
ABSTRACT
Liver fibrosis is a critical health issue in the world due to its rapidly increasing prevalence. It is of great demand to develop effective drugs for the treatment of liver fibrosis. 5-methoxytryptophan (5-MTP) has been reported to play an important role in anti-inflammatory, anti-cancer, myocardial-protective effects. However, the anti-fibrotic effect of 5-MTP is never covered in liver. Here, we investigated anti-fibrotic effects of 5-MTP on liver fibrosis and its underlying mechanism. In vitro, 5-MTP treatment could inhibit TGF-β1-induced elevated levels of collagen I, collagen III, fibronectin and α-smooth muscle actin (SMA) by stimulating autophagy process. Mechanically, the expression of FOXO3a was enhanced by 5-MTP and then repressed the level of miR-21, eventually leading to a restoration of autophagy-related gene ATG5. Furthermore, rescue experiments showed 5-MTP could activate autophagy process and suppress the activation of LX-2 cells by regulating FOXO3a/miR-21/ATG5 pathway. Consistently, 5-MTP significantly attenuated CCl4-induced hepatic fibrosis in rat model. In conclusion, our research discovered that 5-MTP effectively alleviated liver fibrosis in vitro and in vivo, which provided new insights into the application of 5-MTP for liver fibrosis.
Introduction
Hepatic fibrosis is a process of wound healing after the injury in liver which is caused by chronic hepatitis, alcohol, or drugs [Citation1]. When liver tissue is continuously or repeatedly damaged or inflamed, scarring response happens in liver parenchyma and degenerative cells take the place of healthy hepatocytes [Citation1,Citation2]. Cirrhosis is the most common consequence of liver fibrosis, which is the late-stage of liver disease [Citation1]. Although great progress has been achieved in understanding the pathogenesis and molecular mechanism of liver fibrosis in recent years, there is still lack of effective therapeutic drugs developed against hepatic fibrosis and cirrhosis [Citation3]. Hepatic stellate cells (HSCs) play an important role in the development of hepatic fibrosis. HSCs belong to the type of resident mesenchymal cell, which is located in the space between hepatocytes and sinusoidal endothelial cells [Citation4]. When liver injury happens, HSCs will be activated and become proliferative and fibrogenic, which are then accumulated in the extracellular matrix (ECM) and facilitate its remodeling and promote liver fibrosis [Citation5,Citation6]. Therefore, it is very promising to treat liver fibrosis by targeting HSCs.
5-Methoxytryptophan (5-MTP) is a 5-methoxyindole metabolite of tryptophan metabolism [Citation7,Citation8]. 5-MTP has firstly been identified to play a role in the retina’s cyclic metabolism [Citation9]. Meanwhile, previous studies have also demonstrated that 5-MTP could inhibit cancer cell proliferation and migration by regulating inflammatory effectors [Citation7,Citation8]. Another study indicated that 5-MTP could protect heart tissues against ischemia reperfusion injury [Citation10]. Moreover, 5-MTP has been shown to play anti-fibrotic role in kidney disease by attenuating up-regulation of pro-fibrotic proteins collagen I, fibronectin, vimentin and α-SMA [Citation11]. However, the role of 5-MTP in the liver fibrosis is still unclear. Recently, 5-MTP has been reported to up-regulate the expression of FOXO3a [Citation12], which is an important transcription factor and demonstrated to inhibit miR-21 expression [Citation13]. miR-21 is one of the most important microRNAs that plays a critical role in different pathways. Evidences have shown that inhibition of miR-21 could block the activity of TGF-β pathway and attenuate liver fibrosis [Citation14,Citation15], indicating miR-21 as a potential target for liver fibrosis treatment.
Autophagy is a cellular self-digestion process, which is important for the homeostasis maintenance of cell and tissue under stresses [Citation16]. It has also been reported that autophagy is involved in the process of tissue repair and fibrosis [Citation17–19]. Stimulation of autophagy has been shown to repress hepatic fibrosis by blocking HSCs activation [Citation20]. Autophagy-related gene 5 (ATG5) is critical for the formation of autophagosome, which is the hallmark of autophagy [Citation21]. Suppression of ATG5 has been proved to block autophagy process [Citation22]. miR-21 has been predicted to target ATG5 directly, which raises the possibility that 5-MTP could increase autophagy of HSCs by regulating FOXO3a/miR-21/ATG5 pathway and eventually attenuate hepatic fibrosis.
In this study, we demonstrated that 5-MTP treatment could stimulate autophagy and repress the TGF-β1-induced proliferation and fibrosis of LX-2 cells. Meanwhile, FOXO3a was shown to be up-regulated by 5-MTP, which then directly suppressed the expression of miR-21 and restored the level of ATG5 in LX-2 cells. Knockdown of FOXO3a was able to inhibit autophagy induced by 5-MTP treatment, which was then rescued when combined with miR-21 inhibitor. Moreover, 5-MTP significantly attenuated the hepatic fibrosis process in CCl4-induced rat models, highlighting its potential as a therapeutic method for the treatment of hepatic fibrosis.
Materials and methods
Cell culture and reagents
The human hepatic stellate cell line LX-2 was purchased from Sigma-Aldrich (cat. SCC064, MA, USA). LX-2 cells were cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL of penicillin and streptomycin at 37°C in 5% CO2 with 95% humidity. LX-2 cells were trypsin-digested and passaged every two days. TGF-β1 (5 ng/mL, Sigma-Aldrich, USA) treatment for 24 h was used to activate HSCs. 5-Methoxytryptophan (5-MTP) and autophagy inhibitor 3-Methyladenine (3-MA) were purchased from Sigma-Aldrich (MA, USA). LX-2 cells were pretreated with 5-MTP for 12 h prior to the addition of TGF-β1. Cells cultured without any exposure and treatment were used as a control.
Animals and treatment
Male Sprague-Dawley rats (150 ± 10 g, SPF) were provided by the Experimental Animal Center of the first affiliated hospital of Hunan Normal University and were housed under controlled conditions with temperature of 25 ± 2°C, relative humidity of 60 ± 10%, and a 12 h light/dark cycle. The animal experiment was approved by the Institutional Animal Care and Use Committee (Approval Number: 06). Hepatic fibrosis was induced by CCl4 treatment (20% v/v, 2 mL/kg) twice per week for 8 weeks. Animals in the control group were treated with 0.9% saline twice per week for 8 weeks. Animals in the 5-MTP + CCl4 group were treated with 5 mg/kg 5-MTP twice per week for 8 weeks.
Cell transfection
miR-21 inhibitor and miRNA-negative control (NC inhibitor) were purchased from Genepharma (Shanghai, China). Short hairpin RNAs (shRNAs) targeting FOXO3a were designed and synthesized by RiboBio (Guangzhou, China). Transfections were made by Lipofectamine 2000 transfection reagent (cat. 11668019, Invitrogen, USA) according to the manufacturer’s instructions. After 48 h, the cells were used for subsequent experiments.
Total RNA extraction and quantitative PCR
Cells were dissolved in TRIzol reagent (Cat. 15596018, Invitrogen, USA), and total RNA was obtained according to the manufacturer’s protocol. The RNA was then tested for quality and synthesized into cDNA using an iScript cDNA Synthesis Kit (Cat. 1708891, Bio-Rad, USA). qRT-PCR was performed using SYBR Green Supermixes (Cat. 1708882, Bio-Rad, USA). GAPDH and U6 were used as endogenous controls for normalization. Relative levels of expression were normalized and analyzed using the 2−ΔΔCt method. Primer sequences are listed in .
Table 1. Primer sequences for quantitative PCR
Western blot analysis
Cells were washed with cold PBS and incubated with lysis buffer on ice for 30 min. Then, the cells were scraped, and after centrifugation, the supernatant containing the lysate was collected and stored at −80°C. A BCA assay kit (Cat. 5000001, Bio-Rad, USA) was used to determine protein concentrations. Protein samples were denatured and then separated by SDS-PAGE and transferred to PVDF membranes (Cat. IPVH00010, Millipore, USA). After blocking with nonfat milk for 1 h, membranes were incubated overnight at 4°C with the following primary antibodies from Abcam (Cambridge, UK): α-smooth muscle actin (α-SMA, #ab5694), fibronectin (#ab2413), collagen I (#ab90395), collagen III (#ab7778), LC3I (#ab239849), LC3II (#ab48394), Beclin-1 (#ab210498), ATG5 (#ab108327) and β-actin (#ab8227), and all were used at a 1:1000 dilution. After washing three times, the membranes were incubated with HRP-conjugated secondary antibodies (Cell Signaling Technology, USA) at a dilution of 1:3000. The signals were visualized using an ECL detection kit (#32106, Pierce Biotechnology, USA) and analyzed through ImageJ 1.52a (National Institutes of Health, USA).
Cell viability assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay kit was purchased from Sigma-Aldrich (cat. #11465007001, MA, USA) and was used to detect cell viability. Briefly, 1 × 104 cells after indicated treatment were seeded into 96-well plates and treated with different concentrations of 5-MTP (0, 0.25, 0.5, 0.75, 1.25, 2.5 μM) for 24 h. Then, 20 µl of MTT solution was added to each well. The absorbance at 490 nm was measured using a spectrophotometer after 2 h culture.
Bromodeoxyuridine (BrdU) assay
Cell proliferation was measured by BrdU assay kit (cat. #6813, Cell Signaling Technology, USA). In brief, cells were incubated with 20 μM of BrdU and then harvested. After washing, cells were treated with fixation solution at room temperature (RT) for 30 min followed by the incubation of mouse anti-BrdU antibody for 1 h. After washing, goat anti-mouse IgG was added and kept at RT for 1 h. Cells were washed and images were taken and analyzed.
Immunofluorescence and immunohistochemistry
Expressional levels of LC-3 and α-SMA in LX-2 cells were analyzed by immunofluorescence. Briefly, LX-2 cells were washed three times by cold PBS and then fixed in 4% paraformaldehyde/PBS for 10 min. Then the cells were incubated with primary antibodies against LC3 (#ab48394; Abcam) or α-SMA (#ab5694; Abcam) at 4°C overnight. All the antibodies were diluted 1:100 in PBS containing 1% bovine serum albumin. Cells were then incubated by secondary antibodies (Goat Anti-Rabbit IgG H&L Alexa Fluor® 488/555; #ab150077/ab150078; Abcam) at a dilution of 1:1000 for 1 h at 37°C. Images were obtained using a fluorescence microscopy. For immunohistochemistry assay, rat liver tissues embedded in paraffin and tissue slides (4 µm) were then blocked with 5% BSA (bovine serum albumin), incubated with anti-α-SMA antibodies (#ab5694; Abcam) diluted 1:100 at room temperature for 1 h, incubated with a secondary antibody (#ab205718, Abcam) diluted 1:1000 at room temperature for 1 h, and finally developed with diaminobenzidine (DAB) staining.
Dual-luciferase reporter assay
The 293 T cells were pre-seeded into 24-well plates. After 24 h, cells were co-transfected with wild-type or mutant 3′-UTR vectors of ATG5 and miR-21 mimic or miR-negative control using lipofectamine 2000 (Invitrogen). Luciferase activity was measured 48 h after transfection with dual-luciferase reporter gene assay kit (Cat. E1910, Promega, USA) according to manufacturer instruction.
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation assay was conducted as described previously [Citation23]. When LX-2 cell density was estimated to reach about 80% confluence, cells were cross-linked with 1% formaldehyde for 10 min at 37°C and then performed with reference to the EZ-CHIP Reagent kit (Millipore, Billerica, MA). The chromatin fragments of DNA-protein complex were precipitated using a FOXO3a antibody (ab12162, Abcam, USA) or an IgG antibody (Santa Cruz, CA). The DNA was then eluted, extracted with phenol-chloroform, and subjected to PCR. PCR was then performed to detect the FOXO3a binding to the promoter region of miR-21.
Histopathology
Tissue samples of rat livers were preserved in 4% paraformaldehyde/PBS solution. After fixation, all tissues were processed, paraplast-fixed and then sectioned (5 μm thickness). After that, slides were stained with hematoxylin and eosin (H&E) and sirius red staining as described previously [Citation24].
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5. All experiments were conducted at least three times. All data are presented as the mean ± SD. The data were analyzed by one-way ANOVA followed by Tukey’s multiple comparison test or an independent sample t-test. P< 0.05 was considered statistically significant.
Results
5-MTP inhibited the proliferation and fibrosis of LX-2 cells
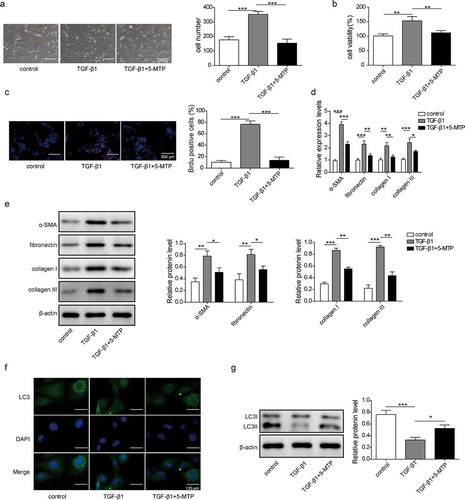
To investigate the effect of 5-MTP on TGF-β1-induced hepatic stellate cell activation, LX-2 cell line was used. CCK-8 results indicated that 0.75 µM 5-MTP was cytotoxic to LX-2 cells (Figure S1). Based on these data, 0.5 µM 5-MTP was used for subsequent experiment. As shown in , after 24 h of TGF-β1 treatment, more cells were observed under microscope, compared to control group. However, when 5-MTP was combined with TGF-β1, the cell number was significantly decreased. Meanwhile, the cell viability measured by MTT assay and proliferation ability represented by BrdU staining were both remarkably enhanced after TGF-β1 treatment when compared to control ones, and then decreased when 5-MTP was added ( and c). The fibrosis of LX-2 cells was further evaluated by testing the expression of hallmark genes. As shown in and e, both mRNA and protein levels of α-SMA, fibronectin,collagen Iand collagen III were significantly up-regulated upon TGF-β1 treatment compared to control, while 5-MTP pre-treatment obviously suppressed TGF-β1-induced cell fibrosis. Moreover, the autophagy status of LX-2 cells was demonstrated by LC3 expression level. Compared to control cells, TGF-β1 treatment dramatically inhibited the expression of LC3. The pre-treatment of 5-MTP blocked the effects of TGF-β1 on LC3 expression ( and g). Taken together, these results suggested that 5-MTP could repress the proliferation and fibrosis of LX-2 cells, but stimulate their autophagy ability.
Figure 1. 5-MTP inhibited the proliferation and fibrosis of LX-2 cells
5-MTP repressed the proliferation and fibrosis of LX-2 cells by inducing autophagy
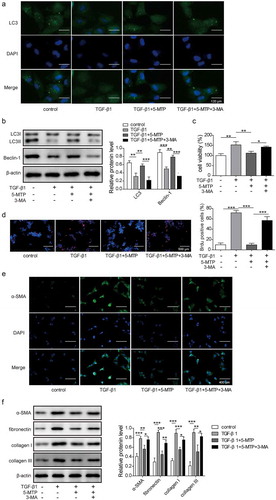
As determined that 5-MTP could inhibit the proliferation and fibrosis, but enhance the autophagy of LX-2 cells led us to think about whether 5-MTP affects proliferation and fibrosis by regulating autophagy. To demonstrate our hypothesis, 3-MA was used to block the autophagy induced by 5-MTP ( and b). Cell viability and proliferation were found to be restored after 3-MA treatment in the LX-2 cells treated by TGF-β1 and 5-MTP ( and d). Consistently, the levels of fibrosis-related protein including α-SMA, fibronectin, collagen I and collagen III in the TGF-β1 exposure were significantly higher than those in the controls. 5-MTP treatment obviously suppressed TGF-β1-induced cell fibrosis, and 3-MA treatment significantly increased the levels of fibrosis-related protein ( and f). These data indicated that 5-MTP inhibited the proliferation and fibrosis abilities of LX-2 cells by inducing autophagy.
Figure 2.. 5-MTP repressed the proliferation and fibrosis of LX-2 cells by inducing autophagy
FOXO3a was upregulated by 5-MTP and suppressed the transcription of miR-21
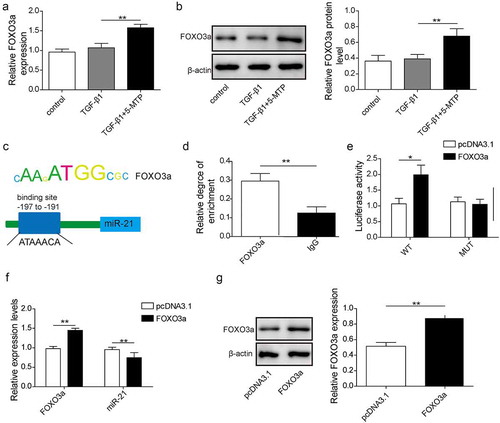
To explore the molecular mechanism of 5-MTP regulating LX2 cell function, the potential downstream target was identified. As shown in and b, TGF-β1 treatment would not affect the expression of FOXO3a in LX-2 cells compared to control ones. But when 5-MTP pre-treatment was applied, there was a significant increase in both mRNA and protein levels of FOXO3a. As a transcription factor, FOXO3a controls quite a few genes’ expression which might affect the autophagy in LX-2 cells. As shown in , the binding sites for FOXO3a in the promoter region of miR-21 were predicted. Then, FOXO3a was found to directly target and bind to the promoter region of miR-21, as determined by ChIP assay (). Furthermore, dual luciferase assay showed that FOXO3a significantly repressed the expression of luciferase gene which contained the wild-type binding sites from miR-21’s promoter, while had no effect on that contained mutant binding site (). Overexpression of FOXO3a remarkably reduced miR-21 expression, whereas enhanced the expression of FOXO3a at both mRNA and protein levels ( and g). In sum, these data strongly demonstrated FOXO3a was stimulated by 5-MTP treatment in LX-2 cells and then directly targeted and repressed the expression of its downstream target miR-21.
Figure 3. FOXO3a was upregulated by 5-MTP and suppressed the transcription of miR-21
miR-21 directly targeted ATG5 and inhibited its expression
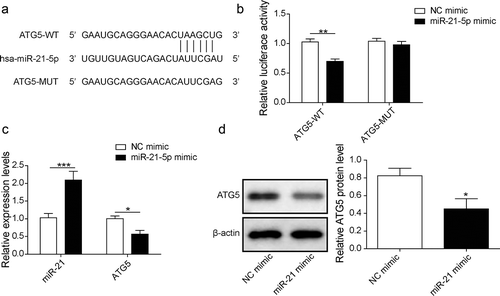
The potential targets of miR-21 were predicted by StarBase database. A putative miR-21 binding site was identified in the ATG5 mRNA sequence, as shown in . Luciferase reporter assay demonstrated that miR-21 was able to target and inhibit luciferase activity which contained wild-type ATG5 3ʹUTR reporter construct, instead of the mutant one (). Moreover, overexpression of miR-21 significantly downregulated ATG5 at both mRNA and protein levels compared to control ones ( and d). Overall, these results demonstrated that ATG5 was a target of miR-21 which could be suppressed by miR-21 directly.
Figure 4. miR-21 directly targeted ATG5 and inhibited its expression
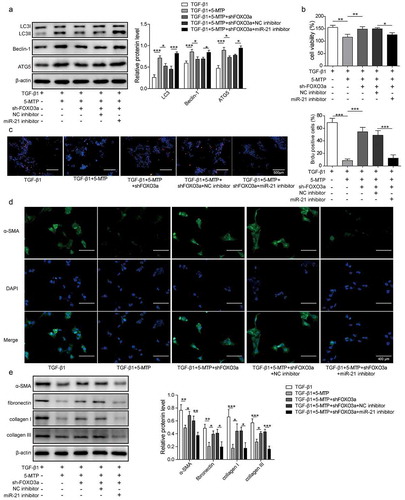
5-MTP suppressed the proliferation and fibrosis of LX-2 cells by modulating FOXO3a/miR-21/ATG5 signaling pathway mediated autophagy
To explore the effects of FOXO3a/miR-21/ATG5 pathway in LX-2 cells function, we conducted this part. FOXO3a shRNA and miR-21 inhibitor were applied to manipulate the pathway activity. As shown in , the 5-MTP-induced autophagy, represented by LC3 and Beclin-1 expression, was inhibited by knockdown of FOXO3a, while the suppression of both FOXO3a and miR-21 made these proteins' expression obviously up-regulated. Meanwhile, the cell viability and proliferation of LX-2 cells were significantly recovered against 5-MTP by inhibition of FOXO3a, but the suppression of both FOXO3a and miR-21 significantly inhibited cell viability and proliferation ( and c). Unsurprisingly, the fibrosis ability of TGF-β1-induced LX-2 cells, which was evaluated by the expression of α-SMA, fibronectin, collagen I and collagen III, also got reduced by 5-MTP pretreatment, while promoted by knockdown of FOXO3a; whereas FOXO3a shRNA combined with miR-21 inhibitor made these fibrosis-related proteins significantly down-regulated ( and e). Taken together, these data indicated that 5-MTP suppressed the proliferation and fibrosis was through regulating FOXO3a/miR-21/ATG5 pathway in LX-2 cells.
Figure 5.. 5-MTP suppressed the proliferation and fibrosis of LX-2 cells by modulating FOXO3a/miR-21/ATG5 signaling pathway mediated autophagy
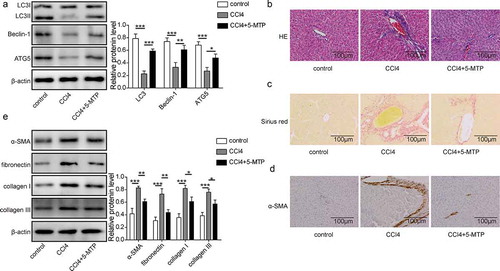
5-MTP alleviated the CCl4-induced liver fibrosis in rat model
To address the therapeutic application of 5-MTP in liver fibrosis, rat model was established by administration of CCl4. Liver tissues were harvested and examined. As shown in , autophagy markers LC3 and Beclin-1 were significantly decreased from CCl4-treated rats when compared to control, together with a down-regulation of ATG5. However, when 5-MTP was applied, the autophagy activity was recovered and the expression of ATG5 was increased against CCl4 treatment. Hematoxylin and eosin (H&E) staining of liver tissues further demonstrated that CCl4 treatment could induce obviously lesions in livers, which then got protected by the treatment of 5-MTP (). Meanwhile, a significantly increased fibrosis in livers, which was examined by sirius red staining, was observed in CCl4-treated rats and then decreased when combined with 5-MTP treatment (). Consistently, the gene markers of fibrosis (α-SMA, fibronectin, collagen I and collagen III) also got significantly enhanced upon CCl4 treatment in liver tissues, which were then suppressed to the similar level of control rats ( and e). These results suggested that 5-MTP could protect liver from CCl4-induced fibrosis in vivo.
Figure 6.. 5-MTP attenuated CCl4-induced hepatic fibrosis in vivo.
Discussion
5-methoxytryptophan (5-MTP) is a newly identified tryptophan metabolite that regulates different cellular processes, such as inflammation [Citation25], cell proliferation and migration [Citation7]. Our results demonstrated that 5-MTP could suppress TGF-β1-induced proliferation and fibrosis of human hepatic stellate cell line LX-2, which were critical processes for hepatic fibrosis. Further evidences have shown that 5-MTP-induced autophagy of LX-2 cells was important for the inhibition of cell proliferation and fibrosis abilities, which could be restored by autophagy inhibitor treatment. Autophagy is a fundamental function of cell that involved in cytoplasmic homeostasis by regulating the recycling of proteins and organelles [Citation26]. It has been reported that activation of autophagy could suppress cell proliferation and affect cell differentiation [Citation27–29]. Several studies have shown that autophagy could activate the HSCs and facilitate liver fibrosis [Citation30,Citation31]. However, there are also other reports about the anti-fibrosis role of autophagy process [Citation17]. In our study, blocking of autophagy could rescue the proliferation and fibrosis abilities of LX-2 cells, which were repressed by 5-MTP treatment, indicating an anti-fibrosis role of autophagy in liver fibrosis.
FOXO3a belongs to the family of Forkhead transcription factors (FOXOs), which are named after the Drosophila melanogaster forkhead genes [Citation32]. FOXO3a has been widely studied and plays an important role in multiple disease, including cancer [Citation33], diabetes [Citation34], and aging [Citation35]. In hepatic fibrosis, FOXO3a has been shown to participate in hepatocyte injury and HSCs activation. In hepatic fibrosis process, HSCs are activated by fibrogenic stimulus and differentiated from a quiescent phenotype to a myofibroblast-like phenotype that can produce collagen and proliferate [Citation36]. FOXO3a was shown to up-regulate P27 expression in HSCs and activate cell cycle arrest, resulting in the inhibition of HSCs proliferation [Citation37]. Meanwhile, FOXO3a has also reported to enhance HSCs’ apoptosis by suppressing c-FLIP expression [Citation38]. Our results have shown that 5-MTP treatment could stimulate the expression of FOXO3a in LX-2 cells. Moreover, knockdown of FOXO3a by shRNA could significantly suppress the 5-MTP-induced cell autophagy and restore cell proliferation and fibrosis, suggesting the critical role of FOXO3a in the 5-MTP-mediated inhibition of hepatic fibrosis.
MicroRNAs (miRNAs) belong to small non-coding RNAs that regulate downstream targets through binding to their 3ʹ-untranslated regions (3ʹUTR) and inducing transcriptional repression or degradation [Citation39]. Several miRNAs have been reported to be involved in the hepatic fibrosis process [Citation40–42]. miR-21 was also shown to facilitate the hepatic fibrosis by enhancing HSCs’ activation and reducing apoptosis via regulation of ERK1 and AP-1 [Citation43], 44]. Our results demonstrated that FOXO3a could directly bind to the promoter region of miR-21 and suppress its expression in LX-2 cells, resulting in the restoration of ATG5 level, which was critical for autophagy process. Meanwhile, knockdown of miR-21 by its inhibitor could abolish the inhibition of HSCs activation induced by FOXO3a suppression after 5-MTP treatment. These results further explained the role of 5-MTP in anti-fibrosis function through regulation of FOXO3a/miR-21/ATG5 pathway. Our in vivo study established by CCl4-induced hepatic fibrosis in rat model also demonstrated that 5-MTP treatment could activate the autophagy process in liver tissues and inhibit fibrotic transition.
In sum, our study determined that 5-MTP treatment could block the fibrosis process of HSCs by inducing autophagy and suppressing HSCs’ activation. FOXO3a was the downstream target of 5-MTP, which could be up-regulated upon its treatment. The expression of FOXO3a was increased then transcriptionally inhibited the expression of pro-fibrosis mediator miR-21 and restored the level of ATG5. Animal study also demonstrated the role of 5-MTP in the attenuation of hepatic fibrosis. Our research identified the molecular mechanism of the anti-fibrosis function of 5-MTP and highlighted the application of 5-MTP as a promising therapeutic method for the treatment of hepatic fibrosis in the future.
Ethics approval
The animal experiment was approved by the Institutional Animal Care and Use Committee.
Supplemental Material
Download TIFF Image (774.2 KB)Disclosure statement
The authors declare that they have no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Friedman SL. Liver fibrosis – from bench to bedside. J Hepatol. 2003;38(Suppl 1):S38–53.
- Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6(1):425–456.
- Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123(5):1887–1901.
- Yin C, Evason KJ, Asahina K, et al. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. 2013;123(5):1902–1910.
- Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275(4):2247–2250.
- Pinzani M, Marra F. Cytokine receptors and signaling in hepatic stellate cells. Semin Liver Dis. 2001; 21(3):397–416.
- Cheng HH, Kuo CC, Yan JL, et al. Control of cyclooxygenase-2 expression and tumorigenesis by endogenous 5-methoxytryptophan. Proc Natl Acad Sci U S A. 2012;109(5):13231–13236.
- Wu KK, Cheng HH, Chang TC. 5-methoxyindole metabolites of L-tryptophan: control of COX-2 expression, inflammation and tumorigenesis. J Biomed Sci. 2014;21(1):17.
- Flight WF, Mans D, Balemans MG. Methoxyindole synthesis in the retina of the frog (Rana esculenta) during a diurnal period. J Neural Transm. 1983;58(3–4):223–230.
- Chou HC, Chan HL. 5-Methoxytryptophan-dependent protection of cardiomyocytes from heart ischemia reperfusion injury. Arch Biochem Biophys. 2014;543:15–22.
- Chen DQ, Cao G, Chen H, et al. Identification of serum metabolites associating with chronic kidney disease progression and anti-fibrotic effect of 5-methoxytryptophan. Nat Commun. 2019;10(1):1476. .
- Chang TC, Hsu MF, Shih CY, et al. 5-methoxytryptophan protects MSCs from stress induced premature senescence by upregulating FoxO3a and mTOR. Sci Rep. 2017;7(1):11133.
- Wang K, Li PF. Foxo3a regulates apoptosis by negatively targeting miR-21. J Biol Chem. 2010;285(22):16958–16966.
- Wu K, Ye C, Lin L, et al. Inhibiting miR-21 attenuates experimental hepatic fibrosis by suppressing both the ERK1 pathway in HSC and hepatocyte EMT. Clin Sci. 2016;130(16):1469–1480.
- Yang F, Luo L, Zhu ZD, et al. Chlorogenic acid inhibits liver fibrosis by blocking the miR-21-regulated TGF-beta1/Smad7 signaling pathway in vitro and in vivo. Front Pharmacol. 2017;8:929.
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721.
- Hidvegi T, Ewing M, Hale P, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329(5988):229–232.
- Liu H, Mi S, Li Z, et al. Interleukin 17A inhibits autophagy through activation of PIK3CA to interrupt the GSK3B-mediated degradation of BCL2 in lung epithelial cells. Autophagy. 2013;9(5):730–742.
- Mi S, Li Z, Yang HZ, et al. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-beta1-dependent and -independent mechanisms. J Iimmunol. 2011;187(6):3003–3014. .
- Zhang XW, Zhou JC, Peng D, et al. Disrupting the TRIB3-SQSTM1 interaction reduces liver fibrosis by restoring autophagy and suppressing exosome-mediated HSC activation. Autophagy. 2020;16(5):782–796.
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27(1):107–132.
- Tekirdag KA, Korkmaz G, Ozturk DG, et al. MIR181A regulates starvation- and rapamycin-induced autophagy through targeting of ATG5. Autophagy. 2013;9(3):374–385.
- Szak ST, Mays D, Pietenpol JA. Kinetics of p53 binding to promoter sites in vivo. Mol Cell Biol. 2001;21(10):3375–3386.
- Yang Q, Wang S, Xie Y, et al. Effect of salvianolic acid B and paeonol on blood lipid metabolism and hemorrheology in myocardial ischemia rabbits induced by pituitruin. Int J Mol Sci. 2010;11(10):3696–3704.
- Wang YF, Hsu YJ, Wu HF, et al. Endothelium-derived 5-methoxytryptophan is a circulating anti-inflammatory molecule that blocks systemic inflammation. Circ Res. 2016;119(2):222–236. .
- Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14(2):207–215.
- Cianfanelli V, Fuoco C, Lorente M, et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol. 2015;17(1):20–30.
- Lam KK, Zheng X, Forestieri R, et al. Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PLoS Pathog. 2012;8(5):e1002691. .
- Phadwal K, Watson AS, Simon AK. Tightrope act: autophagy in stem cell renewal, differentiation, proliferation, and aging. Cell Mol Life Sci. 2013;70(1):89–103.
- Hernandez-Gea V, Friedman SL. Autophagy fuels tissue fibrogenesis. Autophagy. 2012;8(5):849–850.
- Thoen LF, Guimaraes EL, Dolle L, et al. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55(6):1353–1360.
- Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014;39(4):159–169.
- Kumazoe M, Takai M, Bae J, et al. FOXO3 is essential for CD44 expression in pancreatic cancer cells. Oncogene. 2017;36(19):2643–2654. .
- O’Neill BT, Lee KY, Klaus K, et al. Insulin and IGF-1 receptors regulate FoxO-mediated signaling in muscle proteostasis. J Clin Invest. 2016;126(9):3433–3446.
- Kim DH, Park MH, Lee EK, et al. The roles of FoxOs in modulation of aging by calorie restriction. Biogerontology. 2015;16(1):1–14.
- Page A, Paoli PP, Hill SJ, et al. Alcohol directly stimulates epigenetic modifications in hepatic stellate cells. J Hepatol. 2015;62(2):388–397.
- Amabile G, Di Ruscio A, Muller F, et al. Dissecting the role of aberrant DNA methylation in human leukaemia. Nat Commun. 2015;6(1):7091.
- Park SJ, Sohn HY, Yoon J, et al. Down-regulation of FoxO-dependent c-FLIP expression mediates TRAIL-induced apoptosis in activated hepatic stellate cells. Cell Signal. 2009;21(10):1495–1503.
- Jiang X, Tsitsiou E, Herrick SE, et al. MicroRNAs and the regulation of fibrosis. Febs J. 2010;277(9):2015–2021.
- Liang C, Bu S, Fan X. Suppressive effect of microRNA-29b on hepatic stellate cell activation and its crosstalk with TGF-beta1/Smad3. Cell Biochem Funct. 2016;34(5):326–333.
- Yan G, Li B, Xin X, et al. MicroRNA-34a promotes hepatic stellate cell activation via targeting ACSL1. Med Sci Monit. 2015;21:3008–3015.
- Zhang Z, Zha Y, Hu W, et al. The autoregulatory feedback loop of microRNA-21/programmed cell death protein 4/activation protein-1 (MiR-21/PDCD4/AP-1) as a driving force for hepatic fibrosis development. J Biol Chem. 2013;288(52):37082–37093.
- Zhao J, Tang N, Wu K, et al. MiR-21 simultaneously regulates ERK1 signaling in HSC activation and hepatocyte EMT in hepatic fibrosis. PloS One. 2014;9(10):e108005.