its roles in recognizing and binding to viral DNA in the cytoplasm. BAF’s interactions with cytoplasmic viral DNA are well-documented, with evidence showing BAF possessing both pro-viral [Citation75–78] and anti-viral [Citation79–82] activity. In the case of retroviruses, BAF seems to have been evolutionarily co-opted to help form a pre-integration complex in the cytoplasm that prevents suicidal autointegration of the viral DNA. In the case of vaccinia virus, a viral kinase B1 facilitated viral infection by phosphorylating BAF to reduce its inhibitory effects on viral DNA replication and transcription. The current model suggests that BAF’s ability to bind and condense viral DNA relies on a maintaining a predominantly dephosphorylated pool in the cytosol, which is thus “activated” to bind to viral DNA upon entry into the cell. Similarly, the observations that BAF recruitment to NE ruptures is predominantly driven by DNA-binding and is from a cytosolic population suggest a dual function for cytosolic BAF dephosphorylation in detection of pathogenic cytoplasmic DNA and recognition NE rupture. Currently, there are two protein phosphatases with evidence of BAF-dephosphorylation activity. Protein phosphatases (PPs) in the PP2A family, composed of PP2A and PP4, were reported to dephosphorylate BAF in vitro [Citation92,Citation104] and siRNA depletion of PP4 increased cellular levels of phosphorylated BAF [Citation104]. PP2A has been shown to be essential for dephosphorylating BAF to enable its chromatin association during mitotic exit [Citation92]. BAF dephosphorylation is facilitated by the protein Ankle2, a LEM-domain protein of the ER that promotes BAF dephosphorylation by inhibiting VRK1 kinase interactions and acting as an adaptor to promoting association of BAF with PP2A [Citation92]. It is also reported that siRNA depletion of Ankle2 delays re-import of a GFP-NLS reporter following nuclear rupture [Citation46], suggesting a role in efficient NE repair. Indeed, we found that simultaneous knockdown of Ankle2, LEMD2 and Emerin prevented repair of nuclear ruptures; however, Ankle2 depletion alone was insufficient to prevent this repair. It is possible that PP4 and/or PP2A, in coordination with Ankle2, act in NE rupture repair by dephosphorylating cytoplasmic BAF during interphase, thus maintaining a dephosphorylated BAF pool in the cytoplasm “primed” to mobilize to NE ruptures. Additionally, these proteins may also work to counteract potential VRK1/2 phosphorylation at the rupture to promote BAF stability with the exposed chromatin to aid in NE repair.
The observation that proteins involved in recruiting and resealing the NE after mitosis are also key players in repairing the NE during interphase points to a shared evolutionary history between these two cellular mechanisms. It is currently unclear how the origins of BAF-dependent NE reformation and NE rupture repair fit into an evolutionary history. Did BAF-dependent NE repair evolve after the emergence of NE reformation and open mitosis by recycling the post-mitotic NE reformation machinery, or conversely, did BAF-dependent NE rupture repair evolve first in organisms with closed mitosis and was later adapted to facilitate the NE reformation required following open mitosis? It is tempting to speculate that the BAF-dependent NE repair predates open mitosis, and evolved early in the eukaryotic lineage to reseal NE ruptures caused by increased nuclear-cytoskeletal strain in cells that were organized in multicellular configurations but lacking supportive cell walls capable of buffering increased environmental mechanical forces that come with multicellularity.
The current model of NE barrier maintenance in mammalian systems highlights BAF as a critical factor in surveilling the NE for gaps and recruiting repair machinery to reseal the NE barrier. Curiously, budding and fission yeast are still capable of functionally sealing their NE without the presence of a BAF orthologue. Here, the targeting and repair of NE gaps is accomplished by the combined activity of the INM protein LEM2 and cytosolic ESCRT-III adaptor Chm7 (orthologues of mammalian LEMD2 and Chmp7, respectively). With an intact NE, these proteins are confined to their respective compartments on either side of the NE, and only make contact when the cytosolic-nucleoplasmic barrier is broken, leading to activation of ESCRT-III-mediated membrane repair [Citation49,Citation54]. In yeast, LEM2-Chm7 membrane repair is active in NE re-sealing after mitosis [Citation86] and NPC clearance during interphase [Citation55]. Thus, yeast have consigned both NE surveillance and repair duties to LEM2 and ESCRT-III, whereas metazoans appear to have adapted BAF as an upstream repair element, capable of integrating surveillance and membrane repair via DNA-sensing and LEM-domain binding, respectively. Currently, the reasons for the differences in NE repair strategies between yeast and metazoans can only be speculated. Perhaps single-celled yeast may be less susceptible to large mechanically induced NE ruptures, with small NE ruptures created during NPC removal and/or closed mitosis being sufficiently repaired solely by the LEM2-ESCRT-III system. The evolution of metazoans may have required a more robust repair mechanism for larger and/or more frequent ruptures caused by increased nuclear-cytoskeletal strain that accompanies multicellular organization and/or migration between neighboring cells and tissues. BAF may have functionally superseded ESCRT-III in the repair of large NE ruptures as cells adapted to these more mechanically traumatic environments.
The impacts of BAF & NE rupture in human disease
NE rupture imposes a severe challenge to the cell, with the reported consequences of NE rupture including DNA damage [Citation22,Citation35,Citation90], activation of DNA damage pathways [Citation22,Citation33,Citation39], cell cycle arrest [Citation44], senescence [Citation105], altered differentiation [Citation45], and cell death [Citation39]. In light of these findings, it should be unsurprising that NE rupture may participate in the etiology of human disease. There are several reports linking NE rupture to cancer, including as a consequence of the loss of tumor [Citation106], as a driver of DNA damage and genome instability caused by nuclear rupture during metastasis [Citation22], and/or due to defective nuclear lamina [Citation107]. DNA damage and chromothripsis associated with NE rupture in micronuclei can also be an active driver of tumorigenesis and cancer progression [reviewed in Citation108]. Micronuclei have a high propensity for NE rupture, and although BAF is observed to enrich at micronuclei, its presence appears insufficient to stimulate proper NE repair and resealing of these structures. The mechanism behind this apparent repair defect in micronuclei has yet to be completely elucidated, although recent evidence suggests that hyper-stimulated ESCRT-III activity creates aberrant membranes structures that prevent proper resealing [Citation56, Citation57,Preprint]. Discovering whether other NE proteins, such as BAF, are misregulated at micronuclei could give valuable insight into this defective repair mechanism.
Mutations in the genes encoding various NE proteins can lead to a myriad of diverse diseases, collectively called nuclear envelopathies [Citation109–111]. It has recently been proposed that a functional deficiency of A-type lamins, an underlying mechanism for several rare striated muscle diseases, is caused by an increase in NE rupture in myonuclei [Citation38]. Specific to BAF, there are recent reports of an autosomal recessive mutation in BANF1 in two separate unrelated individuals with Nestor–Guillermo Progeria Syndrome (NGPS) [Citation112,Citation113]. Described as a chronic progeria due to prolonged survival and comparatively mild symptoms, NGPS patients share features with the more studied Hutchinson-Guilford Progeria Syndrome (HGPS) predominantly caused by mutations in LMNA, such as failure to thrive, lipoatrophy, osteoporosis, normal cognition and a pseudosenile facial appearance [for review, see Citation114]. However, the NGPS differences are some key aspects from HGPS including lack of atherosclerosis and severe osteolysis. At the cellular level, there is also a shared phenotype of dysmorphic “wrinkled” nuclei for NGPS and HGPS. The NGPS missense mutation of BAF (A12T), despite having no detected effect on protein localization, leads to reduced affinity of BAF to dsDNA [Citation115]. Initial reports of BAF protein instability may instead be due to reduced affinity of the BAF antibody due to the missense mutation [Citation115]. In light of our previously reported role for DNA-binding in the behavior of BAF during nuclear rupture, it is possible that inefficient targeting of BAF to sites of nuclear rupture may delay the repair of nuclear ruptures and could contribute to the disease mechanism in NGPS. It is also possible that perturbation of another DNA-binding-based function of BAF, such as post-mitotic nuclear envelope repair, is the underlying cellular mechanism of NGPS. What is clear is that nuclear rupture and repair cannot be overlooked as a possible mechanism underlying common diseases such as cancer as well as some of the rare nuclear envelopathies. It remains to be seen whether nuclear rupture and repair may also participate in the normal response to tissue injury, or adaptive responses to mechanical stimuli such as observed in cartilage, bone, or muscle.
Acknowledgments
We thank Kelsey Scott and Rhiannon Sears for their review and feedback on the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Burke B, Stewart CL. The nuclear lamins: flexibility in function. Nat Rev Mol Cell Biol. 2013;14:13–24.
- Gerace L, Huber MD. Nuclear lamina at the crossroads of the cytoplasm and nucleus. J Struct Biol. 2012;177:24–31.
- Gruenbaum Y, Foisner R. Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu Rev Biochem. 2015;84:131–164.
- Yanez-Cuna JO, van Steensel B. Genome-nuclear lamina interactions: from cell populations to single cells. Curr Opin Genet Dev. 2017;43:67–72.
- Beck M, Hurt E. The nuclear pore complex: understanding its function through structural insight. Nat Rev Mol Cell Biol. 2017;18:73–89.
- Knockenhauer KE, Schwartz TU. The nuclear pore complex as a flexible and dynamic gate. Cell. 2016;164:1162–1171.
- Schirmer EC, Foisner R. Proteins that associate with lamins: many faces, many functions. Exp Cell Res. 2007;313:2167–2179.
- Schirmer EC, Gerace L. The nuclear membrane proteome: extending the envelope. Trends Biochem Sci. 2005;30:551–558.
- Starr DA. A nuclear-envelope bridge positions nuclei and moves chromosomes. J Cell Sci. 2009;122:577–586.
- Wilson KL, Foisner R. Lamin-binding proteins. Cold Spring Harb Perspect Biol. 2010;2:a000554.
- Kim DI, Birendra KC, Roux KJ. Making the LINC: SUN and KASH protein interactions. Biol Chem. 2015;396:295–310.
- Tzur YB, Wilson KL, Gruenbaum Y. SUN-domain proteins: ‘Velcro’ that links the nucleoskeleton to the cytoskeleton. Nat Rev Mol Cell Biol. 2006;7:782–788.
- Schellhaus AK, De Magistris P, Antonin W. Nuclear reformation at the end of mitosis. J Mol Biol. 2016;428:1962–1985.
- Ungricht R, Kutay U. Mechanisms and functions of nuclear envelope remodelling. Nat Rev Mol Cell Biol. 2017;18:229–245.
- Aoki K, Hayashi H, Furuya K, et al. Breakage of the nuclear envelope by an extending mitotic nucleus occurs during anaphase in Schizosaccharomyces japonicus. Genes Cells. 2011;16:911–926.
- Boettcher B, Barral Y. The cell biology of open and closed mitosis. Nucleus. 2013;4:160–165.
- Hatch E, Hetzer M. Breaching the nuclear envelope in development and disease. J Cell Biol. 2014;205:133–141.
- Hatch EM. Nuclear envelope rupture: little holes, big openings. Curr Opin Cell Biol. 2018;52:66–72.
- Isermann P, Lammerding J. Consequences of a tight squeeze: nuclear envelope rupture and repair. Nucleus. 2017;8:268–274.
- Shah P, Wolf K, Lammerding J. Bursting the bubble - Nuclear envelope rupture as a path to genomic instability? Trends Cell Biol. 2017;27:546–555.
- Broers JL, Peeters EA, Kuijpers HJ, et al. Decreased mechanical stiffness in LMNA-/- cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Hum Mol Genet. 2004;13:2567–2580.
- Denais CM, Gilbert RM, Isermann P, et al. Nuclear envelope rupture and repair during cancer cell migration. Science. 2016;352:353–358.
- Hatch EM, Hetzer MW. Nuclear envelope rupture is induced by actin-based nucleus confinement. J Cell Biol. 2016;215:27–36.
- Irianto J, Pfeifer CR, Bennett RR, et al. Nuclear constriction segregates mobile nuclear proteins away from chromatin. Mol Biol Cell. 2016;27:4011–4020.
- Robijns J, Molenberghs F, Sieprath T, et al. In silico synchronization reveals regulators of nuclear ruptures in lamin A/C deficient model cells. Sci Rep. 2016;6:30325.
- De Vos WH, Houben F, Kamps M, et al. Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum Mol Genet. 2011;20:4175–4186.
- Vargas JD, Hatch EM, Anderson DJ, et al. Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus. 2012;3:88–100.
- Hatch EM, Fischer AH, Deerinck TJ, et al. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell. 2013;154:47–60.
- Cohen S, Behzad AR, Carroll JB, et al. Parvoviral nuclear import: bypassing the host nuclear-transport machinery. J Gen Virol. 2006;87:3209–3213.
- de Noronha CM, Sherman MP, Lin HW, et al. Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science. 2001;294:1105–1108.
- Davidson PM, Sliz J, Isermann P, et al. Design of a microfluidic device to quantify dynamic intra-nuclear deformation during cell migration through confining environments. Integr Biol (Camb). 2015;7:1534–1546.
- Harada T, Swift J, Irianto J, et al. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J Cell Biol. 2014;204:669–682.
- Le Berre M, Aubertin J, Piel M. Fine control of nuclear confinement identifies a threshold deformation leading to lamina rupture and induction of specific genes. Integr Biol (Camb). 2012;4:1406–1414.
- Zhang Q, Tamashunas AC, Agrawal A, et al. Local, transient tensile stress on the nuclear membrane causes membrane rupture. Mol Biol Cell. 2019;30:899–906.
- Chen NY, Kim P, Weston TA, et al. Fibroblasts lacking nuclear lamins do not have nuclear blebs or protrusions but nevertheless have frequent nuclear membrane ruptures. Proc Natl Acad Sci U S A. 2018;115:10100–10105.
- Davidson PM, Lammerding J. Broken nuclei–lamins, nuclear mechanics, and disease. Trends Cell Biol. 2014;24:247–256.
- Houthaeve G, Robijns J, Braeckmans K, et al. Bypassing border control: nuclear envelope rupture in disease. Physiology. 2018;33:39–49.
- Earle AJ, Kirby TJ, Fedorchak GR, et al. Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nat Mater. 2020;19:464–473.
- Raab M, Gentili M, de Belly H, et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science. 2016;352:359–362.
- Maciejowski J, Li Y, Bosco N, et al. Chromothripsis and kataegis induced by telomere crisis. Cell. 2015;163:1641–1654.
- Zhang CZ, Spektor A, Cornils H, et al. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522:179–184.
- Harding SM, Benci JL, Irianto J, et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548:466–470.
- Mackenzie KJ, Carroll P, Martin CA, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461–465.
- Xia Y, Pfeifer CR, Zhu K, et al. Rescue of DNA damage after constricted migration reveals a mechano-regulated threshold for cell cycle. J Cell Biol. 2019;218:2545–2563.
- Smith LR, Irianto J, Xia Y, et al. Constricted migration modulates stem cell differentiation. Mol Biol Cell. 2019;30:1985–1999.
- Halfmann CT, Sears RM, Katiyar A, et al. Repair of nuclear ruptures requires barrier-to-autointegration factor. J Cell Biol. 2019;218:2136–2149.
- Young AM, Gunn AL, Hatch EM. BAF facilitates interphase nuclear membrane repair through recruitment of nuclear transmembrane proteins. Mol Biol Cell. 2020;31:1551-1560.
- Henne WM, Stenmark H, Emr SD. Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb Perspect Biol. 2013;5. DOI:10.1101/cshperspect.a016766
- Lusk CP, Ader NR. CHMPions of repair: emerging perspectives on sensing and repairing the nuclear envelope barrier. Curr Opin Cell Biol. 2020;64:25–33.
- Radulovic M, Stenmark H. ESCRTs in membrane sealing. Biochem Soc Trans. 2018;46:773–778.
- Olmos Y, Hodgson L, Mantell J, et al. ESCRT-III controls nuclear envelope reformation. Nature. 2015;522:236–239.
- Ventimiglia LN, Cuesta-Geijo MA, Martinelli N, et al. CC2D1B coordinates ESCRT-III activity during the mitotic reformation of the nuclear envelope. Dev Cell. 2018;47:547–563 e546.
- Vietri M, Schink KO, Campsteijn C, et al. Spastin and ESCRT-III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature. 2015;522:231–235.
- Thaller DJ, Allegretti M, Borah S, et al. An ESCRT-LEM protein surveillance system is poised to directly monitor the nuclear envelope and nuclear transport system. eLife. 2019;8. DOI:10.7554/eLife.45284
- Webster BM, Thaller DJ, Jager J, et al. Chm7 and Heh1 collaborate to link nuclear pore complex quality control with nuclear envelope sealing. Embo J. 2016;35:2447–2467.
- Vietri M, Schultz SW, Bellanger A, et al. Unrestrained ESCRT-III drivesmicronuclear catasrophe and chromosome fragmentation. Nature Cell Biology.2020;22:856–867. .
- Willan J, Cleasby AJ, Flores-Rodriguez N, et al. ESCRT-III is necessary for the integrity of the nuclear envelope in micronuclei but is aberrant at ruptured micronuclear envelopes generating damage. Oncogenesis. 2019;8:29.
- Penfield L, Wysolmerski B, Mauro M, et al. Dynein-pulling forces counteract lamin-mediated nuclear stability during nuclear envelope repair. Mol Biol Cell. 2018;29:852–868.
- Margalit A, Brachner A, Gotzmann J, et al. Barrier-to-autointegration factor–a BAFfling little protein. Trends Cell Biol. 2007;17:202–208.
- Skoko D, Li M, Huang Y, et al. Barrier-to-autointegration factor (BAF) condenses DNA by looping. Proc Natl Acad Sci U S A. 2009;106:16610–16615.
- Lee KK, Haraguchi T, Lee RS, et al. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J Cell Sci. 2001;114:4567–4573.
- Montes de Oca R, Lee KK, Wilson KL. Binding of barrier to autointegration factor (BAF) to histone H3 and selected linker histones including H1.1. J Biol Chem. 2005;280:42252–42262.
- Mansharamani M, Graham DR, Monie D, et al. Barrier-to-autointegration factor BAF binds p55 Gag and matrix and is a host component of human immunodeficiency virus type 1 virions. J Virol. 2003;77:13084–13092.
- Wang X, Xu S, Rivolta C, et al. Barrier to autointegration factor interacts with the cone-rod homeobox and represses its transactivation function. J Biol Chem. 2002;277:43288–43300.
- Bolderson E, Burgess JT, Li J, et al. Barrier-to-autointegration factor 1 (Banf1) regulates poly [ADP-ribose] polymerase 1 (PARP1) activity following oxidative DNA damage. Nat Commun. 2019;10:5501.
- Ma H, Qian W, Bambouskova M, et al. Barrier-to-autointegration factor 1 protects against a basal cGAS-STING response. mBio. 2020;11. DOI:10.1128/mBio.00136-20
- Shimi T, Koujin T, Segura-Totten M, et al. Dynamic interaction between BAF and emerin revealed by FRAP, FLIP, and FRET analyses in living HeLa cells. J Struct Biol. 2004;147:31–41.
- Haraguchi T, Koujin T, Osakada H, et al. Nuclear localization of barrier-to-autointegration factor is correlated with progression of S phase in human cells. J Cell Sci. 2007;120:1967–1977.
- Nichols RJ, Wiebe MS, Traktman P. The vaccinia-related kinases phosphorylate the N’ terminus of BAF, regulating its interaction with DNA and its retention in the nucleus. Mol Biol Cell. 2006;17:2451–2464.
- Gorjanacz M, Klerkx EP, Galy V, et al. Caenorhabditis elegans BAF-1 and its kinase VRK-1 participate directly in post-mitotic nuclear envelope assembly. Embo J. 2007;26:132–143.
- Haraguchi T, Kojidani T, Koujin T, et al. Live cell imaging and electron microscopy reveal dynamic processes of BAF-directed nuclear envelope assembly. J Cell Sci. 2008;121:2540–2554.
- Haraguchi T, Koujin T, Segura-Totten M, et al. BAF is required for emerin assembly into the reforming nuclear envelope. J Cell Sci. 2001;114:4575–4585.
- Margalit A, Segura-Totten M, Gruenbaum Y, et al. Barrier-to-autointegration factor is required to segregate and enclose chromosomes within the nuclear envelope and assemble the nuclear lamina. Proc Natl Acad Sci U S A. 2005;102:3290–3295.
- Samwer M, Schneider MWG, Hoefler R, et al. DNA cross-bridging shapes a single nucleus from a set of mitotic chromosomes. Cell. 2017;170:956–972 e923.
- Chen H, Engelman A. The barrier-to-autointegration protein is a host factor for HIV type 1 integration. Proc Natl Acad Sci U S A. 1998;95:15270–15274.
- Lee MS, Craigie R. A previously unidentified host protein protects retroviral DNA from autointegration. Proc Natl Acad Sci U S A. 1998;95:1528–1533.
- Lin CW, Engelman A. The barrier-to-autointegration factor is a component of functional human immunodeficiency virus type 1 preintegration complexes. J Virol. 2003;77:5030–5036.
- Suzuki Y, Craigie R. Regulatory mechanisms by which barrier-to-autointegration factor blocks autointegration and stimulates intermolecular integration of Moloney murine leukemia virus preintegration complexes. J Virol. 2002;76:12376–12380.
- Ibrahim N, Wicklund A, Wiebe MS. Molecular characterization of the host defense activity of the barrier to autointegration factor against vaccinia virus. J Virol. 2011;85:11588–11600.
- Wiebe MS, Traktman P. Poxviral B1 kinase overcomes barrier to autointegration factor, a host defense against virus replication. Cell Host Microbe. 2007;1:187–197.
- Ibrahim N, Wicklund A, Jamin A, et al. Barrier to autointegration factor (BAF) inhibits vaccinia virus intermediate transcription in the absence of the viral B1 kinase. Virology. 2013;444:363–373.
- Jamin A, Thunuguntla P, Wicklund A, et al. Barrier to auto integration factor becomes dephosphorylated during HSV-1 infection and can act as a host defense by impairing viral DNA replication and gene expression. PloS One. 2014a;9:e100511.
- Jamin A, Wicklund A, Wiebe MS. Cell- and virus-mediated regulation of the barrier-to-autointegration factor’s phosphorylation state controls its DNA binding, dimerization, subcellular localization, and antipoxviral activity. J Virol. 2014b;88:5342–5355.
- Molitor TP, Traktman P. Depletion of the protein kinase VRK1 disrupts nuclear envelope morphology and leads to BAF retention on mitotic chromosomes. Mol Biol Cell. 2014;25:891–903.
- Birendra K, May DG, Benson BV, et al. VRK2A is an A-type lamin-dependent nuclear envelope kinase that phosphorylates BAF. Mol Biol Cell. 2017;28:2241–2250.
- Gu M, LaJoie D, Chen OS, et al. LEM2 recruits CHMP7 for ESCRT-mediated nuclear envelope closure in fission yeast and human cells. Proc Natl Acad Sci U S A. 2017;114:E2166–E2175.
- Elacqua JJ, McGregor AL, Lammerding J. Automated analysis of cell migration and nuclear envelope rupture in confined environments. PloS One. 2018;13:e0195664.
- Penfield L, Shankar R, Szentgyorgyi E, et al. Regulated lipid synthesis and LEM2/CHMP7 jointly control nuclear envelope closure. J Cell Biol. 2020;219. DOI:10.1083/jcb.201908179
- King MC, Lusk CP. Loss of nuclear envelope integrity? No probLEM-BAF has it covered. J Cell Biol. 2019;218:2077–2079.
- Xia Y, Ivanovska IL, Zhu K, et al. Nuclear rupture at sites of high curvature compromises retention of DNA repair factors. J Cell Biol. 2018;217:3796–3808.
- Lancaster OM, Cullen CF, Ohkura H. NHK-1 phosphorylates BAF to allow karyosome formation in the Drosophila oocyte nucleus. J Cell Biol. 2007;179:817–824.
- Asencio C, Davidson IF, Santarella-Mellwig R, et al. Coordination of kinase and phosphatase activities by Lem4 enables nuclear envelope reassembly during mitosis. Cell. 2012;150:122–135.
- Gorjanacz M. LEM-4 promotes rapid dephosphorylation of BAF during mitotic exit. Nucleus. 2013;4:14–17.
- Snyers L, Erhart R, Laffer S, et al. LEM4/ANKLE-2 deficiency impairs post-mitotic re-localization of BAF, LAP2alpha and LaminA to the nucleus, causes nuclear envelope instability in telophase and leads to hyperploidy in HeLa cells. Eur J Cell Biol. 2018;97:63–74.
- Bar DZ, Davidovich M, Lamm AT, et al. BAF-1 mobility is regulated by environmental stresses. Mol Biol Cell. 2014;25:1127–1136.
- Rempel RE, Anderson MK, Evans E, et al. Temperature-sensitive vaccinia virus mutants identify a gene with an essential role in viral replication. J Virol. 1990;64:574–583.
- Rempel RE, Traktman P. Vaccinia virus B1 kinase: phenotypic analysis of temperature-sensitive mutants and enzymatic characterization of recombinant proteins. J Virol. 1992;66:4413–4426.
- Campillo-Marcos I, Lazo PA. Implication of the VRK1 chromatin kinase in the signaling responses to DNA damage: a therapeutic target? Cell Mol Life Sci. 2018;75:2375–2388.
- Campillo-Marcos I, Lazo PA. Olaparib and ionizing radiation trigger a cooperative DNA-damage repair response that is impaired by depletion of the VRK1 chromatin kinase. J Exp Clin Cancer Res. 2019;38:203.
- Blanco S, Klimcakova L, Vega FM, et al. The subcellular localization of vaccinia-related kinase-2 (VRK2) isoforms determines their different effect on p53 stability in tumour cell lines. Febs J. 2006;273:2487–2504.
- Nichols RJ, Traktman P. Characterization of three paralogous members of the Mammalian vaccinia related kinase family. J Biol Chem. 2004;279:7934–7946.
- Scheeff ED, Eswaran J, Bunkoczi G, et al. Structure of the pseudokinase VRK3 reveals a degraded catalytic site, a highly conserved kinase fold, and a putative regulatory binding site. Structure. 2009;17:128–138.
- Park CH, Ryu HG, Kim SH, et al. Presumed pseudokinase VRK3 functions as a BAF kinase. Biochim Biophys Acta. 2015;1853:1738–1748.
- Zhuang X, Semenova E, Maric D, et al. Dephosphorylation of barrier-to-autointegration factor by protein phosphatase 4 and its role in cell mitosis. J Biol Chem. 2014;289:1119–1127.
- Santaguida S, Richardson A, Iyer DR, et al. Chromosome mis-segregation generates cell-cycle-arrested cells with complex karyotypes that are eliminated by the immune system. Dev Cell. 2017;41:638–651 e635.
- Yang Z, Maciejowski J, de Lange T. Nuclear envelope rupture is enhanced by loss of p53 or Rb. Mol Cancer Res. 2017;15:1579–1586.
- Capo-Chichi CD, Yeasky TM, Smith ER, et al. Erratum to: nuclear envelope structural defect underlies the main cause of aneuploidy in ovarian carcinogenesis. BMC Cell Biol. 2017;18:1.
- Ly P, Cleveland DW. Rebuilding chromosomes after catastrophe: emerging mechanisms of chromothripsis. Trends Cell Biol. 2017;27:917–930.
- Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063.
- Vlcek S, Foisner R. Lamins and lamin-associated proteins in aging and disease. Curr Opin Cell Biol. 2007;19:298–304.
- Wilson KL. The nuclear envelope, muscular dystrophy and gene expression. Trends Cell Biol. 2000;10:125–129.
- Cabanillas R, Cadinanos J, Villameytide JA, et al. Nestor-Guillermo progeria syndrome: a novel premature aging condition with early onset and chronic development caused by BANF1 mutations. Am J Med Genet A. 2011;155A:2617–2625.
- Puente XS, Quesada V, Osorio FG, et al. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am J Hum Genet. 2011;88:650–656.
- Vidak S, Foisner R. Molecular insights into the premature aging disease progeria. Histochem Cell Biol. 2016;145:401–417.
- Paquet N, Box JK, Ashton NW, et al. Nestor-guillermo progeria syndrome: a biochemical insight into barrier-to-autointegration factor 1, alanine 12 threonine mutation. BMC Mol Biol. 2014;15:27.