
ABSTRACT
Clear cell renal cell carcinoma (CC-RCC) remains one of the most deadly forms of kidney cancer despite recent advancements in targeted therapeutics, including tyrosine kinase and immune checkpoint inhibitors. Unfortunately, these therapies have not been able to show better than a 16% complete response rate. In this study we evaluated a cyclin-dependent kinase inhibitor, Dinaciclib, as a potential new targeted therapeutic for CC-RCC. In vitro, Dinaciclib showed anti-proliferative and pro-apoptotic effects on CC-RCC cell lines in Cell Titer Glo, Crystal Violet, FACS-based cell cycle analysis, and TUNEL assays. Additionally, these responses were accompanied by a reduction in phospho-Rb and pro-survival MCL-1 cell signaling responses, as well as the induction of caspase 3 and PARP cleavage. In vivo, Dinaciclib efficiently inhibited primary tumor growth in an orthotopic, patient-derived xenograft-based CC-RCC mouse model. Importantly, Dinaciclib targeted both CD105+ cancer stem cells (CSCs) and CD105− non-CSCs in vivo. Moreover, normal cell lines, as well as a CC-RCC cell line with re-expressed von-Hippel Lindau (VHL) tumor suppressor gene, were protected from Dinaciclib-induced cytotoxicity when not actively dividing, indicating an effective therapeutic window due to synthetic lethality of Dinaciclib treatment with VHL loss. Thus, Dinaciclib represents a novel potential therapeutic for CC-RCC.
Introduction
Clear cell renal cell carcinoma (CC-RCC) is the most common and deadliest form of kidney cancer, accounting for around 70–80% of the projected 13,780 kidney cancer deaths in the US this year [Citation1]. CC-RCC typically involves the loss of function of the von-Hippel Lindau (VHL) tumor suppressor gene, leading to the upregulation of hypoxia-inducible factors (HIFs) and receptor tyrosine kinase (RTK) signaling (reviewed in [Citation2]). Accordingly, RTK pathway inhibitors combined with immune checkpoint inhibitors, represent the current standard first-line treatments for CC-RCC (reviewed in [Citation3]). While the percentage of patients responding to treatment is up to 71%, only 8–16% achieve complete responses [Citation3]. As a result, the 5-year survival rate of metastatic CC-RCC remains dismal (13.9%) [Citation1], making the development of novel targeted therapies a significant unmet medical need.
Cyclin-dependent kinases (CDKs) are a family of serine/threonine kinases that act as master regulators of the cell cycle and transcription (reviewed in [Citation4] and [Citation5], respectively). CDK4 and CDK6 initiate phosphorylation of the retinoblastoma associated protein 1 (Rb), which is enhanced by CDK2. Rb phosphorylation releases E2F transcription factors, which control expression of genes required for entry into the S phase of the cell cycle. CDK1 acts at a later stage of the cell cycle and is responsible for G2/M progression. CDK3 and CDK5 were shown to regulate cell cycle in some cell systems, but their functions remain elusive. CDKs 7, 9, 12, and 13, do not directly affect the cell cycle, and instead promote RNA polymerase II-dependent transcription.
Since almost all tumors have defects in the regulation of cell cycle and proliferation, CDKs have long been attractive targets for cancer therapy with many CDK inhibitors developed and tested (reviewed in [Citation6]). However, the use of CDK inhibitors in the clinic, to date, have had variable success, ranging from the failure of broad spectrum CDK inhibitors – Flavopiridol and Roscovitine, to FDA approval of CDK4/6 inhibitor – Palbociclib – for treatment of hormone-receptor positive breast cancer in combination with hormone therapy. Multiple selective CDK inhibitors targeting cell cycle- and/or transcription-regulating CDKs are currently being developed and tested. So far, only a few CDK inhibitors were assessed as potential therapeutics for CC-RCC. Flavopiridol was tested in advanced CC-RCC in a Phase II clinical trial with no [Citation7] and modest [Citation8] anti-tumor activity and notable normal tissue toxicity. Recently, two studies have shown that CDK 4/6 inhibitors – Ribociclib [Citation9] and Palbociclib [Citation10] – had an anti-proliferative and pro-apoptotic effect on CC-RCC in vitro and in vivo, as single agents or in combination with chemotherapy [Citation9], immunotherapy [Citation9], or a HIF2 inhibitor [Citation10]. In addition, two studies showed synthetic lethality of CDK4/6 inhibition [Citation10] or CDK4 inhibition [Citation11] with VHL deficiency, indicating that such inhibition would selectively target CC-RCC and provide a therapeutic window.
Importantly, in addition to CDK4, CDK6, and their partner – cyclin D1 – being dysregulated in CC-RCC [Citation12,Citation13], CDK1 [Citation14,Citation15], CDK2 [Citation14,Citation16], and CDK9 [Citation17] appear to be dysregulated, providing rationale for their evaluation as potential therapeutic targets. Adding to this rationale are the studies using genetic and pharmacologic inhibition of CDK1 [Citation18,Citation19] and CDK9 [Citation20–22] to effectively target multiple types of c-Myc-driven cancers in vivo. The c-Myc pathway is activated in most CC-RCC tumors either through genomic amplification of chromosome 8q.24 [Citation23,Citation24], or through the loss of function of the VHL tumor suppressor, leading to stabilization of HIF2α (positive regulator of c-Myc) [Citation25,Citation26], making CC-RCC a candidate cancer for testing CDK1 and CDK9 inhibitors.
Accordingly, in this study, we sought to evaluate a CDK inhibitor Dinaciclib (also known as MK-7965 and SCH727965) as a potential therapeutic for CC-RCC treatment, as it is simultaneously capable of inhibition of CDK 4/6 (critical for survival of VHL-deficient tumors) and CDK1/9 (critical for survival of tumors relying on c-Myc activation). Initially identified as a CDK 1, 2, 5, and 9 inhibitor [Citation27] and further shown to block CDK 3, 4, 6 and 12 [Citation28,Citation29], Dinaciclib was shown to be potent and effective in preclinical models, alone and in combination with other agents [Citation27,Citation29–34]. While Dinaciclib has been mostly ineffective as a monotherapy in clinical trials for previously treated advanced breast cancer [Citation35] and non-small cell lung cancer [Citation36], it has shown anti-tumor activity in relapsed multiple myeloma [Citation37] and leukemia [Citation38] with manageable normal tissue toxicity, warranting its further testing in other cancer types.
The cancer stem cell (CSC) hypothesis postulates that a certain population of tumor cells has tumor-initiating potential, while other populations lack this potential (reviewed in [Citation39,Citation40]). CSCs have been reported to be slow cycling and evading therapeutic targeting, thus, driving tumor recurrence, which may cause their resistance to Dinaciclib. However, CC-RCC CSCs have been shown to upregulate c-Myc expression [Citation41,Citation42], which may render them sensitive to CDK1 or CDK9 inhibition [Citation18–22]. Thus, in this study we addressed the effect of Dinaciclib on CSC population in vivo using CD105 as a cell surface marker of CC-RCC CSCs as previously reported [Citation43].
Here we show that Dinaciclib has a potent anti-proliferative and pro-apoptotic effect both in vitro, using CC-RCC cell lines, and in vivo, using an orthotopic patient-derived xenograft (PDX)-based CC-RCC mouse model. Importantly, we identified a therapeutic window for Dinaciclib as normal cell lines expressing VHL and CC-RCC cell lines with re-expressed VHL, appear protected from Dinaciclib-induced cytotoxicity when not actively dividing. We have further shown that Dinaciclib targets both CSCs and non-CSCs. These data support the use of Dinaciclib as a therapeutic for CC-RCC.
Materials and methods
Cell culture and reagents
Cancer cell lines used in this study were grown in Dulbecco’s Modified Eagle’s Medium (DMEM; Genesee Scientific, San Diego, CA, #25-500) (786-O, RCC4, UM-RC2, MDA-MB-231, MCF-7), RPMI 1640 (Thermo Fisher Scientific, Waltham, MA, #A1049101) (TK-10, T-47D), McCoy’s 5A (Genesee Scientific, #25-518) (Caki-1, Caki-2), or Eagle’s Minimum Essential Medium (EMEM; Sigma-Aldrich, St. Louis, MO, #M4655) (MDCK) supplemented with 10% Fetal Bovine Serum (FBS; Genesee Scientific, #25-514) and 1% Penicillin/Streptomycin (P/S; Genesee Scientific, #25-512) in 5% CO2 at 37°C. RPTEC cells were grown in DMEM/F12 medium with 36 ng/mL hydrocortisone (Thermo Fisher Scientific, #SH30261.01, #AC352450010), 5 μg/mL insulin, 5 μg/mL apo-transferrin, 5 ng/mL sodium selenite (Sigma-Aldrich, #I0516-5ML, #T1147-100 MG, #S5261-10 G), 50 ng/mL human EGF (PeproTech, Cranbury, NJ #AF-100-15), 1% P/S, and 100 μg/mL G418 (Selleck Chemicals, Houston, TX, #S3028). Dinaciclib and Q-VD-OPh (MedChem Express, LLC, Monmouth Junction, NJ, #HY-10492, #HY-12305) were diluted in Dimethyl Sulfoxide (DMSO) and serially diluted for each experiment. ABT-263 (BioVision Inc., Milpitas, CA, #2467-5), Staurosporine (BioVision Inc., #1048-01) were diluted in DMSO and used at concentrations indicated in the manuscript.
Cell Titer Glo viability assays
Cells were plated at low density (≤5% confluency) in 96-well tissue culture plates, and serial dilutions of experimental compounds were added the next day. Cell Titer Glo (CTG; Promega, Madison, WI, #G7572) assay was performed according to the manufacturer’s protocol: 4–5 days following the addition of experimental compounds, CTG reagent was added to the plate and luminescence was detected on a Synergy Biotek HT plate reader. For 3D viability assays, cells were embedded in 50% Matrigel (Corning, Glendale, AZ, #356237)/50% culture medium on ice or in Cultrex PathClear Basement Membrane Extract, Type 2 (Thermo Fisher Scientific, Waltham, MA, #3532-005-02) in 96-well plates. The matrix was allowed to harden at 37°C for 30 minutes, and experimental compounds were added in culture medium to the indicated final concentration; 5 days following the addition of experimental compounds, CTG reagent was added.
Western blotting
After treatments, cells were lysed in ice-cold lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM Na4P2O7, 1 mM β-glycerophosphate, 1 mM Na3VO4) supplemented with protease (Fisher Scientific, Pittsburgh, PA, #P1-88266) and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO, #04906845001), and Western blot was performed as previously described [Citation44]. Membranes were incubated with primary antibodies pRb T821/826 #271930 (1:200), Rb #73598 (1:200), Cleaved Caspase 3 #9661 (1:1,000), Cleaved PARP #5625 (1:1,000), c-Myc #5605 (1:1,000), MCL-1 #4572 (1:1,000) (Cell Signaling Technology, Danvers, MA), VHL #564183 (1:500) (BD Biosciences, San Jose, CA), HIF-2α #NB100122 (1:1,000, Novus Biologicals, Littleton, CO), and β-actin #A5441 (1:5,000, Sigma Aldrich, St. Louis, MO) in 5% BSA/PBS/0.05%Tween20 (PBS-T) overnight at 4°C, and secondary antibody incubation with Goat anti-Rabbit IgG #31460 (1:5,000) (Thermo Fisher Scientific, Waltham, MA) and Goat anti-Mouse IgG #31430 (1:2,000) (Thermo Fisher Scientific, Waltham, MA) in 5% nonfat milk/PBS-T blocking buffer for 1 hour at room temperature (RT). The HRP signal was developed using ECL WB substrate (GE Healthcare, Scottsdale, AZ, USA; #RPN2236). Images were acquired on the Bio-Rad ChemiDoc XRS+ imaging system (BioRad, Hercules, CA).
TUNEL assay
Cells were plated on cover glasses (Fisher Scientific, #22-293232), grown as a monolayer in 5% CO2 at 37°C, and treated with DMSO, 40 nM Dinaciclib and 40 nM Dinaciclib+20uM Q-VD-OPh for 24 h, or 2uM staurosporine for 4 h. Cells were fixed in 4% paraformaldehyde in PBS for 15 min at RT, permeabilized in 0.25% Triton X-100 in PBS for 20 min at RT, and TUNEL analysis was conducted using the Click-iT™ Plus TUNEL Assay (Thermo Fisher Scientific, Waltham, MA, #C10617) following the manufacturer’s protocol. Cover glasses were mounted on microscope slides (VWR, #16004–368) with Vecta shield (Vector Laboratories, #H-1000) and analyzed on Keyence BZ-X800 microscope under 10x magnification.
Cell cycle analysis
Cells were plated and treated as described in the text, then dissociated with TrypLE reagent (Thermo Fisher Scientific, Waltham, MA, #12604021). Cells were centrifuged at 200x g for 5 min, washed twice with PBS, fixed with ice-cold 70% ethanol for 30 min at 4°C, and washed once with PBS. The pellet was resuspended in 25 μg/mL propidium iodide + 10 μg/mL RNase A in 1% BSA/PBS. Cells were analyzed on a BD Fortessa flow cytometer. Dean Jett Fox modeling of cell cycle distribution was performed in FlowJo version 10.
Crystal violet staining
Adherent cells were washed twice with PBS, fixed and stained in 0.1% Crystal Violet solution in 0.3% Acetic Acid and 95% Ethanol for 15 minutes, then washed once with PBS and allowed to air dry.
Plasmids, lentivirus production and infection
The following plasmids were used for virus production: pBabe_HA-VHL (Addgene, Cambridge, MA, #19234) and pFUluc2-EGFP. HEK-293 T cells were transfected with a retroviral/lentiviral plasmid along with packaging plasmids, pVSVG and ΔR8.2 at 1:1:1 ratio using Lipofectamine/Plus (Life Tech, Carlsbad, CA; #18324–012 and #11514–015). Virus collection and infection of target cells were carried out accordingly to previously published protocol [Citation45]. Two to three days post infection transduced 786-O cells were selected in medium supplemented with 1 μg/mL puromycin for 2 weeks. For infection of PDX tumor cells with pFUluc2-EGFP lentivirus, a cell-dissociated freshly harvested tumor was infected with a 1:1 lentivirus:culture medium (DMEM+10%FBS) with 6 μg/mL polybrene on ultra-low adhesion 96-well tissue culture plates under centrifugation at 1,000x g for 90 minutes at 32°C. The cells were then washed twice with PBS and re-implanted in NSG mice subcutaneously for expansion.
Cell dissociation, flow cytometry and cell sorting
Freshly harvested tumors were chopped thoroughly with a razor blade and digested in 2 mg/mL Collagenase Type IV (Sigma Aldrich, St. Louis, MO, #C2139-100 MG) in DMEM/F12 medium (Thermo Fisher Scientific, Waltham, MA, #SH30261.01) in 50 mL tube for 60 min at 37°C on a shaker. The resulting cell suspension was centrifuged for 5 minutes at 400x g, washed once with DMEM/F12, the pellet resuspended in 10 mL of TrypLE, and incubated at 37°C for 10 min. The cells were washed once with DMEM/F12, resuspended in 1 mL DNAse I (20 U/mL Sigma-Aldrich, St. Louis, MO, #4716728001 in DMEM/F12), incubated for 5 min at RT, washed once in DMEM/F12, filtered through a 70 μM cell strainer, washed once in DMEM/F12, and resuspended in FACS buffer (1% BSA in PBS). One million cells were stained in Zombie NiR™ viability dye (1:1,000; BioLegend, San Diego, CA, #423105) for 15 min at RT, washed once in PBS, resuspended in CD105-APC and CD298-PE marking human cells (both 1:40, BioLegend, San Diego, CA, #323207, #341704) antibody mixture in FACS buffer, and incubated for 30 min at 4°C. The cells were then washed once with FACS buffer and analyzed on a BD Fortessa Flow Cytometer (BD Biosciences, San Jose, CA). To separate CD105hi and CD105lo cells, all cells were first gated on CD298-positive cells (cells of human origin) and Zombie NiR-negative cells (viable cells). Then CD105hi cells were gated as the top ~10% of CD105 expressing cells, separated by >1 log fluorescent intensity from the CD105lo population representing the bottom ~20% of cells analyzed. To enrich for pFUluc2-EGFP-labeled cells, after the s.c. tumors formed from pFUluc2-EGFP-infected cells, they were cell dissociated as described above, EGFP+ cells sorted into DMEM containing 10% FBS, washed once in PBS, 2 × 105 cells resuspended in a 1:1 matrigel: PBS mixture, and injected s.c. into NSG mice (NOD.Cg-PrkdcscidIl2-rgtm1Wjl/SzJ, The Jackson Laboratory).
Animal experiments
All animal research in this study was approved by the Institutional Animal Care and Use Committee of the University of California, Irvine. NSG mice were allowed to acclimatize to the animal facility for at least one week after shipment. PDX tumor pieces were obtained from the NIH Patient-Derived Models Repository (PDMR) and passaged sc in NSG mice. Freshly harvested, low-passage tumors labeled with EGFP and luciferase (as described above) were cut to pieces of ~1 mm3 and individually implanted under the renal capsule of 18 6–8 week old male NSG mice in a survival surgery. After 3 weeks, mice were injected i.p. with 150 mg/kg D-Luciferin (GoldBio, St. Louis, MO, #LUCK-1 G) and bioluminescence assessed on a Xenogen IVIS Lumina system. Mice were separated into vehicle and Dinaciclib treatment groups to equally distribute small and large tumors between the groups. Mice were injected i.p. 3 times a week for 4 weeks with 30 mg/kg Dinaciclib or vehicle (20% hydroxypropyl-β-cyclodextrin). Bioluminescent imaging was performed once a week. At sacrifice, tumors were photographed, weighed, and subjected to further analysis including Western blot, and FACS for CD105 expression.
Quantitative RT-PCR
RNA isolation, cDNA synthesis, and QRT-PCR were performed as described in [Citation46]. Amplification was conducted using Power SYBR Green (Thermo Fisher #43-687-06) and 200 nM forward and reverse GAPDH primers (human [5ʹggagcgagatccctccaaaat3’, 5ʹggctgttgtcatacttctcatgg3’] and mouse [5ʹaggtcggtgtgaacggatttg3’, 5ʹggggtcgttgatggcaaca3’]) on an QuantStudio 6 Flex machine (Applied Biosystems).
Immunohistochemistry
Tumors were processed, embedded, sectioned, and H&E stained as previously described [Citation47]. Pictures were taken at 20x magnification using a Keyence BZ-X810 widefield microscope.
TCGA RNA expression analysis
The TCGA CC-RCC (KIRC) dataset was retrieved from the UCSC Xena Browser (PMID:32444850). The IlluminaHiSeq gene expression dataset was downloaded in its normalized format. RNA-Seq values were grouped between matched normal and tumor samples (72 total patients).
TCGA Kaplan overall survival curves
Kaplan overall survival analysis based on CDK expression in CC-RCC TCGA database (KIRC dataset) with Bonferroni p values were generated using R2: Genomics Analysis and visualization Platform (http://r2.amc.nl).
Results
CC-RCC cell lines are sensitive to Dinaciclib treatment
To assess the effect of CDK inhibition on CC-RCC cell proliferation and viability in vitro, we seeded a panel of CC-RCC cell lines at low density to allow for proliferation and treated them with Dinaciclib. Dinaciclib treatment was effective at reducing the proliferation and/or viability of all 5 CC-RCC cell lines tested, measured by ATP content using the Cell Titer Glo (CTG) assay at day 5 ()). We next harvested, plated and treated cells from 3 CC-RCC PDX tumors grown subcutaneously (s.c.) in mice with Dinaciclib. Similar to established CC-RCC cell lines, all 3 PDX-derived tumor cell types were sensitive to Dinaciclib treatment ()). To compare Dinaciclib sensitivity across different tumor types, we tested 3 breast cancer cell lines along with our CC-RCC cells. The triple-negative breast cancer cell-line MDA-MB-231 was sensitive to Dinaciclib, while the two-estrogen receptor (ER)+ cell lines T-47D and MCF-7 were partially and fully resistant, respectively ()). The IC50 values for all CC-RCC cells ranged from 5–16 nM, while breast cancer cells had a wider range of 6 – >40 nM (); no IC50 was reached in MCF-7 cells).
Figure 1. CC-RCC cell lines and cells isolated from PDX tumors are sensitive to Dinaciclib treatment in 2D and 3D cultures in vitro. Both CC-RCC cell lines (a) and CC-RCC cells isolated from PDX tumors (b) are sensitive to Dinaciclib treatment in 2D. (c) MDA-MB-231, T-47D and MCF-7 breast cancer cell lines are sensitive, partially resistant, and fully resistant to Dinaciclib treatment in 2D, respectively. (a-c) Cells were plated in 96-well plates, treated for 5 days with Dinaciclib or vehicle (DMSO), and viable cell numbers assessed by Cell Titer Glo (CTG) assay. (d) Box plot showing average IC50 values grouped by cell type based on (a-c). MCF-7 cells did not reach an IC50 (shown as “+”). (e) 786-O cell line and two CC-RCC cells isolated from PDX tumors are sensitive to Dinaciclib treatment in 3D. Cells were plated in 50%/50% Matrigel: growth medium in ultra-low adhesion 96-well plates; Dinaciclib treatments and CTG assay were conducted as in (a-c). (f) Representative images of organoids from (e). Arrowheads mark abnormal organoids that show characteristics of cell death. Pictures were taken under 10x magnification. The results in a-e were normalized to vehicle-treated cells. n = 3, error bars correspond to SEM.
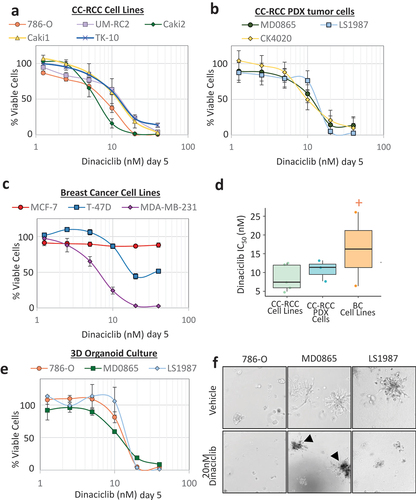
To further test the sensitivity of the PDX tumor-derived cells in a physiologically relevant environment, we set up 3D organoid cultures based on tumor cell suspensions embedded in a 50%/50% mixture of matrigel and growth medium. Two (MD0865, LS1987) out of the 3 PDX tumor cell suspensions formed robust organoid-like structures in 3D along with the 786-O cell line. Cell proliferation/viability of 3D organoids measured by a CTG assay was sharply reduced in the presence of Dinaciclib, with similar IC50s to the 2D culture (). In short, Dinaciclib treatment was effective at reducing the proliferation/viability of all CC-RCC cell lines tested (5 cell lines and 3 PDX cell-dissociated tumors), and was effective in both 2D and 3D cell culture assays.
Dinaciclib reduces proliferation and viability of CC-RCC cells in vitro
To ensure that Dinaciclib effectively inhibited CDK activity, we selected phosphorylated Rb (pRb, mediated by CDK4, 6, and 2) [Citation4], and the expression of MCL-1 (mediated by CDK9) as CDK downstream targets for analysis [Citation20,Citation21]. We treated CC-RCC cell lines with 40 nM Dinaciclib for 24 h and observed an effective decrease of pRb at T821/826 by western blot ()). The ability of Dinaciclib to decrease breast cancer cell line proliferation/viability ()) was directly correlated with its ability to negatively affect Rb phosphorylation. Indeed, in fully Dinaciclib-resistant MCF-7 cells pRb was unchanged, and in partially resistant T-47D cells pRb was only partially reduced ()). The expression of MCL-1, a key pro-survival protein, was decreased by Dinaciclib treatment in all cell lines except MCF-7 cells, which were Dinaciclib-resistant (), )). c-Myc expression was reported to depend on CDK9 activity, first going up (24 h) and then going down (>72 h) after CDK9 inhibition [Citation48]. Our data are consistent with the 24 h short-term CDK9 inhibition ()). These results confirm that Dinaciclib targets pRb and MCL-1 expression and indicate that both proliferation and cell death contribute to Dinaciclib-mediated reduction of viable cell numbers as shown in .
Figure 2. Dinaciclib treatment induces apoptosis and/or cell cycle arrest in Dinaciclib-sensitive cell lines. (a) Dinaciclib treatment consistently decreases Rb phosphorylation (marker of cell cycle progression) and MCL-1 expression (pro-survival marker) in Dinaciclib-sensitive cell lines, and not in Dinaciclib-resistant MCF7. c-Myc expression increases. Dinaciclib treatment increases markers of apoptosis (cleaved PARP and caspase 3) in a subset of cell lines. Cells were plated and treated at 50% confluence with 40 nM Dinaciclib for 24 hours and analyzed by western blot. (b-d) Dinaciclib treatment causes apoptosis in 786-O cells. (b) Dinaciclib-stimulated reduction in viable cell numbers is partially rescued by treatment with pan-caspase inhibitor Q-VD-Oph. 786-O cells were plated in 96-well plates, treated for 5 days with 10–20 nM Dinaciclib or vehicle (DMSO) with or without 20uM Q-VD-Oph, and viable cell numbers were assessed by a CTG assay. The results were normalized to vehicle-treated cells. n = 3, error bars correspond to SEM. ***p < 0.001. (c) Dinaciclib-stimulated DNA fragmentation is rescued by treatment with Q-VD-Oph. 786-O cells were plated in 24-well plates, treated for 24 h with 20 nM Dinaciclib or vehicle (DMSO) with or without 20uM Q-VD-Oph, and TUNEL apoptosis assay conducted. Treatment with Staurosporine was used as a positive control. (d) Western blot showing that Dinaciclib treatment increases cleaved PARP and caspase 3 in 786-O cells, which is reversed by treatment with Q-VD-Oph. Treatment with a pro-apoptotic compound ABT-263 was used as a positive control. Cells were plated and treated as in (a). (e) Dinaciclib treatment causes a decrease in the S phase population in all Dinaciclib-sensitive cell lines and an increase in sub-G1 apoptotic population and G2 population in 786-O, Caki2, and MDA-MB-231 cell lines. Cells were treated with 40 nM Dinaciclib for 24 hours, stained with PI and analyzed by FACS. Cell cycle distributions were calculated using Dean Jett Fox model on FlowJo version 10. Average percentage of cells in each cell cycle phase is shown. FACS analysis for each cell line was repeated 2–3 times, and error bars represent SEM. Significant changes upon Dinaciclib treatment within certain cell cycle parameters are marked with “*”. *p < 0.05. Significance in b and e was calculated using unpaired student’s t test.
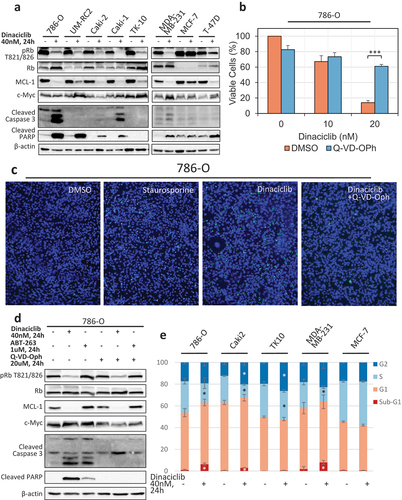
Since the CTG assay does not distinguish between inhibition of cellular proliferation and cell death, we assessed the cleavage of caspase 3 and PARP (poly-ADP ribose polymerase) apoptotic markers by western blot. Cleavage products of either caspase 3 and/or PARP were found in 4 out of 5 CC-RCC and 2 out of 3 breast cancer cell lines ()). Dinaciclib-sensitive TK-10 CC-RCC cells ()), expressing wild-type VHL, showed neither of these apoptotic markers, indicating that these cells were likely undergoing cell cycle arrest rather than apoptosis.
To confirm that Dinaciclib treatment induces apoptosis, we treated 786-O cells with 10–20 nM Dinaciclib with or without 20uM Q-VD-Oph pan-caspase inhibitor for 5 days and conducted a CTG assay. The results in ) show that Dinaciclib caused a reduction in the percent of viable cells at 20 nM, which was partially rescued by the inhibition of caspases. We further confirmed the Dinaciclib-induced apoptosis by a terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay ()). We treated 786-O cells with Dinaciclib, with or without Q-VD-Oph, and observed Dinaciclib-induced DNA fragmentation, which was then rescued by inhibition of the caspases. Staurosporine treatment was used as a positive control for DNA fragmentation. ) provides confirmation that Q-VD-Oph rescues Dinaciclib-induced cleavage of caspase 3 and PARP in 786-O cells. A pro-apoptotic compound ABT-263 [Citation49] was used as a positive control for caspase 3 and PARP cleavage.
To examine Dinaciclib’s effect on the cell cycle, a panel of cell lines was stained with propidium iodide (PI) and analyzed by FACS. Dinaciclib treatment at 40 nM for 24 h caused a significant decrease in S phase population in all 3 CC-RCC cell lines tested (786-O, Caki2, and TK10) as well as Dinaciclib-sensitive breast cancer cell line, MDA-MB-231, indicating a decrease in proliferation (), Supplementary Figure 1). Dinaciclib caused a significant increase in the sub-G1 population in 786-O, Caki2, and MDA-MB-231 cells, indicating an induction of cell death. Dinaciclib did not, however, cause an increase in sub-G1 population in Dinaciclib-sensitive TK-10, indicating that their sensitivity is caused by cell cycle arrest, rather than cell death. Dinaciclib did not cause cell cycle changes in Dinaciclib-resistant MCF7. Importantly, the appearance of a sub-G1 population correlated well with the observance of cleaved caspase 3 and PARP shown in ). Together, these results indicate that Dinaciclib causes either cell cycle arrest or both cell cycle arrest and apoptosis in Dinaciclib-sensitive cell lines.
Limited cell divisions of normal cells protect them from dinaciclib-induced cell death
To address the effect of Dinaciclib treatment on cancer vs normal cells, we conducted further experiments on the 786-O empty vector (EV)-transduced CC-RCC cell line together with two normal non-transformed renal epithelial cell lines: Renal Proximal Tubule Epithelial Cells (RPTEC) and Madin Darby Canine Kidney (MDCK) cells. We also included 786-O HA-VHL cells stably transduced with an HA-tagged VHL expression construct. Our rationale for the existence of a therapeutic window is the reported synthetic lethality of CDK inhibition with VHL deficiency [Citation10,Citation11] and with c-Myc overexpression/overactivation [Citation18–22] (see details in introduction). Additionally, our prior synthetic lethality screen in RCC4 CC-RCC cells ±VHL had revealed that VHL re-expression mildly protected CC-RCC from cell death caused by SU 9516, Indirubin-3’-oxime, and Kenpaullone CDK inhibitors (unpublished).
As normal cells in most tissues are in a quiescent state and do not undergo cell divisions, we decided to compare the effect of Dinaciclib treatment on sparse (dividing) and monolayer (confluent, non-dividing) cell cultures. ) provides evidence that RPTEC, MDCK, and 786-O HA-VHL were protected against Dinaciclib-induced cell death when seeded in monolayer, while 786-O EV cells were sensitive in both monolayer and sparse conditions as shown by a Crystal Violet assay. The synthetic lethality interaction between VHL deficiency and Dinaciclib treatment was further supported in experiments conducted in a second pair of CC-RCC ± VHL: RCC4 (Supplementary Figure 2). ) complements the findings for RPTEC, MDCK, and 786-O cells by CTG assay. Western blot analysis confirmed cell cycle inhibition in monolayer cultures (low to undetectable pRb) and showed that Dinaciclib treatment caused a decrease in pRb in sparse cultures ()).
Figure 3. Dinaciclib has a therapeutic window under cell culture conditions inhibiting cell divisions. (a) Inhibition of cell division rate by plating the cells at high cell density (monolayer) protects two normal cell lines–RPTEC (non-transformed Renal Proximal Tubule Epithelial Cells) and MDCK (Madin-Darby Canine Kidney cells)–as well as 786-O HA-VHL cancer cell line with re-introduced VHL, but not VHL-deficient 786-O, from Dinaciclib cytotoxicity. Cells were plated at low (sparse, ~5% confluency) or high (monolayer, 100% confluency) density in 6 cm plates, treated for 5 days with 40 nM Dinaciclib or vehicle (DMSO), followed by staining with Crystal Violet. (b) Confirmation of results in (a) by cell viability CTG assay. Cells were plated in 96-well plates and treated for 5 days with 10 nM and 20 nM Dinaciclib or vehicle (DMSO). The results were normalized to vehicle-treated cells. n = 3 (786–0 and RPTEC), n = 2 (MDCK), error bars correspond to SEM. Significance was calculated using unpaired student’s t test. *p < 0.05. (c) Dinaciclib induces expression of apoptosis markers only in sparse 786-O cells, but not in sparse RPTEC cells. Dinaciclib decreases MCL-1 and c-Myc expression only in monolayer 786-O cells, but not in monolayer RPTEC cells. Cells were treated with 40 nM Dinaciclib for 24 hours and analyzed by western blot. Monolayer cell culture conditions effectively decreased Rb phosphorylation in both cell lines as compared to sparse conditions, indicating cell cycle arrest. (d) Dinaciclib induces expression of apoptosis markers only in sparse 786-O cells, but not in sparse 786-O HA-VHL cells. Dinaciclib treatment decreases Rb phosphorylation and MCL-1 expression in both 786-O and 786-O HA-VHL cell lines. VHL re-expression caused downregulation of its target protein HIF2α. Cells were plated and treated as in (c).
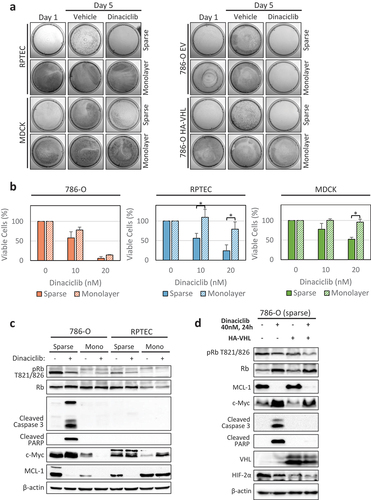
The apoptotic markers appeared to be different for sparse versus monolayer cultures when Dinaciclib treatment was applied. In sparse conditions Dinaciclib induced cleavage of caspase 3 and PARP apoptosis markers in 786-O cells, but not in normal RPTEC cells. Dinaciclib treatment decreased MCL-1 expression in both cell lines. In monolayer conditions, Dinaciclib decreased expression of MCL-1 and c-Myc in 786-O cells, but not in the RPTEC cells ()). When apoptotic markers were compared between 786-O EV and 786-O HA-VHL cells by western blot, Dinaciclib induced cleavage of caspase 3 and PARP in sparse 786-O EV cells, but not in sparse 786-O HA-VHL cells ()); however, Dinaciclib treatment decreased Rb phosphorylation and MCL-1 expression in both cell lines. These results suggest that Dinaciclib treatment preferentially triggers cell death in VHL-deficient CC-RCC cells and not normal cells, providing a potential therapeutic window.
Dinaciclib reduces tumor growth and induces cell death in an orthotopic PDX mouse model of CC-RCC
To develop a relevant to human disease mouse model characterized by tumor heterogeneity and the presence of a cancer stem cell (CSC) population, we acquired 3 low passage (p1 or p2) CC-RCC PDX tumor pieces from the NIH Patient-Derived Model Repository (PDMR) and implanted them s.c. in NSG mice. CD105 (Endoglin) was used as a CSC marker for CC-RCC, as previously reported [Citation41,Citation43]. FACS analysis of this marker’s cell surface expression showed that MD0865 and LS1987 had higher numbers of CD105+ cells than CK4020 CC-RCC PDX tumors (Supplementary Figure 3). Consistent with these findings, the CK4020 PDX which had low numbers of CD105+ cells grew the slowest out of three in vivo, taking more than double the time required for palpable tumors to form (data not shown).
Based on the above, we chose the MD0865 PDX for our in vivo study. For tumor imaging purposes, we optimized a cell dissociation and lentiviral infection protocol (total time in vitro <4 h, see “materials and methods”) and labeled MD0865 tumor cells with a lentivirus expressing firefly luciferase and enhanced green fluorescent protein (luc2-EGFP). Infected tumor cell suspensions were implanted s.c. Once a tumor was formed, it was cell dissociated, EGFP+ cells sorted by FACS, and re-implanted s.c. to enrich for labeled cells. Tumor pieces were used for experiments described below and cryo-banked (p4).
To evaluate the anti-tumor effects of Dinaciclib, we implanted ~1 mm3 pieces of luciferase-EGFP-labeled PDX tumors under the renal capsule of NSG mice. As expected, these implantations produced variable luminescence based on luciferase imaging at 3 weeks post-implantation due to tumor heterogeneity coupled with labeling variability. To allow for tumor growth comparisons in the course of the experiment, we assigned a similar distribution of luminescence to Vehicle and Dinaciclib treatment groups and formed tumor pairs with similar luminescence (Supplementary Figure 4(a)).
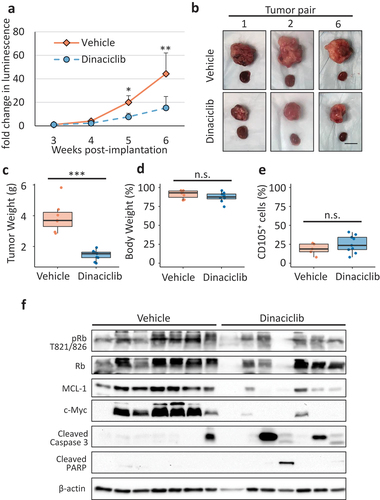
The mice were treated with 30 mg/kg Dinaciclib or Vehicle intraperitoneally (i.p.) 3 times a week for 4 weeks (weeks 3–7 post-implantation). The Dinaciclib treatments significantly decreased the tumor growth kinetics (), Supplementary Figure 4(b)) and decreased the tumor size and weight at sacrifice (). One mouse in the Dinaciclib-treated group died during week 5 of the study of unclear causes (no signs of toxicity by gross examination of the organs), thus this animal pair was eliminated from analysis. During week 7 tumor-bearing mice from pairs #3, 4, 5, 7, 8 either died or exhibited oversaturated luminescence signal, thus, only a subset of samples is included in Supplementary Figure 4(b) and . Similar to the previous in vivo studies reporting no normal tissue toxicity of Dinaciclib [Citation27,Citation50,Citation51], gross examination of the organs of Dinaciclib-treated mice did not show signs of toxicity. The mouse weight was also similar in the two groups throughout the duration of the study ()).
Figure 4. Dinaciclib treatment reduces tumor growth in a CC-RCC MD0865 PDX-based orthotopic mouse model. (a) Tumor growth curves based on imaging of firefly luciferase luminescence. Imaging and Dinaciclib treatment started at 3 weeks post-implantation. The data is the normalized average derived from Supplemental Figure 4. Week 3 luminescence for each tumor was set to “1” and then each subsequent measurement was divided by week 3 measurement to get a normalized value. (b) Representative images of paired tumors in Dinaciclib- and Vehicle-treated groups at sacrifice (7 weeks post-implantation). The non-cancerous kidney is shown for reference. Measure bar = 1 cm. (c) Box plot showing that Dinaciclib treatment significantly reduced tumor weight. Each point represents a single tumor. (d) Box plot showing that Dinaciclib- and Vehicle-treated groups have similar body weight. For each mouse, the weight at week 6 was normalized by the weight at week 3. Each point represents a single mouse. (e) Box plot showing that Dinaciclib treatment did not enrich for immunophenotypically defined (CD105+) cancer stem cells. Tumors were cell-dissociated and analyzed for CD105 expression by FACS. Each point represents a single tumor. Significance in (a, c-e) was calculated using unpaired student’s t test. *p < 0.05, **p < 0.001, ***p < 0.001. Error bars represent SD. (f) Western blot showing that tumors from Dinaciclib-treated group had a tendency for decreased MCL-1 and c-Myc expression and increased cleavage of caspase 3 and PARP. Rb phosphorylation remained largely unchanged.
We also evaluated metastasis to the lungs in both groups by isolating RNA and conducting quantitative RT-PCR (QRT-PCR) with the primers annealing to only mouse or only human GAPDH transcript. Only a few mice had high amounts of human transcripts in the lungs, all being from the Vehicle-treated group; the averaged data showed a tendency for lower metastatic burden in Dinaciclib-treated mice but did not reach statistical significance (Supplementary Figure 5(a)). Additionally, we assessed the metastatic burden in the H&E-stained liver tissue. While there was no difference in the numbers of micrometastases, Dinaciclib-treated animals tended to have less macrometastases in the livers than Vehicle-treated animals (Supplementary Figure 5(b,c)).
To address the effect of Dinaciclib on CSC population, we assessed the relative numbers of CD105+ CSCs present in cell-dissociated tumors resected from Vehicle- and Dinaciclib-treated animals by FACS. shows no difference in CSC percentages between the two groups, indicating that Dinaciclib treatment did not enrich for immunophenotypically defined CSCs. Thus, Dinaciclib treatment is not expected to cause expansion of the CSC population as has been reported in response to other therapeutic treatments [Citation39,Citation40].
Finally, we analyzed tumor tissue lysates by western blot, which showed that tumors from the Dinaciclib-treated group had a tendency for decreased MCL-1 and c-Myc expression and increased caspase 3 and PARP cleavage relative to the Vehicle-treated group ()). The reduction of c-Myc expression in response to Dinaciclib in vivo was in agreement with our in vitro experiments conducted using 786-O monolayer cell cultures (), suggesting that monolayer cell culturing conditions are more representative of the signaling pathways occurring in vivo. The level of Rb phosphorylation in vivo was similar in mice from both groups. These results provide evidence that Dinaciclib affects at least two signaling pathways expected to be affected in tumors of treated animals in vivo. Together, these data indicate that Dinaciclib offers potential therapeutic benefit for CC-RCC patients with VHL deficiency.
Discussion
In this study, using multiple cell lines and an orthotopic CC-RCC PDX-based mouse model, we have shown that CDK inhibitor, Dinaciclib, is a potential therapeutic for CC-RCC. We have also provided evidence that Dinaciclib spares VHL-expressing cells, including normal cells undergoing limited cell divisions, providing a therapeutic window for the drug.
Dinaciclib has been reported to inhibit multiple CDKs – CDK1-6, 9 and 12 [Citation27–29]. In Supplementary Figure 6 we show that RNA expression of CDK1-5, 9 and 12 is elevated in tumors of CC-RCC patients compared to matched adjacent normal tissue (TCGA KIRC dataset). Kaplan curves shown in Supplementary Figure 7 also support a better overall survival probability for CC-RCC patients with low CDK1-4, 6, and 9 expression than with high expression (TCGA KIRC dataset). Thus, further investigation is required to determine which of these kinases is/are important for CC-RCC proliferation/survival and are responsible for Dinaciclib-induced cytotoxicity. It would also be important to assess potential off-target effects of this drug. These investigations will provide an initial means for stratification of CC-RCC patients into groups projected to be sensitive or resistant to Dinaciclib treatment based on expression of CDK/s. It will also enable testing of drugs targeting specific CDK subsets or specific CDKs, which have been recently developed [Citation4,Citation5].
In this study, we used a PDX-based mouse model of CC-RCC, which allowed us to assess Dinaciclib’s effects on human tumor cells with preserved heterogeneity, including the presence of CSCs. Importantly, Dinaciclib has been reported to induce immunogenic cell death [Citation33], justifying further in vivo experiments conducted in syngeneic mouse models of CC-RCC [Citation52,Citation53], especially when combining Dinaciclib with immune checkpoint inhibitors, which represent a first-line therapy for CC-RCC [Citation3].
mTOR inhibitors represent a second-line therapy for CC-RCC [Citation3]. It has been recently reported that CC-RCC cells that developed resistance to mTOR inhibitors upregulate CDK2 and Cyclin A expression [Citation54], indicating the need for a CDK2 inhibitor as part of a combination therapy. Although a specific CDK2 inhibitor is not currently available [Citation55], Dinaciclib inhibits CDK2 [Citation27], as does Fadraciclib, a recently developed CDK2/9 inhibitor [Citation55,Citation56]. Thus, a combination treatment consisting of an mTOR inhibitor and Dinaciclib or Fadraciclib might represent a powerful second-line regimen.
CC-RCC cells are notoriously resistant to apoptosis due to impairments in intrinsic and extrinsic apoptotic pathways, making them difficult to target (reviewed in [Citation57]). The mechanisms of resistance include the HIF pathway (e.g. ARC [Citation58], VEGF), NFκB, Bcl2 family, and immune checkpoint proteins, among others. Since Dinaciclib induced apoptotic cell death in CC-RCC, it represents a potential cytotoxic therapeutic for this disease. In this study we did not investigate the mechanism of Dinaciclib-mediated sensitization of CC-RCC to apoptosis, and it is likely complex. Dinaciclib causes robust downregulation of MCL-1 as seen in this study and by others [Citation20,Citation59]. Although genetic and pharmacological studies have shown that MCL-1 inhibition leads to tumor growth inhibition in some animal models, mainly hematological malignancies [Citation60–63], most of the studies suggest combining MCL-1 inhibitors with other therapeutic agents, mainly BCL-family inhibitors, for achieving cancer cell death (reviewed in [Citation64]). At the same time, MCL-1 was not systematically evaluated as a therapeutic target in CC-RCC on its own. Interestingly, one of the studies investigating Dinaciclib-induced cell death concluded that genetic inhibition of MCL-1 in lung adenocarcinoma did not phenocopy Dinaciclib-mediated apoptosis [Citation65], but genetic inhibition of survivin (CDK1 phosphorylation target [Citation66] and possible CDK9 target [Citation65,Citation67]) did, making it another candidate protein to study.
Although our in vitro and in vivo data clearly show that c-Myc expression goes down upon long-term Dinaciclib treatment, it is unlikely that c-Myc downregulation causes Dinaciclib-mediated apoptosis of CC-RCC cells. The experiments involving switching on and off c-Myc expression in c-Myc-driven transgenic mouse models of RCC, show that turning c-Myc off does not induce apoptosis in vivo [Citation52]; however, loss of c-Myc does have a potent anti-proliferative effect. Since c-Myc is a master-regulator of the cell cycle and has been reported to directly regulate transcription of multiple cyclins and CDKs (including 1, 2, 4, and 6) [Citation68], it is likely that its downregulation by Dinaciclib blocks CC-RCC proliferation.
Since this study shows synthetic lethality of Dinaciclib treatment with VHL loss, and the main consequence of VHL loss is an upregulation of the HIF transcriptional program (reviewed in [Citation2,Citation69]), it would be important to conduct studies manipulating HIF activity in CC-RCC and compare sensitivity to Dinaciclib. VHL loss causes upregulation of a positive (HIF2) and a negative (HIF1) regulator of c-Myc [Citation25,Citation26], thus genetic studies manipulating HIF1α and HIF2α expression and also comparing CC-RCC naturally expressing both isoforms or just HIF2α in terms of Dinaciclib sensitivity need to be conducted. Interestingly, several studies suggest that the mechanism of synthetic lethality shown here might be HIF-dependent. First, CDK1 was reported to phosphorylate and enhance the formation of EZH2/ZMYND8 complex in CC-RCC, where ZMYND8 is upregulated by hypoxia mimetics and by VHL loss, contributing to gene expression changes and tumor progression [Citation70]. Second, CDK1 and CDK4/6 were reported to stabilize HIF1α by a VHL-independent mechanism [Citation71]. Accordingly, our data in ) shows that Dinaciclib treatment leads to lower HIF2α expression. Also, the VHL interactor database [Citation72] suggests that VHL forms a complex with a subset of CDKs and their regulators: CDK inhibitors 2A and 1B, CDK2, CDK4, CDK6, and Cyclin-A1, which provides a direct link between the VHL and CDK pathways.
In summary, this study shows preclinical efficacy of Dinaciclib in CC-RCC in vivo, targeting both CSCs and non-CSCs, and discusses the rationale for combining Dinaciclib with first- and second-line treatments.
Author contributions
LJN and OVR designed the study. LJN, KEC, BX, JMT, NBD, JL, JW, KRK and OVR performed the experiments and acquired the data. LJN, KEC, BX, JMT, JW, KRK and OVR analyzed the data. KEC and OVR wrote the manuscript.
Supplemental Material
Download PDF (1,020 KB)Supplemental Material
Download PDF (990.3 KB)Acknowledgments
The project was supported by an ACS RSG-170151-01-CDD to OVR, and an NIH ICTS UL1 TR001414 to LJN. The authors would like to thank Dr. Inder Verma for providing pVSVG and ΔR8.2 plasmids, Dr. Jennifer Prescher (UC Irvine) for providing pFUluc2-EGFP plasmid, Brynn Thayer for technical assistance, Dr. David Fruman (UC Irvine) for providing ABT-263 compound, NIH Patient-Derived Model Repository (PDMR) for providing frozen PDX chunks, and the Stem Cell Research Center UC Irvine for use of the FACS and animal facilities.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Cancer Stat Facts: cancer of the Kidney and Renal Pelvis Cancer National Institute of Health: National Cancer Institute; [cited 2021]. Available from: https://seer.cancer.gov/statfacts/html/kidrp.html
- Shen C, Kaelin WG Jr. The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol. 2013 Feb;23(1):18–25.
- Geynisman DM, Maranchie JK, Ball MW, et al. A 25 year perspective on the evolution and advances in an understanding of the biology, evaluation and treatment of kidney cancer. Urol Oncol. 2021 Jun 4;39:548–560.
- Asghar U, Witkiewicz AK, Turner NC, et al. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015 Jan 30;14:130.
- Chou J, Quigley DA, Robinson TM, et al. Transcription-Associated cyclin-dependent kinases as targets and biomarkers for cancer therapy. Cancer Discov. 2020 Mar;10(3):351–370.
- Zhang M, Zhang L, Hei R, et al. CDK inhibitors in cancer therapy, an overview of recent development. Am J Cancer Res. 2021;11(5):1913–1935.
- Stadler WM, Vogelzang NJ, Amato R, et al. Flavopiridol, a novel cyclin-dependent kinase inhibitor, in metastatic renal cancer: a University of Chicago Phase II Consortium study. J Clin Oncol. 2000 Jan;18(2):371–375.
- Van Veldhuizen PJ, Faulkner JR, Lara PN Jr., et al. A phase II study of flavopiridol in patients with advanced renal cell carcinoma: results of Southwest Oncology Group Trial 0109. Cancer Chemother Pharmacol. 2005 Jul;56(1):39–45.
- Chen D, Sun X, Zhang X, et al. Inhibition of the CDK4/6-Cyclin D-Rb pathway by ribociclib augments chemotherapy and immunotherapy in renal cell carcinoma. Biomed Res Int. 2020;2020:9525207.
- Nicholson HE, Tariq Z, Housden BE, et al. HIF-independent synthetic lethality between CDK4/6 inhibition and VHL loss across species. Sci Signal. 2019 Oct 1;12(601). DOI:https://doi.org/10.1126/scisignal.aay0482.
- Bommi-Reddy A, Almeciga I, Sawyer J, et al. Kinase requirements in human cells: III. Altered kinase requirements in VHL-/- cancer cells detected in a pilot synthetic lethal screen. Proc Natl Acad Sci U S A. 2008 Oct 28;105(43):16484–16489.
- Xiao H, Zeng J, Li H, et al. MiR-1 downregulation correlates with poor survival in clear cell renal cell carcinoma where it interferes with cell cycle regulation and metastasis. Oncotarget. 2015 May 30;6(15):13201–13215.
- Atkins DJ, Gingert C, Justenhoven C, et al. Concomitant deregulation of HIF1alpha and cell cycle proteins in VHL-mutated renal cell carcinomas. Virchows Arch. 2005 Sep;447(3):634–642.
- Hongo F, Takaha N, Oishi M, et al. CDK1 and CDK2 activity is a strong predictor of renal cell carcinoma recurrence. Urol Oncol. 2014 Nov;32(8):1240–1246.
- Li Y, Quan J, Chen F, et al. MiR-31-5p acts as a tumor suppressor in renal cell carcinoma by targeting cyclin-dependent kinase 1 (CDK1). Biomed Pharmacother. 2019 Mar 1;111:517–526.
- Wang X, Chen X, Han W, et al. miR-200c targets CDK2 and suppresses tumorigenesis in renal cell carcinoma. Mol Cancer Res. 2015 Dec;13(12):1567–1577.
- Xiao H, Xiao W, Cao J, et al. miR-206 functions as a novel cell cycle regulator and tumor suppressor in clear-cell renal cell carcinoma. Cancer Lett. 2016 Apr 28;374(1):107–116.
- Goga A, Yang D, Tward AD, et al. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat Med. 2007 Jul;13(7):820–827.
- Horiuchi D, Kusdra L, Huskey NE, et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J Exp Med. 2012 April 9;209(4):679–696.
- Gregory GP, Hogg SJ, Kats LM, et al. CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia. 2015 Jun;29(6):1437–1441.
- Hashiguchi T, Bruss N, Best S, et al. Cyclin-Dependent Kinase-9 Is a therapeutic target in MYC-Expressing diffuse large B-Cell lymphoma. Mol Cancer Ther. 2019 Sep;18(9):1520–1532.
- Huang C-H, Lujambio A, Zuber J, et al. CDK9-mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma. Genes Dev. 2014 Aug 15;28(16):1800–1814.
- Beroukhim R, Brunet J-P, Napoli AD, et al. Patterns of gene expression and copy-number alterations in von-Hippel Lindau Disease-Associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009 Jun 1;69(11):4674–4681.
- Sato Y, Yoshizato T, Shiraishi Y, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013 Aug;45(8):860–867.
- Gordan JD, Bertovrt JA, Hu C-J, et al. HIF-2α promotes hypoxic cell proliferation by enhancing c-Myc transcriptional activity. Cancer Cell. 2007 April;11(4):335–347.
- Gordan JD, Lal P, Dondeti VR, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008 Dec 9;14(6):435–446.
- Parry D, Guzi T, Shanahan F, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. 2010 Aug 1;9(8):2344–2353.
- Chen P, Lee NV, Hu W, et al. Spectrum and Degree of CDK Drug Interactions Predicts Clinical Performance. Mol Cancer Ther. 2016 Oct;15(10):2273–2281.
- Johnson SF, Cruz C, Greifenberg AK, et al. CDK12 inhibition reverses De Novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep. 2016 Nov 22;17(9):2367–2381.
- Alagpulinsa DA, Ayyadevara S, Yaccoby S, et al. A cyclin-dependent kinase inhibitor, dinaciclib, impairs homologous recombination and sensitizes multiple myeloma cells to PARP inhibition. Mol Cancer Ther. 2016 Feb;15(2):241–250.
- Baker A, Gregory GP, Verbrugge I, et al. The CDK9 inhibitor dinaciclib exerts potent apoptotic and antitumor effects in preclinical models of MLL-Rearranged acute myeloid leukemia. Cancer Res. 2016 Mar 1;76(5):1158–1169.
- Chen -X-X, Xie -F-F, Zhu X-J, et al. Cyclin-dependent kinase inhibitor dinaciclib potently synergizes with cisplatin in preclinical models of ovarian cancer. Oncotarget. 2015 Mar 30;6(17):14926–14939.
- Hossain DMS, Javaid S, Cai M, et al. Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. J Clin Invest. 2018 Feb 1;128(2):644–654.
- Hu C, Dadon T, Chenna V, et al. Combined inhibition of cyclin-dependent kinases (Dinaciclib) and AKT (MK-2206) blocks pancreatic tumor growth and metastases in patient-derived xenograft models. Mol Cancer Ther. 2015 Jul;14(7):1532–1539.
- Mita MM, Joy AA, Mita A, et al. Randomized Phase II trial of the cyclin-dependent kinase inhibitor Dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin Breast Cancer. 2014 Jun 1;14(3):169–176.
- Stephenson JJ, Nemunaitis J, Joy AA, et al. Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer. Lung Cancer. 2014 Feb 1;83(2):219–223.
- Kumar SK, LaPlant B, Chng WJ, et al. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood. 2015 Jan 15;125(3):443–448.
- Gojo I, Walker A, Cooper M, et al. Phase II study of the cyclin-dependent kinase (CDK) inhibitor Dinaciclib (SCH 727965) in patients with advanced acute leukemias. Blood. 2010 Nov 19;116(21): 3287–3287.
- Diehn M, Cho RW, Clarke MF. Therapeutic implications of the cancer stem cell hypothesis. Semin Radiat Oncol. 2009 Apr;19(2):78–86.
- Peired AJ, Sisti A, Romagnani P. Renal cancer stem cells: characterization and targeted therapies. Stem Cells Int. 2016;2016:1–12.
- Hu J, Guan W, Liu P, et al. Endoglin is essential for the maintenance of self-renewal and chemoresistance in renal cancer stem cells. Stem Cell Reports. 2017 Aug 8;9(2):464–477.
- Hu J, Guan W, Yan L, et al. Cancer stem cell marker Endoglin (CD105) induces epithelial mesenchymal transition (EMT) but not metastasis in clear cell renal cell carcinoma. Stem Cells Int. 2019;2019:9060152.
- Bussolati B, Bruno S, Grange C, et al. Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J. 2008 July 9;22(10):3696–3705.
- Nelson LJ, Wright HJ, Dinh NB, et al. Src kinase is biphosphorylated at Y416/Y527 and activates the CUB-Domain containing protein 1/protein kinase c delta pathway in a subset of triple-negative breast cancers. Am J Pathol. 2020 Feb;190(2):484–502.
- Razorenova OV, Ivanov AV, Budanov AV, et al. Virus-based reporter systems for monitoring transcriptional activity of hypoxia-inducible factor 1. Gene. 2005 Apr 25;350(1):89–98.
- Wright HJ, Hou J, Xu B, et al. CDCP1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc Natl Acad Sci U S A. 2017 Aug 8;114(32):E6556–E6565.
- Thompson JM, Alvarez A, Singha MK, et al. Targeting the mevalonate pathway suppresses VHL-Deficient CC-RCC through an HIF-Dependent mechanism. Mol Cancer Ther. 2018 Aug;17(8):1781–1792.
- Bragelmann J, Dammert MA, Dietlein F, et al. Systematic kinase inhibitor profiling identifies CDK9 as a synthetic lethal target in NUT midline carcinoma. Cell Rep. 2017 Sep 19;20(12):2833–2845.
- Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008 May 1;68(9):3421–3428.
- Howard D, James D, and Murphy K, et al. Dinaciclib, a bimodal agent effective against endometrial cancer. Cancers (Basel). 2021 Mar 6;13(5):1135.
- Mita MM, Mita AC, Moseley JL, et al. Phase 1 safety, pharmacokinetic and pharmacodynamic study of the cyclin-dependent kinase inhibitor dinaciclib administered every three weeks in patients with advanced malignancies. Br J Cancer. 2017 Oct 24;117(9):1258–1268.
- Shroff EH, Eberlin LS, Dang VM, et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc Natl Acad Sci U S A. 2015 May 26;112(21):6539–6544.
- Bailey ST, Smith AM, Kardos J, et al. MYC activation cooperates with Vhl and Ink4a/Arf loss to induce clear cell renal cell carcinoma. Nat Commun. 2017 Jun 8;8:15770.
- Juengel E, Nowaz S, Makarevi J, et al. HDAC-inhibition counteracts everolimus resistance in renal cell carcinoma in vitro by diminishing cdk2 and cyclin A. Mol Cancer. 2014 Jun 16;13(1):152.
- Tadesse S, Caldon EC, Tilley W, et al. Cyclin-Dependent Kinase 2 Inhibitors in Cancer Therapy: an Update. J Med Chem. 2019 May 9;62(9):4233–4251.
- Poon E, Liang T, Jamin Y, et al. Orally bioavailable CDK9/2 inhibitor shows mechanism-based therapeutic potential in MYCN-driven neuroblastoma. J Clin Invest. 2020 Nov 2;130(11):5875–5892.
- Lai Y, Zeng T, Liang X, et al. Cell death-related molecules and biomarkers for renal cell carcinoma targeted therapy. Cancer Cell Int. 2019;19:221.
- Razorenova OV, Castellini L, Colavitti R, et al. The apoptosis repressor with a CARD domain (ARC) gene is a direct hypoxia-inducible factor 1 target gene and promotes survival and proliferation of VHL-deficient renal cancer cells. Mol Cell Biol. 2014 Feb;34(4):739–751.
- Li L, Pongtornpipat P, Tiutan T, et al. Synergistic induction of apoptosis in high-risk DLBCL by BCL2 inhibition with ABT-199 combined with pharmacologic loss of MCL1. Leukemia. 2015 Aug;29(8):1702–1712.
- Glaser SP, Lee EF, Trounson E, et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012 Jan 15;26(2):120–125.
- Yasuda Y, Ozasa H, Kim YH, et al. MCL1 inhibition is effective against a subset of small-cell lung cancer with high MCL1 and low BCL-XL expression. Cell Death Dis. 2020 Mar 9;11(3):177.
- Derenne S, Monia B, Dean NM, et al. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood. 2002 Jul 1;100(1):194–199.
- Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. 2002 Mar 15;99(6):1885–1893.
- Bolomsky A, Vogler M, Kose MC, et al. MCL-1 inhibitors, fast-lane development of a new class of anti-cancer agents. J Hematol Oncol. 2020 Dec 11;13(1):173.
- Paliouras AR, Buzzetti M, Shi L, et al. Vulnerability of drug-resistant EML4-ALK rearranged lung cancer to transcriptional inhibition. EMBO Mol Med. 2020 Jul 7;12(7):e11099.
- Tsukahara T, Tanno Y, Watanabe Y. Phosphorylation of the CPC by Cdk1 promotes chromosome bi-orientation. Nature. 2010 Oct 7;467(7316):719–723.
- Yang L, Zhou F, Zhuang Y, et al. Acetyl-bufalin shows potent efficacy against non-small-cell lung cancer by targeting the CDK9/STAT3 signalling pathway. Br J Cancer. 2021 Feb;124(3):645–657.
- Bretones G, Delgado MD, Leon J. Myc and cell cycle control. Biochim Biophys Acta. 2015 May;1849(5):506–516.
- Gossage L, Eisen T, Maher ER. VHL, the story of a tumour suppressor gene. Nat Rev Cancer. 2015 Jan;15(1):55–64.
- Tang B, Sun R, and Wang D, et al. ZMYND8 preferentially binds phosphorylated EZH2 to promote a PRC2-dependent to -independent function switch in hypoxia-inducible factor-activated cancer. Proc Natl Acad Sci U S A. 2021 Feb 23;118(8):e2019052118.
- Zhao S, El-Deiry WS. Identification of Smurf2 as a HIF-1alpha degrading E3 ubiquitin ligase. Oncotarget. 2021 Sep 28;12(20):1970–1979.
- Tabaro F, Minervini G, Sundus F, et al. VHLdb: a database of von Hippel-Lindau protein interactors and mutations. Sci Rep. 2016 Aug 11;6:31128.