
ABSTRACT
The cyclin D-CDK4/6 complex has two distinct functions. Its kinase-dependent role involves its ability to act as serine/threonine kinase, responsible for phosphorylation of substrates required for cell cycle transitions, while its kinase-independent function involves its ability to act as a reservoir for p27Kip1. This association sequesters p27 from cyclin E-CDK2 complexes, allowing them to remain active. The aim of this current study is two-fold: to understand the contribution of the kinase-dependent and kinase-independent functions of CDK4 and CDK6 in epithelial cells and to directly compare CDK4 and CDK6 in a simple model system, TGF-β treatment, where arrest is initiated by the expression of p15Ink4b. Cells that overexpressed a catalytically inactive, p15-insensitive CDK6 variant (p27 sequestration only mutant) were able to overcome TGF-β-mediated arrest by maintaining CDK2 activity, while cells expressing the identical mutations in CDK4 were not. This result can be partially explained by the presence of a previously unidentified cyclin D-CDK6 dimer, which serves as a sink for free p27 during TGF-β treatment, enabling CDK2 to remain inhibitor free. The use of the TGF-β model system and the characterization of CDK pool dynamics and p27 switching is relevant to the CDK4/6 specific inhibitors, such as palbociclib, whose mechanism of action may resemble that of p15.
Introduction
The mammalian cell cycle is an ordered process where the entire genome of a cell is duplicated and then distributed equally between two daughter cells. The progression between cell cycle phases is regulated by the cyclin-CDKs, where cyclin D-associated CDK4 or CDK6 starts the cycle, causing release from G0 [Citation1].The presence of mitogens leads to the synthesis of the D-type cyclins, which partner with either CDK4 or CDK6. The cyclin D-CDK4 or cyclin D-CDK6 complexes phosphorylate substrates, like the retinoblastoma (Rb) protein, which then allows the transcription of cyclin E. Cyclin E-CDK2 complexes fully phosphorylate RB and complete the G1-S phase transition. Antiproliferative responses, such as growth to confluence or TGF-β treatment, cause inhibition of cyclin D-CDK4/6 complexes by the induction and association of cyclin-dependent kinase inhibitors (CKIs), such as p15Ink4b, p27Kip1, or p21 Cip1.
The association of p27 with cyclin D-CDK4 is not straightforward, as it binds to this complex in two different confirmations: an active, ON conformation, where p27 is phosphorylated on tyrosine 74, 88 or 89 and alternatively an inactive, OFF conformation, where p27 is not tyrosine phosphorylated [Citation2–5]. p27 is required to assemble and stabilize cyclin D-CDK4, as the dimeric complex is inherently unstable and is not detected in vivo. Thus, p27 assembles the complex, but its phosphorylation serves as a “switch” required to turn the assembled, but inactive cyclin D-CDK4 complex on. The three-dimensional structure of the p27-cyclin D-CDK4 ternary complex confirms that p27 functions like a door: when it is tyrosine phosphorylated (pY), the door is open and CDK4 is on, but when it is not phosphorylated, the door is closed and CDK4 is off [Citation2,Citation3].
However, in addition to cyclin D-CDK4’s ability to phosphorylate substrates like Rb, its association with p27 serves an important secondary function. The cyclin D-CDK4/6 complex acts as an essential reservoir for p27, sequestering it away from CDK2, as all association of p27 with CDK2 is inhibitory [Citation2].The “switching” of p27 from CDK4 complexes to CDK2 complexes is a potent cell cycle inhibitory mechanism. For example, TGF-β treatment of epithelial cells rapidly causes the transcriptional induction of p15, which binds exclusively to CDK4/6, inhibiting catalytic activity, but p15 also displaces p27 from CDK4, allowing free p27 to bind to and inhibit CDK2 [Citation6]. This coordinated inhibition of both CDK4/6 and CDK2 is required for TGF-β-mediated arrest and is likely involved in the growth arrest of a variety of other inhibitors, including CDK4 small molecule inhibitors (CDK4i), such as palbociclib.
Palbociclib has roughly 100-fold more inhibitory activity toward CDK4 and CDK6 than CDK2 [Citation7,Citation8]. Recent studies have suggested that palbociclib is only associated with the monomeric, non-active form of CDK4 [Citation3]. While it initially seemed surprising that a kinase inhibitor does not associate with the active form of its cognate kinase, this suggests that palbociclib may function more like p15. The creation of palbociclib-CDK4 dimers reduces the formation of the active p27-cyclin D-CDK4 complex, releasing p27 to inhibit CDK2. Thus, to achieve growth arrest, CDK4i may mimic TGF-β, and cause p27 redistribution as a way to inhibit both CDK4 and CDK2 kinases.
Overexpression of cyclin D, CDK4 or CDK6 might cause cells to become resistant to TGF-β mediated arrest and this has been demonstrated for CDK4 expression in Mv1Lu (mink lung epithelial) cells [Citation9,Citation10]. This also suggests that the same might be true for palbociclib: increases in cyclin D, CDK4 or CDK6 would increase cyclin D-associated catalytic activity and the level of the cyclin D-CDK4/6 sink [Citation5]. While overexpression of cyclin D and CDK6 has been detected clinically in CDK4i resistant breast cancer patients [Citation7,Citation8], overexpression of CDK4 has not. In this study, we sought to understand the contribution of the kinase and sequestration functions of CDK4/6 in epithelial cells, by separating them using specific, well-characterized CDK4 and CDK6 mutants. TGF-β treatment of Mv1Lu cells has been well characterized biochemically and by gel filtration [Citation10] for p27-cyclin D-CDK4 pool formation and dissolution and thus provides a clear model system to follow the catalytic and sequestration requirements of cyclin D-CDK4/6. We found that a catalytically inactive, sequestration-only mutant of CDK6 was able to rescue TGF-β-arrested cells by sequestering p27 to maintain CDK2 activity, while the homologous CDK4 mutant was not. Gel filtration analysis of these cells revealed the presence of a previously unidentified cyclin D-CDK6 dimer, which enables the titration of p27, promoting CDK2 activity and proliferation.
Materials and methods
Plasmids: Constructs encoding human HA-CDK4WT, HA-CDK6WT, HA-CDK4D158N/R24C, CDK6D163N/R31C were generated by direct cloning into pUHD 10–3 hygromycin or using PCR mutagenesis as described [Citation11].
Cell Culture: Mv1Lu, Mv1Lu-tTA cells [Citation11] and their derivatives were cultured in Minimal Essential Media supplemented with 10% fetal bovine serum. Media for the tetracycline-regulated cell lines also contained 2 µg/ml tetracycline, 500 µg/ml G418, and 300 µg/ml hygromycin. Tet-regulated lines expressing the CDK4 and CDK6 variants were generated by transfecting tTA cells with 25 µg of the appropriate DNA using Lipofectin (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. Individual colonies generated during selection with hygromycin were grown in the absence of tetracycline and screened for exogenous protein expression. To test the effects of exogenous protein expression, the cell lines were grown in the presence of tetracycline (referred to in each figure as the OFF condition) or in the absence of tetracycline (referred to in each figure as the ON condition). Where indicated, the cells were treated with 100 pM TGF-β (R&D Systems, Minneapolis, MN) for 24 hours. For cell cycle analysis, cells were harvested and analyzed by flow cytometry as previously described [Citation12].
Antibodies: The antibodies used in this study were as follows: anti-HA (Y-11), anti-CDK4 (C-22), anti-cyclin E (C-19), anti-cyclin D1 (M-20 and H-295), anti-cyclin D2 (C-17), anti-p27 (M-197, M-20 and C-19), and anti-CDK6 (C-21) (all from Santa Cruz, Santa Cruz, CA), anti-cyclin D1 (DCS-6) and anti-cyclin D2 (DCS-11) (both from Biosource), anti-p27 (C-57), and anti-mouse p27, anti-mink CDK4, anti-mouse CDK4 and anti-mouse CDK2 (rabbit polyclonal, generous gifts from Dr Joan Massagué), anti-CDK6 (DCS-90) anti-β actin (both from Sigma, St. Louis, MO), protein A-HRP (Millipore, Billerica, MA), goat anti-mouse-HRP (Bio-Rad, Hercules, CA), goat anti-mouse (Southern Biotech), and goat anti-rabbit (Jackson Labs).
Immunoblot Analysis, Immunoprecipitations, and Kinase Assays: For immunoblot analysis, cells were lysed in TNT buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100) supplemented with protease inhibitor cocktail V (Calbiochem, San Diego, CA) and 1 mM phenylmethylsulfonyl fluoride (PMSF). For all immunoprecipitations, cells were lysed in Tween-20 buffer as previously described [Citation7]. All immunoprecipitations, immunoblot analysis, and in vitro kinase assays using Rb or histone H1 as a substrate were performed as previously described [Citation12].
Gel Filtration: Lysates were separated by size-exclusion chromatography (gel filtration) as previously described [Citation12]. Cells were lysed in a Tween-20 buffer and the lysates were cleared by sonication followed by ultracentrifugation at 50,000 rpm. 4.5 mg of extract was fractionated on a Superdex 200 10/30 column (GE-Amersham) and fractions #15-42 were collected from each run. Fractions were either run on SDS-PAGE gel and immunblotted as described, or immunoprecipitated followed by immunoblot analysis.
Results
Expression of sequestration-only CDK6, but not CDK4, allows cells to overcome TGF-β- mediated growth arrest.
Mv1Lu cells expressing variants of human CDK4 (Tet-CDK4WT, Tet-CDK4D158N/R24C) or human CDK6 (Tet-CDK6WT, and Tet-CDK6D163N/R31C), in a tetracycline-repressible manner (,), were generated to assess the requirement for CDK4 and CDK6 kinase and kinase-independent activities during TGF-β treatment. The D158N and D163N mutations specifically target the catalytic core of CDK4 and CDK6, respectively, creating kinase inactive proteins that retain their ability to bind to cyclin D, p27, and p15 [Citation13–15]. The R24C and R31C mutation do not affect catalytic activity, still permit p27 binding, but prevent binding and inhibition by Ink4 proteins (p15, p16, p18, p19) [Citation13,Citation16]. These mutations were identified in familial melanoma patients and specifically act as a p15-resistant form of CDK4 or CDK6 in our system. Therefore, the double mutants, CDK4D158N/R24C and CDK6D163N/R31C lack catalytic activity and cannot be inhibited by p15, but retain the ability to bind p27 [Citation13]. These are p27 sequestration-only mutants, and in these cells treated with TGF-β, we predicted that all CDK4 and CDK6 kinase activity, derived from the endogenous proteins, would be inhibited by p15 expression.
Figure 1. Expression of sequestration-only CDK6, but not CDK4, allows cells to overcome TGF-β- mediated growth arrest. (a, b). Tet-CDK4WT and Tet-CDK4D158N/R24C cells (a) or Tet-CDK6WT and Tet-CDK6D163N/R31C cells (b) were grown for 20 h + (OFF) or – (ON) Tet, before lysates were analyzed by immunoblot using anti-CDK4 or anti-CDK6 antibodies. (c, d) Tet-CDK4WT and Tet-CDK4D158N/R24C cells grown OFF or ON and treated ± TGF-β (c) or Tet-CDK6WT and Tet-CDK6D163N/R31C cells grown OFF or ON and treated ± TGF-β (d) were analyzed for DNA content by flow cytometry. The percentage of cells in S/G2/M in each line treated + Tet/- TGF-β was set to 100%. The results shown are representative of three independent experiments
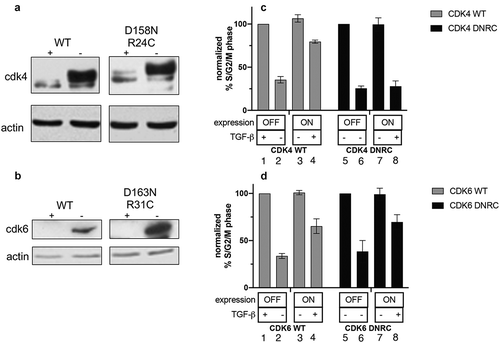
To confirm the expression of the exogenous proteins, each line was grown in the presence of tetracycline (Tet) (the OFF condition, no exogenous protein expression) or in the absence of Tet (the ON condition, exogenous protein expressed) for 24 h. and the resulting lysates were subjected to immunoblot analysis (, b). Cell lines were grown in the OFF or ON conditions for 24 h. and TGF-β was added for an additional 24 h. before the cells were harvested for cell cycle analysis (). The percentage of cycling cells (S/G2/M content) of the OFF/-TGF-β condition was set to 100%. TGF-β arrested these cells, resulting in a reduced S/G2/M content (). Expression of CDK4WT or CDK6WT partially rescued this arrest (, D, lanes 4). The CDK6D163N/R31C cells were also able to overcome TGF-β-mediated growth arrest (, lane 8). As CDK6D163N/R31C cannot bind p15 and is catalytically inactive [Citation17], its only function should be to sequester p27. In contrast, the expression of the CDK4D158N/R24C mutant did not rescue TGF-β-mediated growth arrest (, lane 8).
Expression of CDK6D163N/R31C rescues CDK2 activity in the presence of TGF-β
Because the CDK6D163N/R31C mutant cannot bind p15, endogenous CDK4, and CDK6 kinase activity should be inhibited by p15 expression in the presence of TGF-β. As the CDK6D163N/R31C mutant is catalytically inactive, we hypothesized that these cells were proliferating due to preservation of CDK2 catalytic activity. To address this, we examined the expression of cell cycle-related proteins and their activity in the double mutant cells in the presence and absence of TGF-β by immunoblot and immunoprecipitation analysis (). In the ON condition, CDK4D158N/R24C or CDK6D163N/R31C were expressed (, CDK4, CDK6, lanes 3, 4, 7, 8). The level of cyclin D1 does not significantly change when either CDK4D158N/R24C or CDK6D163N/R31C was induced (, cyclin D1). p27 levels increased in the presence of TGF-β (, p27).
Figure 2. Expression of CDK6D163N/R31C rescues CDK2 activity in the presence of TGF-β Tet-CDK4D158N/R24C and Tet-CDK6D163N/R31C cells were grown in the presence (OFF) or absence of Tet (ON) and treated ± TGF-β for 24 h. Lysates from untreated Tet-CDK4D158N/R24C (lanes 1,2) and Tet- CDK6D168N/R31C (lanes 5, 6) and TGF-β treated Tet-CDK4D158N/R24C (lanes 3, 4) and Tet-CDK6D168N/R31C (lanes 7,8) cells were immunoblotted for cyclin D1 (row 1), CDK4 (row 2), CDK6 (row 3), CDK2 (row 4) and p27 (row 5). The same extracts were also immunoprecipitated with anti-cyclin D1 and anti-p27 antibodies and immunoblotted with anti-CDK4 (rows 6,7) or anti-CDK6 antibodies (rows 8,9). Lysates incubated with normal rabbit serum (NRS) were included as a control. A sample from each of the same lysates was immunoprecipitated with anti-CDK2 antibodies (row 10) or anti-p27 antibodies (row 11) and used in Rb in vitro kinase assays. The results shown are representative of three independent experiments
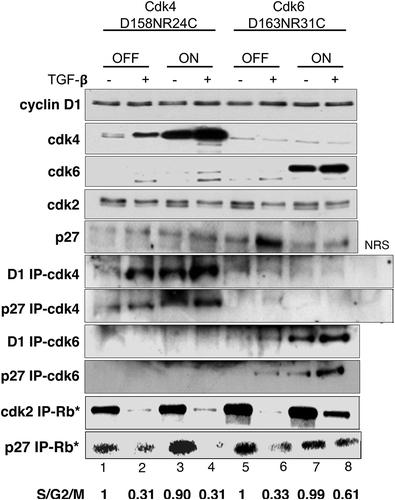
Increased levels of CDK4 or CDK6 were associated with cyclin D1 in the ON conditions, as detected by cyclin D1 immunoprecipitation (, D1 IP-CDK4, D1 IP-CDK6). More CDK4 or CDK6 was associated with p27 in the ON conditions as detected by p27 immunoprecipitation (, p27 IP-CDK4, p27 IP-CDK6). This suggested that the expression of the exogenous CDK4D158N/R24C or CDK6D163N/R31C increased ternary complexes formation.
p27 was immunoprecipitated from lysates and 32P-ATP and recombinant Rb was added to perform in vitro kinase assays (, p27 IP-RB*). We used p27-associated kinase activity as a surrogate marker for CDK4 and CDK6 activity, as previously demonstrated [Citation12]. When TGF-β was added to uninduced cells, p27 kinase activity was decreased, consistent with p15’s disruption of CDK4 and CDK6 complexes (, p27 IP-RB*, lanes 2, 6). When TGF-β was added to the cells in the presence of CDK6D163N/R31C or CDK4D158N/R24C, p27 kinase activity was also decreased, although the low activity remained in the CDK6D163N/R31C expressing cells (, p27 IP-RB*, lanes 4, 8). As the CDK4 and CDK6 mutants cannot bind p15, this suggested that the endogenous catalytically active CDK4 and CDK6 were inhibited by the binding of p15.
CDK2 is frequently detected as a doublet due to an artifact of gel electrophoresis, with the faster migrating band signifying the active CDK2 form [Citation18]. When the uninduced cells are treated with TGF-β, the fast migrating form disappears and is chased into the slower migrating inactive form (, CDK2, lanes 2, 6). When CDK4D158N/R24C is expressed in the presence of TGF-β, only the slower migrating inactive form is detected, consistent with the growth arrest seen in these conditions (, CDK2, lane 4). However, when CDK6D163N/R31C is expressed in the presence of TGF-β, the faster migrating form persists, suggesting that CDK2 is still active (, CDK2, lane 8). This was directly confirmed using a CDK2 in vitro kinase assay, where CDK2 was immunoprecipitated followed by the addition of 32P-ATP and recombinant Rb (, CDK2 IP-Rb*). Whereas the CDK2-associated kinase activity is lost in the presence of TGF-β in the CDK4D158N/R24C cells (, CDK2 IP-Rb*, lane 4), CDK2 kinase activity is still detected in the CDK6D163N/R31C cells (, CDK2 IP-Rb*, lane 8), suggesting that at least some CDK2 kinase activity persisted.
CDK6 can form a dimer with cyclins D1/D2
Gel filtration chromatography allowed us to analyze the distribution of CDK4, CDK6 and associated proteins in the lysates from untreated and TGF-β treated cells (). Equal mg of lysate was separated by size on the gel filtration column. Next, equal volume of each fraction was immunoprecipitated and/or immunoblotted in all panels shown. Due to the nature of this technique, the band intensity cannot be compared between the different panels, only within each panel, except for , panels 8 and 13 which were transferred and developed on the same membrane .
Figure 3. Endogenous CDK6 exists in a dimer with cyclins D1/D2. Tet-CDK6WT cells were grown in the presence of Tet (OFF) and, where indicated, treated with TGF-β. Lysates from these cells were separated by gel filtration and the resulting fractions were subjected to direct immunoblot analysis or immunoprecipitation-immunoblot analysis. Fractions from Tet-CDK6WT cells grown in the presence of Tet (CDK6 OFF) were subjected to direct immunoblot analysis using anti-CDK4 (row 1), anti-CDK6 (row 2) and anti-p27 (row 3) antibodies. Fractions were immunoprecipitated with anti-cyclin D1 and anti- cyclin D2 antibodies and the resulting complexes were analyzed by immunoblotting with anti-CDK4 (row 4), anti-CDK6 (row 5), anti-cyclin D1/anti-cyclin D2 (row 6) and anti-p27 (row 7) antibodies. Fractions from parental Mv1Lu cells were either immunoprecipitated with anti-cyclin D1/cyclin D2 antibodies and immunoblotted for CDK6 (row 8) or immunoprecipitated with anti-CDK6 antibodies and used in a Rb in vitro kinase assay (row 9). Fractions from TGF-β treated parental Mv1Lu cells were subjected to cyclin D1 immunoprecipitation and used in an anti-CDK6 immunoblot (row 13). Lysates TGF-β treated Tet- CDK6WT cells (OFF) were separated by gel filtration and the resulting fractions were processed by direct immunoblot analysis using anti-CDK4 (row 10), anti-CDK6 (row 11) and anti-p27 (row 12) antibodies
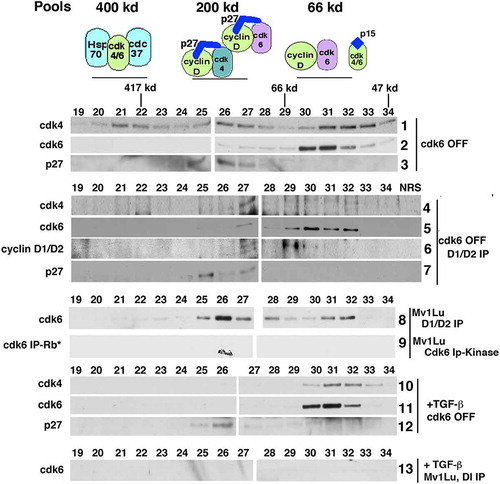
In Tet-CDK6WT cells grown in the OFF condition, CDK4 was present equally in three distinct pools, which have been described previously: 400 kD, 200 kD, and 66 kD (, panel 1, fractions 20–22, 25–27 and 30–33, respectively) [Citation10,Citation12,Citation17,Citation19–21]. The 400 kD pool contains CDK4 complexed with molecular chaperones, such as cdc37 and Hsp 90. The 200 kD pool contains cyclin D-CDK4-p27 ternary complexes. In continuously proliferating cells, this is the only complex that has CDK4-, cyclin D- or p27-associated kinase activity [Citation12]. The 66 kD pool contains monomeric, catalytically inactive CDK4 or CDK4 associated with any residual INK4 proteins, such as p15 [Citation10,Citation12]. These cannot be separated on this column. p27 is not detected as a monomer in epithelial cells, and it is detected at 200 KD (, panel 3), where it was previously shown to be associated entirely with CDK4, CDK6, or CDK2 complexes [Citation10,Citation12].
CDK6, on the other hand, partitioned into only two complexes (, panel 2). While there was a low but detectable 200 kD complex, the majority of CDK6 was detected at 66 kD.
To further examine this complex, we co-immunoprecipitated cyclin D1/cyclin D2 complexes from the Tet-CDK6WT cells grown in the OFF condition, followed by CDK4, CDK6, cyclin D1/D2 and p27 immunoblot analysis (, panels 4–7). Mv1Lu cells do not express cyclin D3 [Citation10]. As previously demonstrated, cyclin D1-CDK4 complexes are found only in the 200 kD pool and contain p27, (, panel 4, fractions 25–27) [Citation10,Citation12,Citation19]. This was consistent with observations that cyclin D cannot complex with CDK4 in the absence of p27 or another assembly factor [Citation12]. In contrast, we found that CDK6 was bound to cyclins D1/D2 in the 200 kD pool (, panel 5, fractions 25–27) and in the 66 kD pool (, panel 5, fractions 30–32). This suggests that CDK6 was able to dimerize with cyclin D in the absence of p27. p27- associated cyclin D1/D2 complexes were only detected in the 200kD complexes (, panel 7, fractions 25–27), consistent with previous studies [Citation10,Citation12]. The presence of a putative cyclin D-CDK6 dimer in our system distinguishes CDK6 from CDK4, which is not detected in a dimeric form. Additionally, we found that CDK6 formed dimers with both cyclin D1 and cyclin D2 individually (data not shown). The presence of the cyclin D-CDK6 dimer was confirmed in parental Mv1Lu cells (, panel 8), as well as in other epithelial cell types (MCF10A and HaCaT, data not shown). However, this cyclin D-CDK6 dimer had no catalytic activity when immunoprecipitated from parental Mv1Lu lysate fractions and used in an in vitro kinase assay.
Catalytic activity was only detected in the 200 KD fraction (, panel 9).
When epithelial cells are treated with TGF-β, which induces the expression of p15, the cyclin D-CDK4-p27 complex is dissociated and CDK4 is only detected in the 66Kd pool. A similar result was seen when the CDK6WT OFF cells were treated with TGF-β: CDK4 and CDK6 are only detected in the lower molecular weight pool (, panel 10–11) [Citation10,Citation12,Citation17,Citation19–21]. p27 continues to be detected at 200 kD, presumably now associated with CDK2, as previously described [Citation10] (, panel 12). When TGF-β-treated lysates were immunoprecipitated with cyclin D1/D2 antibodies before being subject to gel filtration, all cyclin D-CDK6 (200 kD and 66kD) were lost (, compare panels 8 and 13). Thus, the induction of p15 by TGF-β destroys the 200 kD p27 cyclin D-CDK6 complexes and the 66 kD cyclin D-CDK6 dimer.
Overexpressed CDK6 also exists in a dimer with cyclins D1/D2 and can provide a reservoir to titrate p15 during TGF-β treatment.
Gel filtration analysis was used to further understand the role of CDK6 in cyclin D pool dynamics. Lysates from CDK6WT cells grown in the absence of tetracycline to induce transgene expression were subjected to gel filtration chromatography and immunoblot analysis. As expected, an increase in CDK6 was detected (, panel 2). p27 continued to be detected only at 200 kD (, panel 3), and the previously described three pools of CDK4 were maintained (, panel 1). When cyclin D1/D2 were immunoprecipitated from these induced cells (, panels 5–8), CDK6 was detected in both the 200 kD p27-associated pool and the 66kD CDK6, p27-free dimeric pool (, panel 6). CDK4 pool distribution was not disrupted (, panel
Figure 4. Overexpressed CDK6 exists in a dimer with cyclins D1/D2 and can provide a reservoir to titrate p15 during TGF-β treatment. Tet-CDK6WT cells were grown in the absence of Tet (ON) and lysates were separated by gel filtration chromatography (rows 1–8). The resulting fractions were subjected to direct immunoblot analysis with anti-CDK4 (row 1), anti-CDK6 (row 2) and anti-p27 (row 3) and anti-cyclin D1 (row 4) antibodies. Fractions were immunoprecipitated with anti-cyclin D1 and anti-cyclin D2 antibodies and analyzed by immunoblotting with anti-CDK4 (row 5), anti-CDK6 (row 6), anti-cyclin D1/anti-cyclin D2 (row 7) and anti-p27 (row 8) antibodies. Fractions from TGF-β treated Tet-CDK6WT cell (ON) lysates were separated by gel filtration and immunoprecipitated with anti-cyclin D1 and anti-cyclin D2 antibodies. The resulting immunoblots were probed with anti-CDK4 (row 9), anti- CDK6 (row 10) and anti-p27 (row 11) antibodies. Fractions from Tet-CDK6D168N/R31C cell lysates treated ±TGF- β were also separated by gel filtration and the resulting fractions were immunoprecipitated with anti-cyclin D1/anti-cyclin D2 antibodies, followed by immunoblot analysis with anti-CDK6 antibodies (rows 12, 13)
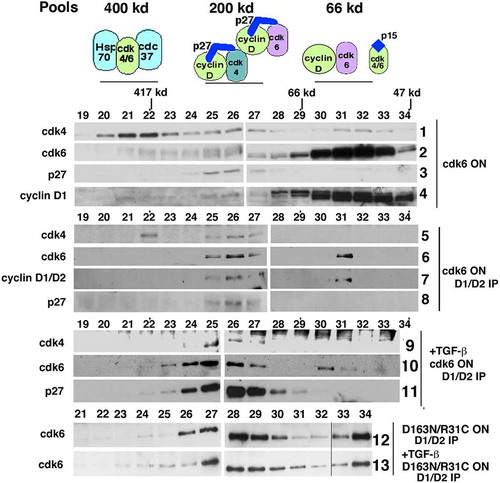
5). As shown previously, CDK6 ON cells are not arrested by TGF-β treatment (, panel 4). Lysates from these cells were separated by gel filtration and immunoprecipitated with cyclin D1 and D2 antibodies to look for associated proteins (, panels 9–11), CDK4 was detected in the 200 kD pool, suggesting that it was able to continue to exist in a p27-associated complex, even in the presence of p15 (, panel 9). CDK6 was detected in the 200 kD p27-associated pool and as a cyclin D-CDK6 dimer (, panel 10). Additionally, cyclins D1/D2 remain bound to p27 (. panel 11). In uninduced cells, p27 is no longer associated with cyclin D in the presence of TGF-β [Citation11]. However, in the presence of the CDK6 transgene, p27 remains associated with cyclin D, suggesting that p27 switching to CDK2 has not occurred and active CDK4 and CDK6 complexes persisted, accounting for the ability of these cells to overcome TGF-β-mediated arrest (, panel 4).
CDK6D163N/R31C ON cells are also not arrested by TGF-β treatment, even though the transgene expressed has no catalytic activity and is unable to bind p15 (, lane 8). We assumed this was because CDK2 remained active in these conditions (, CDK2 IP-Rb*, lane 8). When lysates from CDK6D163N/R31C ON cells treated -/+ TGF-β were analyzed by gel filtration followed by cyclin D1/D2 immunoprecipitation and immunoblot analysis (, panels 12, 13), both CDK6 pools were still detected. Because the CDK6D163N/R31C protein is not able to bind to p15, this TGF-β-induced p15 must be sequestered by the endogenous CDK4 and CDK6 proteins. However, the CDK6D163N/R31C protein still binds to p27, resulting in the persistence of the 200 kD cyclin D-CDK6-p27 ternary complex. This would result in maintaining p27-free CDK2 complexes and continued CDK2 activity.
Discussion
Our study demonstrates that non-catalytic, Ink4-insensitive CDK6 is able to sequester p27, maintain CDK2 activity, and rescue TGF-β-induced growth arrest, while the homologous CDK4 variant did not. While possible that subtle differences in the expression level of CDK6 relative to CDK4 may contribute, we believe that the ability of CDK6 to partition into an inactive dimer with cyclin D, available to titrate CDK inhibitors p27 or p15, may account for these differences. While we and others have not detected a similar CDK4-cyclin D dimer in epithelial cells even in the presence of overexpressed CDK4, our observations that a cyclin D-CDK6 dimer can exist in vivo is consistent with observations that inherent structural and stability differences exist between CDK4 and CDK6 [Citation22]. This supports previous data that suggest that cyclin D-CDK4 complexes are unstable and inactive without p21 or p27 to act as a stabilizing factor [Citation3,Citation12]. In contrast, CDK6, with its larger cyclin D interface domain, has been readily crystallized and studied in association with a Kaposi Sarcoma viral cyclin V, p16, p18 and p19 [Citation23]. However, we find that this dimer is catalytically inactive, consistent with data that suggest that p27’s association and subsequent tyrosine (Y) phosphorylation are required to convert the complex into an active form.
We chose to examine the roles of cyclin D-CDK4/6 during TGF-β treatment as a means to understand the role of p27 sequestration in the absence of kinase activity. Our system mimics the action of the small-molecule CDK4 inhibitor, palbociclib, where its mechanism seems dependent at least in part on the disassembly of the cyclin D-CDK4/6 complex [Citation3]. TGF-β treatment of mink epithelial cells is a well-characterized biological system, where p27-cyclin D-CDK4/6 pool dynamics and activity are disrupted solely by the binding of p15 [Citation10,Citation11]. We used this system to model CDK pool dynamics and p27 redistribution, as it has particular relevance in the design of targeted therapies against G1 cell cycle components, such as CDK4/6 and CDK2. The coupling and inhibition of CDK4/6 and CDK2 is directly linked to p27, and adaptive resistance mechanisms frequently feedback to p27 regulation. For example, increases in cyclin D expression in response to palbociclib in pancreatic ductal adenocarcinoma [Citation24] or CDK4i-resistant melanoma cells [Citation25] will increase the net amount of CDK4/6 activity, but again will also result in the redistribution of p27 into newly formed cyclin D-CDK4/6 complexes. In breast cancer, the overexpression of CDK6 as a result of amplification or by loss of the tumor suppressor FAT, is also a CDK4i resistance mechanism [Citation26]. While this may increase the net amount of CDK6 kinase activity to evade pharmacological levels of palbociclib, it would also cause p27 redistribution from cyclin E-CDK2 to cyclin D-CDK6, resulting in maintained CDK2 activity.
Thus, the simplistic view might be that these inhibitors merely target CDK4/6 kinase activity, but the ability to cause p27 redistribution to inhibit CDK2 may be the factor upon which success depends.
In epithelial cells, CDK4 is the dominant kinase and Ink4 proteins are only expressed under specific conditions [Citation27]. Conversely, in hematopoietic cells CDK6 is the dominant kinase and Ink4 proteins are constitutively expressed. In these cells, the cyclin D-CDK6 dimer was not detected [Citation17]. We posit that in epithelial cells, the pool distribution of the cyclin-CDK complexes is dynamic and dependent upon the intracellular concentration of the different components (Ink4s, Cip/Kips, cyclin D, CDK4, and CDK6). In these cells, where Ink4s are normally not expressed, CDK6 may be required to sequester cyclin D in an inactive pool until needed. This appears to be a CDK6-specific phenomenon, which may have implications in those cancer cells where CDK6 is overexpressed or Ink4 expression is lost. Overexpression of CDK4 has not been detected as a CDK4i resistance mechanism in breast cancer to date, consistent with the idea that
these kinases are not truly redundant, especially in terms of tissue distribution and structure and now the ability to form a cyclin D-CDK6 dimeric sink.
Our data also suggest that epithelial cells are able to proliferate in the absence of significant cyclin D-associated kinase activity, provided that CDK2 kinase activity was maintained, which is again consistent with the drug resistance that develops in the presence of extended CDK4 inhibition [Citation28]. Previous attempts to probe the requirements for CDK4 and CDK6 have used knockout or knockdown strategies, which would reduce both the catalytic and sequestration functions of these kinases. For example, loss of cyclin D1 in knockout animals causes the loss of both the catalytic and sequestration functions of CDK4 and CDK6 [Citation29]. These animals have neurological impairments, abnormal retinal development and decreased mammary epithelial cell proliferation during pregnancy. These phenotypes can be rescued by the expression of the cyclin D1K112E allele, which binds to CDK4 or CDK6 but cannot induce catalytic activity [Citation30,Citation31]. Cyclin D1-CDK4 complexes isolated from the retina of these knock-in mice retain the ability to bind p27, suggesting that it is the kinase-independent functions that are involved in the development of this tissue. In contrast, MMTV-ErbB-2 mice do not develop breast cancer when crossed with cyclin D1K112E/K112E animals, suggesting that catalytic activity is specifically involved in tumor formation. This current work supports the idea that as we develop additionally targeted therapies, it will be important to further understand the differences between CDK4 and CDK6 and ensure that drugs target both their kinase dependent and independent roles.
Acknowledgments
We wish to thank Drs. Sander van den Heuvel and Charles Sherr for the kind gifts of CDK4 and CDK6 plasmids and Dr. Joan Massagué for the kind gift of antibodies. We wish to thank Dina Leznova, Dr. Ting-Chung Suen, Dr. Nuria Ferrandiz-Diaz, Sadia Bhuyan, Jared Macklin and Irfan Azam for technical assistance.
Disclosure statement
SWB is the co-founder and equity holder in Concarlo Holdings, LLC. There are no competing interests with this current work.
Additional information
Funding
References
- Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 2016;6(4):353–367.
- Blain SW. Switching cyclin D-Cdk4 kinase activity on and off. Cell Cycle. 2008;7(7):892–898.
- Guiley KZ, Stevenson JW, Lou K, et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science. 2019;366(6471):6471.
- Patel P, Asbach B, Shteyn E, et al. Brk/protein tyrosine kinase 6 phosphorylates p27KIP1, regulating the activity of cyclin D-cyclin-dependent kinase 4. Mol Cell Biol. 2015;35(9):1506–1522. .
- Sherr CJ. Surprising regulation of cell cycle entry. Science. 2019;366(6471):1315–1316.
- Zhang Y, Alexander PB, Xf W. TGF-β family signaling in the control of cell proliferation and survival. Cold Spring Harb Perspect Biol. 2017;9(4):a022145.
- Ingham M, Schwartz GK. Cell-cycle therapeutics come of age. J Clin Oncol. 2017;35(25):2949–2959.
- Guarducci C, Bonechi M, Boccalini G, et al. Mechanisms of resistance to CDK4/6 inhibitors in breast cancer and potential biomarkers of response. Breast Care (Basel). 2017;12(5):304–308. .
- Ewen ME, Sluss HK, Whitehouse LL, et al. TGFß inhibition of CDK4 synthesis is linked to cell cycle arrest. Cell. 1993;74(6):1009–1020.
- Warner BJ, Blain SW, Seoane J, et al. Myc downregulation by transforming growth factor beta required for activation of the p15(Ink4b) G(1) arrest pathway.. Mol Cell Biol. 1999;19(9):5913–5922.
- Reynisdottir I, Polyak K, Iavarone A, et al. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev. 1995;9(15):1831–1845.
- James MK, Ray A, Leznova D, et al. Differential modification of p27Kip1 controls its cyclin D-cdk4 inhibitory activity. Mol Cell Biol. 2008;28(1):498–510.
- Grossel MJ, Baker GL, Hinds PW. cdk6 can shorten G(1) phase dependent upon the N-terminal INK4 interaction domain. J Biol Chem. 1999;274(42):29960–29967.
- Matsushime H, Ewen ME, Strom DK, et al. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71(2):323–334. .
- Van Den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262(5142):2050–2054.
- Wölfel T, Hauer M, Schneider M, et al. A p16INK4A-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in human melanoma.. Science. 1995;269(5228):1281–1284. .
- Parry D, Mahony D, Wills K, et al. CyclinD-cdk subunit arrangement is dependent on the availability of competing Ink4 and p21 class inhibitors. Mol Cell Biol. 1999;19:1775–1783.
- Gu Y, Rosenblatt J, Morgan DO. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 And Tyr15. Embo J. 1992;11(11):3995–4005.
- Mahony D, Parrry DA, Active EL. CDK6 complexes are predominantly nuclear and represent only a minority of the CDK6 in T cells. Oncogene. 1998;16(5):603–11.
- McConnell BB, Gregory FJ, Stott FJ, et al. Induced expression of p16Ink4a inhibits both CDK4- and CDK2-associated kinase activity by reassortment of cyclin-cdk inhibitor complexes.. Mol Cell Biol. 1999;19(3):1981–1989.
- Stepanova L, Leng X X, Sg P, et al. Mammalian p50cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes CDK4. Genes Dev. 1996;10(12):1491–1502.
- Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15(6):122.
- Wood DJ, Endicott JA. Structural insights into the functional diversity of the CDK-cyclin family. Open Biol. 2018;8(9):180112.
- Knudsen ES, Kumarasamy V, Ruiz A, et al. Cell cycle plasticity driven by MTOR signaling: integral resistance to CDK4/6 inhibition in patient-derived models of pancreatic cancer. Oncogene. 2019 May;38(18):3355–3370
- Katlinskaya YV, Katlinski KV, Yu Q, et al. Suppression of type I interferon signaling overcomes oncogene-induced senescence and mediates melanoma development and progression. Cell Rep. 2016;15(1):171–180. .
- Li Z, Razavi P, Li Q, et al. Loss of the FAT1 tumor suppressor promotes resistance to cdk4/6 inhibitors via the hippo pathway. Cancer Cell. 2018;34(6):893–905 e8. .
- Tsutsui T, Hesabi B, Moons DS, et al. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(kip1) activity. Mol Cell Biol. 1999;19(10):7011–7019.
- Álvarez-Fernández M, Malumbres M. Mechanisms of sensitivity and resistance to CDK4/6 inhibition. Cancer Cell. 2020;37(4):514–529.
- Sicinski P, Donaher JL, Parker SB, et al. Cyclin D1 provides a link between development and oncogenesis in the retina and breast.. Cell. 1995;82(4):621–630. .
- Yu Q, Sicinska E, Geng Y, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9(1):23–32. .
- Landis WM, Pawlyk SB, Li T, et al. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell. 2006;9(1):13–22.