
ABSTRACT
WT1 has been reported to function as an oncogene and a tumor suppressor in acute myeloid leukemia (AML). The molecular mechanisms have not yet been fully elucidated. Here, we report that p53, served as a tumor suppressor, plays a critical role in regulating the function of WT1 in AML. For details, we performed a meta-analysis on 1131 AML cases, showing that WT1 gene mutation and TP53 gene exhibited a mutually exclusive predisposition in AML. p53 can be recruited to the promoter region of WT1’s target genes to modulate their expression by physically interacting with WT1. The AML-derived p53 mutation (p53R248Q) can disrupt the interaction between WT1 and p53, resulting in the loss of modulation of WT1’s target genes. Furthermore, wild-type p53 maintained the anti-proliferation activity of WT1 in AML cells. In contrast, WT1 promoted AML cell proliferation in the absence of p53 (or mutated p53). In conclusion, we demonstrated a novel explanation of the controversial function of WT1 in AML. These results provided a mechanism by which WT1 inhibited AML cell proliferation in a p53-dependent manner.
KEYWORDS:
Introduction
Acute myeloid leukemia (AML) is a rapidly progressing blood malignancy characterized by unlimited proliferation of myeloid cells [Citation1]. To further delve into the pathogenesis of AML, we focused on its genetic characterization. Current genetic studies have reported multiple molecular mutations in AML patients, including the WT1 (Wilms’ tumor 1) and TP53. WT1 is a transcription factor that regulates genes involved in nephrogenesis and hematopoiesis [Citation2–5]. Previous studies have demonstrated that WT1 was mutated in ∼6% to 15% of AML and functioned as a tumor suppressor [Citation6–8]. However, WT1 was also reported to be overexpressed in AML patients and contributed to leukemogenesis, suggesting WT1 is an oncogene [Citation9]. These controversial findings hinder the further understanding of AML pathogeny [Citation10]. WT1 has previously been demonstrated to interact with p53 [Citation11], a tumor suppressor encoded by TP53 gene in humans. TP53 mutations occur in more than half of cancer and the frequency of TP53 mutation in AML is reported 5–10% (9% in TCGA) [Citation12]. When we studied the WT1 and TP53 mutations in AML, we noted that WT1 mutation and TP53 mutation performed a mutually exclusive predisposition in AML, prompting us to hypothesize that a shared functional role for WT1 and p53 in AML. Our study demonstrated that p53 maintains the tumor suppressor activity of WT1 through interaction with WT1. When TP53 mutated, WT1 would turn out to be an oncogene contributing to leukemogenesis.
Results
P53 modulates WT1-target gene expression
To investigate the mutation landscape of WT1 and TP53 genes in AML, we performed a meta-analysis of 1131 AML cases with detailed high throughput sequencing data obtained from http://www.cbioportal.org (OHSU, Nature 2018; TCGA, NEJM 2013; TCGA, PanCancer Atlas; TCGA, Provisional). One hundred and eighty-two AML cases (16.1%) carried at least one gene mutation, including 84 WT1-mutated cases (7.4%) and 100 p53-mutated cases (8.8%) ()). Notably, only two cases (0.177%) carried both gene mutations, suggesting WT1 mutation and TP53 mutation exhibited a mutually exclusive predisposition in AML (P= 0.0491). Given the bioinformatics analysis above, we speculated a shared functional role for WT1 and p53 in AML.
Figure 1. P53 modulates WT1-target gene expression. (a) Somatic variants in WT1 and p53 were identified in a total of 1131 AML cases, in which 184 cases carried at least one mutation; (b) p53 was overexpressed in HEK293T cells, and the mRNA expression of indicated WT1-target genes was determined by quantitative real-time PCR; (c) (left) HEK293T cells were transduced with retrovirus expressing different shRNAs against WT1, and the WT1 knockdown efficiency was verified by both qRT-PCR and western blot. (right) Overexpression of p53 in HL-60 cells with or without WT1 knockdown. HL-60 cells were transduced with retrovirus expressing different shRNAs against WT1 and retrovirus expressing Flag-tagged full-length p53. The expression of WT1 and p53 proteins was determined by western blot; (d) and (e) p53 was overexpressed in HEK293T cells as treated in Figure 1(c) (left), and the mRNA expression of indicated WT1-target genes was determined by qRT-PCR; (f) HCT116 (p53−/-) and HCT116 (p53+/+) cells were transfected with WT1, and the mRNA expression of indicated WT1-target genes was determined by qRT-PCR; (g) Stable HL-60 cells were generated as described in Figure 1(c) (right), and the mRNA expression of WT1-target genes was determined by quantitative real-time PCR. Shown are average values of triplicated results with SD; *p < 0.05; **p < 0.01; ***p < 0.001 for the indicated comparison
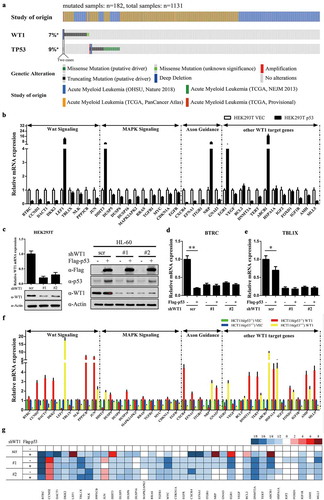
To determine this speculation, we first examined whether p53 is involved in WT1-regulated gene expression. qRT-PCR results demonstrated that overexpressed p53 saliently altered the expression profiles of WT1-regulated genes in HEK293T cells ()). Furthermore, p53-mediated modulation of WT1-regulated genes was abolished (i.e. BTRC and TBL1X, two known direct WT1-target genes [Citation13], ) when WT1 was knocked down by two shRNAs targeting WT1 in HEK293T cells () left). Next, HCT116 (p53−/-) and HCT116 (p53+/+) cells were transfected with WT1 expression vector, respectively. We found that the most upregulated target genes of WT1 in HCT116 (p53−/-) were downregulated in HCT116 (p53+/+), and vice versa ()).
To verify that this mechanism existed in AML, HL-60 cells were transfected with Flag-p53 expressing vectors () right). p53 overexpression indeed changed the expression profiles of WT1-regulated genes in HL-60 cells ()). Once WT1 was depleted by shRNAs, the p53-mediated alteration of WT1-regulated gene expression was abolished ()). In all, p53 takes part in modulating WT1-regulated genes.
P53 directly binds to WT1
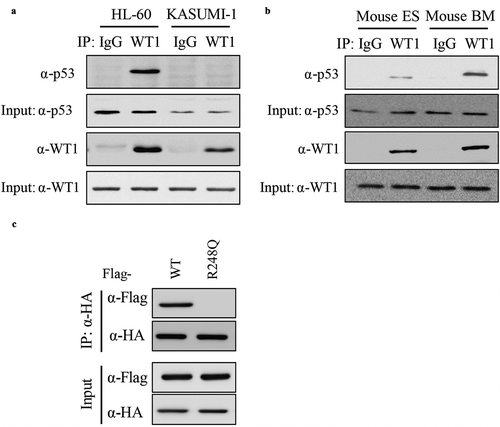
To investigate the molecular mechanism that p53 modulated WT1-regulated gene, IP-western was performed to determine the endogenous WT1-p53 interaction in HL-60, KASUMI-1 ()), mouse embryonic stem cells (ESCs) and primary bone marrow cells (BMCs) ()). These results demonstrated that the WT1-p53 interaction was readily detected in HL-60 cells, ESCs, and BMCs, but not in p53-mutated KASUMI-1 cells, suggesting that the interaction between p53 and WT1 indeed exists and could be disrupted by p53 mutation. To determine the binding sites of WT1 with p53, HA-WT1 was ectopically expressed with Flag-p53 (FL) or Flag-p53R248Q in HEK293T cells. We found that WT1 binds to full-length p53, but not with p53R248Q ()). Collectively, these data demonstrate that p53 directly binds to WT1 and AML-derived p53R248Q mutation disrupts its binding to WT1.
Figure 2. P53 directly binds to WT1. (a) Endogenous WT1 protein was immunoprecipitated from two human AML cell lines (i.e. HL-60 and KASUMI-1), followed by western blot to detect p53. Normal rabbit IgG was used as a negative control; (b) Endogenous WT1 protein was immunoprecipitated from mouse ESCs or BMCs cells, followed by western blot to detect p53. Normal rabbit IgG was used as a negative control; (c) Wild-type p53 and p53R248Q were co-expressed with WT1 in HEK293T cells. Protein–protein interaction was examined by IP-western using the indicated antibodies
WT1 recruits p53 to the promoter region of its target genes
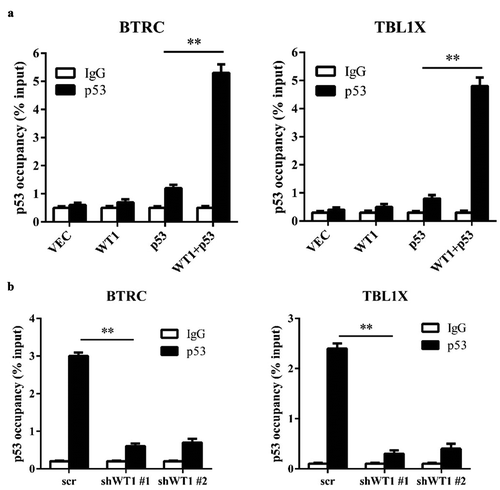
To verify how p53 and WT1 collaboratively regulated WT1-target genes, ChIP-qPCR was performed in HEK293T cells. It was demonstrated that ectopically expressed p53 could bind to the promoter region of WT1-regulated genes, and the binding was substantially increased when WT1 was co-expressed in HEK293T cells ()). We conducted the same experiment in HL-60 cells. Consistently, we found that p53 was significantly enriched in the promoters of BTRC and TBL1X ()). Moreover, the p53 occupancy on the promotors of BTRC and TBL1X was significantly decreased when endogenous WT1 was knocked down in HL-60 cells ()). In sum, the results above show that WT1 recruits p53 to the promoter region of its target genes.
Figure 3. WT1 recruits p53 to its target genes promoter region. (a) p53 was transiently expressed either singularly or with WT1 in HEK293T cells. The occupancy of p53 at the promoter regions of WT1-target genes was determined by ChIP-qPCR. Mouse IgG was included as negative controls for ChIP-qPCR; (b) HL-60 cells were transduced with retrovirus expressing different shRNAs against WT1. The occupancy of p53 on the promoter regions of indicated WT1-target genes was determined by ChIP-qPCR. Mouse IgG was included as a negative control. Shown are average values of triplicated results with SD; *p < 0.05; **p < 0.01; ***p < 0.001 for the indicated comparison
WT1 Inhibits AML cell proliferation in a p53-dependent manner
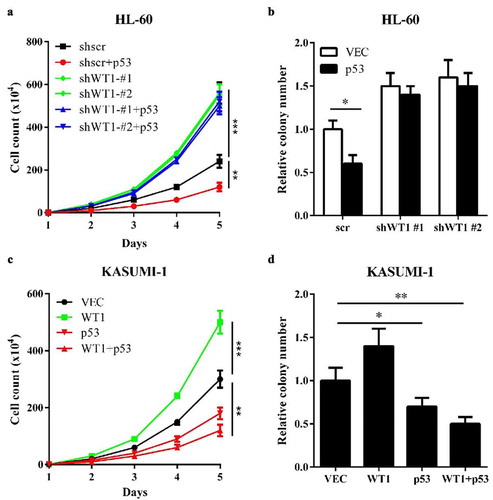
Based on the above results, we investigated the impact of WT1-p53 interaction on AML. First, we overexpressed p53 in HL-60 cells and examined the effect of p53 on cell proliferation and colony formation. The results indicated that p53 inhibited HL-60 cells proliferation ()) and reduced HL-60 cells colony formation ()) compared to control. When WT1 was knocked down in HL-60, the inhibitory effects were abolished ()). Then, we performed the same experiments in p53-mutated KASUMI-1 cells. The overexpression of WT1 promoted proliferation ()) and increased colony formation ()) in KASUMI-1 cells, suggesting that WT1 is a pro-proliferation protein in p53-mutated AML cells. Interestingly, wild-type p53, co-overexpression with WT1 into KASUMI-1 cells, substantially inhibited KASUMI-1 cells proliferation and colony formation. Collectively, the experiments demonstrate that the molecular status of p53 (wild-type or mutated) significantly influences the role of WT1 on AML cell proliferation.
Figure 4. WT1 inhibits AML cell proliferation in a p53-dependent Manner. (a) and (b) Cell proliferation (a) and colony formation (b) of stable HL-60 cells overexpressing full-length p53 with or without WT1 knockdown were determined by cell counting and colony-forming assay, respectively; (c) and (d). KASUMI-1 cells were transiently expressed with WT1, or p53, or both, and cell proliferation (c) and colony formation (d) were determined by cell counting and colony-forming assay, respectively. Shown are average values of triplicated results with SD; *p < 0.05; **p < 0.01; ***p < 0.001 for the indicated comparison
High-frequency, AML-derived, WT1-binding-defective p53R248Q fails to inhibit AML cell proliferation
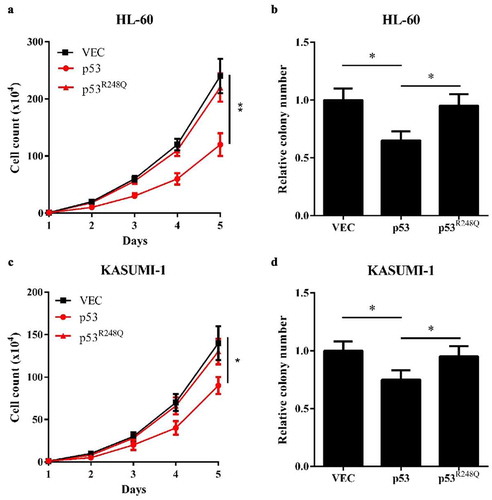
Finally, we screened out a high-frequency AML-derived mutated p53, p53R248Q, which lost the binding activity to WT1. The biological experiments showed that overexpression of wild-type p53, but not p53R248Q in HL-60 cells decreased cell proliferation and reduced colony formation ()). Similar results were displayed in KASUMI-1 cells ()). Taken together, our results suggest that the inhibitory effects of WT1 on AML cell proliferation is p53-dependent and that the AML-derived, WT1-binding-defective p53 mutation loses the inhibitory effects on WT1.
Figure 5. AML-derived, WT1-binding-defective p53R248Q fails to inhibit AML cell proliferation. (a) and (b) Cell proliferation (a) and colony formation (b) of HL-60 cells overexpressing wild-type p53 or p53 (R248Q) were determined by cell counting and colony-forming assay, respectively; (c) and (d) Cell proliferation (c) and colony formation (d) of KASUMI-1 cells overexpressing wild-type p53 or p53 (R248Q) were determined by cell counting and colony-forming assay, respectively. Shown are average values of triplicated results with SD; *p < 0.05; **p < 0.01; ***p < 0.001 for the indicated comparison
Discussion
WT1 was first identified as a predisposition gene for Wilms’ tumor [Citation14] with loss-of-function mutations in ∼15% of sporadic Wilms’ tumor cases [Citation15]. In hematologic malignancies, WT1 is mutated in ∼6% to 15% of cases of de novo AML [Citation6–8]. Meanwhile, it is also demonstrated that WT1 is overexpressed in AML and implicates in leukemogenesis [Citation16]. This is a particular challenge in AML, in which WT1 has been shown to function as an oncogene and a tumor suppressor [Citation2,Citation8,Citation17,Citation18].
Here, we demonstrated a novel understanding that p53 is involved in the WT1’s functional changes in AML. p53 is a well-known tumor suppressor in AML [Citation19]. In our study, we found that p53 was recruited to the promoter region of WT1-regulated genes by directly binding to WT1. These results demonstrated that once WT1 was not mutated, the status of p53 (wild-type or mutated) would direct the regulation of WT1-targeted genes. Hence, reports of overexpressed WT1 functioned as an oncogene may be the indirect influence of p53 mutation.
Further study shows that the binding of p53 to these WT1-regulated genes depends on the presence of WT1 protein and the binding of p53 to WT1-regulated gene is functional. The inhibitory effects of WT1 on AML cell proliferation and colony formation need the presence of p53. When wild-type p53 is replaced with mutated p53 that disrupt the interaction with WT1, WT1 will turn into an oncogene that contributes to leukemogenesis.
Except for AML, WT1 and p53 genes are also reported to mutate in many other cancers, such as hepatocellular carcinoma [Citation20,Citation21], gastrointestinal stromal tumor [Citation22], breast invasive carcinoma [Citation23], lung adenocarcinoma [Citation24], and prostate cancer [Citation25]. Thus, it is possible that the WT1/p53 complex is involved not only in AML but also in other types of cancer.
In conclusion, this study provided a novel understanding of the mutual exclusivity of WT1 and p53 mutations and explored the potential mechanism of p53 in maintaining the anti-proliferation activity of WT1 in AML, which was of great significance for improving the diagnosis and treatment of AML.
Materials and methods
Plasmids construction
The full-length cDNA of human WT1 was cloned into pcDNA3-HA. The full-length cDNA of human p53 was cloned into pcDNA3-Flag. Point mutations were generated as described previously [Citation26] by site-directed mutagenesis using a QuickMutation™ Site-Directed Mutagenesis Kit (Beyotime). Plasmids for shRNA expression vectors, pMKO.1-puro and pMKO.1-hyg, were purchased from Addgene and pMKO.1-puro-shWT1 and pMKO.1-hyg-shp53 were constructed. All plasmids were verified by DNA sequencing.
Cell culture and transfection
HEK293T was maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) in the presence of penicillin, streptomycin, and 8 mM L-glutamine (Invitrogen). HCT116 (p53−/-) and HCT116 (p53+/+) cells were maintained in McCoy’s 5A Medium (Gibco) supplemented with 10% FBS in the presence of penicillin, streptomycin. ES-E14TG2a from mouse embryo (blastocyst) were purchased from Sigma (08021401). Moreover, HL-60 and KASUMI-1 (p53-mutated) cells were maintained in RPMI-1640 medium supplemented with 20% FBS, 2 mmol/L L-glutamine, 100 U/mL penicillin/streptomycin. Plasmid transfection was carried out using lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
RNA isolation and quantitative real-time PCR
Total RNA was isolated from cultured cells using Trizol reagent (Invitrogen) following the manufacturer’s instructions. RNA was reverse transcribed and preceded by real-time PCR with gene-specific primers (TaKaRa). β-actin was used as control. Primer sequences are presented in Table S1.
Immunoprecipitation and western blotting
Cells were lysed in ice-cold NP-40 buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.1% NP-40] containing protease inhibitor cocktail (PIC, Roche). Immunoprecipitation was carried out either by incubating Flag/HA beads (Sigma) at 4°C with lysate for 3 h or by incubating appropriate antibody with cell lysate for 1 h, followed by incubating with Protein-A beads (Upstate) for another 2 h at 4°C before beads were washed for three times with ice-cold NP-40 buffer. Standard western blot protocols were performed. Antibodies against p53 (ab1101, Abcam), WT1 (sc-7385X, Santa Cruz), HA (sc-7392X, Santa Cruz), Flag (14,793, CST), mouse IgG (Santa Cruz), and rabbit IgG (Santa Cruz) were purchased commercially.
Cell proliferation array
Cell proliferation was measured using Cell Counting Kit-8 (CCK-8; Beyotime) assays. Briefly, HL-60 or KASUMI-1 cells were seeded in 96-well plates, 4 wells for each group, with 3–5 × 104 cells. At 0, 24, 48, 72, 96 h, 10 μl of CCK-8 reagent was added to each well, and the cells were incubated for 1 h at 37°C. Finally, the absorbance at 450 nm was measured using the ELx808™ Absorbance Microplate Reader (BioTek).
Colony formation assay
HL-60 or KASUMI-1 cells were plated (1000 cells per 35 mm dish) in methylcellulose-based MethoCult medium (Stem Cell Technologies). Culture dishes were incubated at 37°C in a humid atmosphere with 5% CO2. 6 days later, colonies (>50 μm diameter) were counted using ImageJ software.
Generation of stable WT1 and/or p53 knockdown cell pools
pMKO.1-puro-shWT1 and pMKO.1-hyg-shp53 were constructed to generate stable WT1 and/or p53 knockdown cell pools. Briefly, the shRNA construct was co-transfected with vectors expressing the gag and vsvg genes into HEK293T cells. The supernatant was harvested 36 h after transfection, and mixed with 8 mg/ml polybrene to increase the infection efficiency. Cells were infected with the retrovirus and selected in 1 mg/ml puromycin or 200 mg/ml hygromycin B for 1 week.
ChIP-qPCR assays
ChIP-qPCR assays were performed as described previously [Citation27]. Briefly, cells were crosslinked with 1% paraformaldehyde and sonicated (Bioruptor UCD-200, Bioruptor). Solubilized chromatin was immunoprecipitated with antibodies against WT1, Flag, HA, or negative control IgG. Antibody-TF-chromatin complexes were pulled down using protein A-sepharose (Millipore), washed, and then eluted. After crosslink reversal and proteinase K treatment, IPed DNA was extracted with phenol-chloroform and ethanol precipitated. The DNA fragments were further analyzed by qPCR.
Statistical analysis
Data were described with mean ± SD. Statistical significance was performed using SPSS 20.0 (SPSS, Chicago, IL, USA). A two-tailed Student’s t-test was used for a significance analysis, in which p < 0.05 was set as the threshold.
Authors’ contributions
PL and LZ conceived and designed the work. YY and XW performed research and collected data. LZ wrote the paper. All authors read and approved the final manuscript.
Supplemental Material
Download Zip (9.6 KB)Acknowledgments
We would like to thank the authors of ‘WT1 Recruits TET2 to Regulate Its Target Gene Expression and Suppress Leukemia Cell Proliferation (Molecular Cell, 2015)’ for the advice of experimental design.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- Rose D, Haferlach T, Schnittger S, et al. Subtype-specific patterns of molecular mutations in acute myeloid leukemia. Leukemia. 2017;31:11–17.
- Yang L, Han Y, Suarez Saiz F, et al. A tumor suppressor and oncogene: the WT1 story. Leukemia. 2007;21:868–876.
- Rivera MN, Haber DA. Wilms’ tumour: connecting tumorigenesis and organ development in the kidney. Nat Rev Cancer. 2005;5:699–712.
- Huff V. Wilms’ tumours: about tumour suppressor genes, an oncogene and a chameleon gene. Nat Rev Cancer. 2011;11:111–121.
- Pronier E, Bowman RL, Ahn J, et al. Genetic and epigenetic evolution as a contributor to WT1-mutant leukemogenesis. Blood. 2018;132:1265–1278.
- Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278.
- Cancer Genome Atlas Research N, Ley TJ, Miller C, et al.. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074.
- Becker H, Marcucci G, Maharry K, et al. Mutations of the Wilms tumor 1 gene (WT1) in older patients with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood. 2010;116:788–792.
- Hosen N, Shirakata T, Nishida S, et al. The Wilms’ tumor gene WT1-GFP knock-in mouse reveals the dynamic regulation of WT1 expression in normal and leukemic hematopoiesis. Leukemia. 2007;21:1783–1791.
- Rampal R, Alkalin A, Madzo J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014;9(5):1841–1855.
- Zhan Q, Chen IT, Antinore MJ, et al. Tumor suppressor p53 can participate in transcriptional induction of the GADD45 promoter in the absence of direct DNA binding. Mol Cell Biol. 1998;18:2768–2778.
- Quintas-Cardama A, Hu C, Qutub A, et al. p53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia. 2017;31:1296–1305.
- Kim MK, McGarry TJ, Broin PO, et al. An integrated genome screen identifies the Wnt signaling pathway as a major target of WT1. Proc Natl Acad Sci U S A. 2009;106:11154–11159.
- Call KM, Glaser T, Ito CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus. Cell. 1990;60(3):509–520.
- Charlton J, Pritchard-Jones K. WT1 mutation in childhood cancer. Methods Mol Biol. 2016;1467:1–14.
- Miwa H, Beran M, Saunders GF. Expression of the Wilms’ tumor gene (WT1) in human leukemias. Leukemia. 1992;6:405–409.
- Hou HA, Huang TC, Lin LI, et al. WT1 mutation in 470 adult patients with acute myeloid leukemia: stability during disease evolution and implication of its incorporation into a survival scoring system. Blood. 2010;115:5222–5231.
- Ellisen LW, Carlesso N, Cheng T, et al. The Wilms tumor suppressor WT1 directs stage-specific quiescence and differentiation of human hematopoietic progenitor cells. EMBO J. 2001;20:1897–1909.
- Hayashi Y, Goyama S, Liu X, et al. Antitumor immunity augments the therapeutic effects of p53 activation on acute myeloid leukemia. Nat Commun. 2019;10:4869.
- Berasain C, Herrero JI, Garcia-Trevijano ER, et al. Expression of Wilms’ tumor suppressor in the liver with cirrhosis: relation to hepatocyte nuclear factor 4 and hepatocellular function. Hepatology. 2003;38:148–157.
- Qin Q, Lin YW, Zheng XY, et al. RNAa-mediated overexpression of WT1 induces apoptosis in HepG2 cells. World J Surg Oncol. 2012;10:11.
- Kang GH, Kim KM, Noh JH, et al. WT-1 expression in gastrointestinal stromal tumours. Pathology. 2010;42:54–57.
- Craig DW, O’Shaughnessy JA, Kiefer JA, et al. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol Cancer Ther. 2013;12:104–116.
- Xu C, Wu C, Xia Y, et al. WT1 promotes cell proliferation in non-small cell lung cancer cell lines through up-regulating cyclin D1 and p-pRb In Vitro and In Vivo. PLoS One. 2013;8:e68837.
- Brett A, Pandey S, Fraizer G. The Wilms’ tumor gene (WT1) regulates E-cadherin expression and migration of prostate cancer cells. Mol Cancer. 2013;12:3.
- Hartkamp J, Carpenter B, Roberts SG. The Wilms’ tumor suppressor protein WT1 is processed by the serine protease HtrA2/Omi. Mol Cell. 2010;37:159–171.
- Lan F, Collins RE, De Cegli R, et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448:718–722.