
ABSTRACT
The mammary gland of mammals possesses the specific function of synthesizing, secreting, and delivering milk. Notably, mammary epithelial cells are considered to be central to control the expansion and remodeling of mammary gland into a milk-secretory organ. And the biological function of mammary gland is mainly regulated by the endocrine system, especially for estrogen. G protein-coupled receptor 30 (GPR30), an estrogen membrane receptor, mediates estrogen-induced functions of physiology and pathophysiology. However, the relationship between estrogen/GPR30 signaling and proliferation of goat mammary epithelial cells (gMECs) is still unclear. Herein, estrogen promoted cell proliferation than control, as evidence by upregulation of cell numbers, BrdU-positive cell counts, and cell viability. Of note, these activities were all obviously reduced by treatment with GPR30 antagonist G15, yet GPR30 agonist G1 increased cell proliferation than control. Further, GPR30 silencing inhibited cell proliferation than negative control. This inhibition was accompanied by a G2/M phase arrest and downregulation of cell cycle regulators. Meanwhile, estrogen increased the phosphorylation of ERK1/2 and AKT. Further, the protein level of p-ERK1/2 and p-AKT was enhanced by GPR30 agonist G1 but inhibited by GPR30 antagonist G15 and GPR30 silencing. Importantly, MEK inhibitor and PI3K inhibitor decreased the expression of cell cycle regulators, and repressed estrogen-induced and G1-driven promotion of cell proliferation, suggesting that estrogen regulated cell proliferation of gMECs through mechanisms involving cell cycle, dependent of GPR30 and MEK/ERK and PI3K/AKT signaling pathway. This may provide a strong theoretical basis for researching estrogen sustained-release drugs promoting breast development and improving lactation performance.
Abbreviations: gMECs, goat mammary epithelial cells; E2, 17β-estradiol; GPR30, G protein-coupled receptor 30; shRNA, small hairpin RNA; CDK, cyclin-dependent kinase; PI3K, phosphatidylinositol 3-kinase; AKT, proteinkinase B; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase kinase; ERK1/2, extracellular signal-regulated kinase 1/2.
1. Introduction
Goat milk act as a pivotal role in milk consumption of human diet, which is a good supplier for providing exogenous protein due to its high protein production and small size of fat globule that is highly digestible and less allergic [Citation1,Citation2]. The mammary gland of mammals possesses the specific function of synthesizing, secreting, and delivering milk [Citation3,Citation4]. During a pregnancy/lactation cycle, mammary epithelial cells proliferate rapidly to form ducts and secretory alveoli through hormonal regulation, responsible for expansion and remodeling of mammary gland into a milk-secretory organ [Citation5,Citation6]. And the biological function of mammary gland is mainly regulated by the endocrine system, especially for estrogen (17β-estradiol) [Citation7,Citation8]. G protein-coupled receptor 30 (GPR30), an estrogen membrane receptor, mediates estrogen-induced functions of physiology and pathophysiology [Citation9,Citation10].
Normal cell cycle progression is a crucial event for every multicellular organism, due to it deciding body size and shape, tissue renewal and senescence, and is also important for reproduction [Citation11], which is closely related to cell proliferation. Promotion of cell proliferation was accompanied by an abundance of S and G2 phase [Citation12,Citation13] and upregulation of cell cycle checkpoint regulators [Citation14]. However, the arrest of cell cycle in the G2 phase contributed to suppressing cell survival ability [Citation15,Citation16]. On the other hand, studies revealed that GPR30 was associated with cell cycle progression, and effects of GPR30 activation on cell cycle were not the same [Citation17].
Estrogen membrane receptor GPR30 is widely expressed in reproductive tissues, such as mammary gland [Citation8]. After binding with estrogen, GPR30 mediates the rapid non-genomic actions by stimulating its downstream signaling, including MAPK and PI3K/AKT [Citation18–20]. Estrogen and GPR30 agonist promoted cell proliferation via MEK/ERK signaling, on the contrary, GPR30 antagonist caused a reverse. Moreover, GPR30 silencing further confirmed the inhibition of cell proliferation via MEK/ERK signaling inactivity [Citation21–23]. On the other hand, PI3K/AKT signaling was also reported to regulate estrogen-stimulated and GPR30-mediated promotion of cell proliferation, yet GPR30 antagonist inhibited the effect induced by estrogen [Citation24,Citation25]. Moreover, GPR30 silencing abolished estrogen-induced PI3K/AKT signaling activity and led to the suppression of cell proliferation [Citation26]. Based on previous studies, we hypothesized that estrogen binding to GPR30 may regulate the proliferation of goat mammary epithelial cells (gMECs).
In this study, we investigated the relationship among 17β-estradiol, GPR30, and proliferation of gMECs. The results showed that 17β-estradiol binding to GPR30 contributed to the proliferation of gMECs by driving cell cycle progression, dependent of MEK/ERK and PI3K/AKT signaling pathway.
2. Materials and methods
2.1. Goat mammary epithelial cell isolation and culture
Goat mammary epithelial cells were isolated from mammary gland tissues from a 2-y-old lactating Guanzhong dairy goat who was reared in Shaanxi Province (China), as described previously [Citation27]. Briefly, tissue pieces were finely minced and put into a 24-well culture plates, one piece for each hole, and cultured in the complete basal medium containing Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (phenol red-free, BasalMedia, China, L340KJ), 10% KnockOut Serum Replacement (Gibco, USA, 10828028), and 10 ng/mL epidermal growth factor (Sigma, USA, E5036). The medium was replaced every 3 d, until cells had spread across the bottom of the plate. All cells were cultured in a 37°C incubator with 5% CO2.
2.2. Immunofluorescence
The procedure of immunofluorescence was as previous description with some modifications [Citation28]. Briefly, the gMECs were fixed in 4% paraformaldehyde (Solarbio, China, P1110) for 30 min, permeabilized with 0.2% Triton-X 100 (Sigma, T8532) for 15 min, and then incubated with a blocking buffer (1% fetal bovine serum diluted by PBS, Gibco, 10270106) for 2 h at room temperature. Subsequently, the cells were incubated with primary antibody against cytokeratin 18 (Abcam, USA, ab668) or GPR30 (Abcam, ab39742) overnight at 4°C. The nuclei were stained with Hoechst 33342 (Sigma, B2261) in dark for 5 min at room temperature. Images were captured using a fluorescence microscope (Olympus, Japan).
2.3. Construction and identification of shGPR30 lentiviral vectors
To knockdown the expression of GPR30 gene (GenBank: XM_018039918), short hairpin RNAs against GPR30 (shGPR30) and negative control (shRNA) were synthesized by Sangon Biotech (Shanghai) Co., Ltd. All sequences are as described in Table S1. The method of constructing short hairpin RNA lentivirus vectors and packaging virus were performed as previous description [Citation29]. gMECs were incubated with lentivirus of shGPR30 or shRNA for 18 h before removing. After transfection, cells were allowed to grow, and then gained for protein extraction. Western blot was used to evaluate the knockdown efficiency of GPR30.
2.4. Cell counting assay
Cell proliferation was tested by cell counting assay. Briefly, 1 × 104 cells/well were seeded in 24-well plates and cultured in the complete basal medium with different treatments for 5 d . Cell numbers were determined using a hemocytometer.
2.5. BrdU incorporation assay
Proliferation of gMECs was detected using the bromodeoxyuridine (BrdU) incorporation assay. In detail, 1 × 104 cells/well were seeded in 24-well plates and cultured in the complete basal medium with different treatments for 5 d . Then, the gMECs were co-cultured with BrdU (100 µg/mL, Sigma, B5002) for 2 h. Next, cells were fixed in 4% paraformaldehyde (Solarbio) for 30 min, permeabilized with 0.2% TritonX-100 (Sigma) for 15 min, treated with 1 M HCl for 15 min at room temperature, and then incubated with an anti-BrdU antibody (Sigma, SAB4700630) overnight at 4°C. The nuclei were stained by Hoechst 33342 (Sigma) in dark for 5 min at room temperature. BrdU-positive cells were observed and counted through an inverted fluorescence microscope (Olympus, Japan).
2.6. Cell viability assay
Cell viability was evaluated using the Cell Counting Kit-8 kit (BOSTER, China, AR1160). Briefly, 6 × 103 cells/well were seeded in 96-well plates and cultured in the complete basal medium with different treatments for 5 d. Then, fresh complete basal medium containing CCK-8 solution (Vcck-8: Vmedium, 1: 10) was added into each well and incubated for 2 h. And the absorbance value was detected at 450 nm.
2.7. Cell cycle analysis
Flow cytometry analysis was used to measure cell cycle distribution. The gMECs were harvested with 0.25% (w/v) trypsin, fixed in 70% ice-cold ethanol overnight at 4°C, incubated with RNase A (20 µg/mL, CWbio, China, CW0600S) for 30 min at room temperature, and stained with propidium iodide (50 µg/mL, Solarbio, China, C0080) in dark for 20 min at room temperature. The cell cycle data were assayed using the flow cytometry (BD Biosciences, USA).
2.8. Western blot
The total protein of mammary epithelial cells was collected using High-efficiency RIPA buffer (Solarbio, R0010) including protease and phosphatase inhibitors according to manufacturer’s instructions. The equal amounts of protein were applied to SDS-PAGE and then transferred to a PVDF membrane. The nonspecific binding was blocked using the 5% nonfat milk. Whereafter, the membranes were incubated overnight at 4°C with the specific primary antibody against β-casein (Santa Cruz Biotechnology, USA, sc-166530), GPR30 (Abcam, ab39742), cyclin D1 (Sangon Biotech, China, D160236), CDK2 (Sangon Biotech, D120395), cyclin B1 (Cell Signaling Technology, USA, 4135), CDK1 (Cell Signaling Technology, 77055), ERK1/2 (Cell Signaling Technology, 4695), p-ERK1/2 (Cell Signaling Technology, 4370), AKT (BOSTER, BM4390), p-AKT (BOSTER, BM4390), or β-actin (BOSTER, BM0627). Finally, the protein bands were detected with a chemiluminescence kit (Biotanon, China, 180–5001).
2.9. Statistics
The data were presented as mean ± SD from three independent experiments. Statistical analyses were performed with SPSS (version 20.0; SPSS Inc., Chicago, IL, USA), which were checked using Tukey’s test. Probability (p) value <0.05 was considered statistically significant.
3. Results
3.1. Morphology and characteristics of goat mammary epithelial cells
In the present study, gMECs were insolated from mammary gland tissues (Figure S1a), and displayed the characteristic as reported for mammary epithelial cells [Citation28,Citation30,Citation31]. gMECs presented the typical epithelial cobblestone morphology (Figure S1b) and formed growing islands at low density (Figure S1c). After postconfluent cultures for 2 d to 3 d, gMECs formed dome-like structures (Figure S1d). To further characterize the gMECs, the expression of cell-type specific cytoskeletal markers and milk-protein specific genes [Citation30,Citation32] was detected in gMECs. As the results showed, gMECs were positive staining for mammary epithelium maker cytokeratin 18 (Figure S1e), and expressed the protein of milk-protein specific gene β-casein (P3–P9) (Figure S1f).
3.2. 17β-estradiol promotes the proliferation of gMECs
In this study, we first investigated the effect of 17β-estradiol (E2, 0.01, 0.1 , and 1 µM) on the proliferation of gMECs. As shown in , 0.01 µM 17β-estradiol did not increase cell numbers, BrdU-positive cell counts, and cell viability, yet 0.1 and 1 µM 17β-estradiol increased cell numbers, BrdU-positive cell counts, and cell viability compared to control (0 µM) and there was no statistical significance between 0.1 and 1 µM 17β-estradiol (), suggesting that 17β-estradiol promoted the proliferation of gMECs, and 0.1 µM 17β-estradiol was chosen to perform in the following experiments.
Figure 1. GPR30 mediates 17β-estradiol-induced cell proliferation of gMECs. Cell counting assay (a), BrdU incorporation assay (b), and CCK-8 assay (c) were performed to detect the effect of 17β-estradiol (E2, 0.01, 0.1, and 1 μM) for 5 d on cell proliferation. Determination of GPR30 expression by immunofluorescence (d) and Western blot (e). Cell counting assay (f), BrdU incorporation assay (g), and CCK-8 assay (h) were utilized to detect the effect of 17β-estradiol (0.1 μM), G1 (0.1 μM), and 17β-estradiol (0.1 μM) + G15 (1 μM) for 5 d on cell proliferation, respectively. β-Actin serves as a loading control. P3, P6, P9, the 3, 6, and 9 generation of gMECs. Scale bar = 50 µm. nsp > 0.05, *p < 0.05, **p < 0.01.
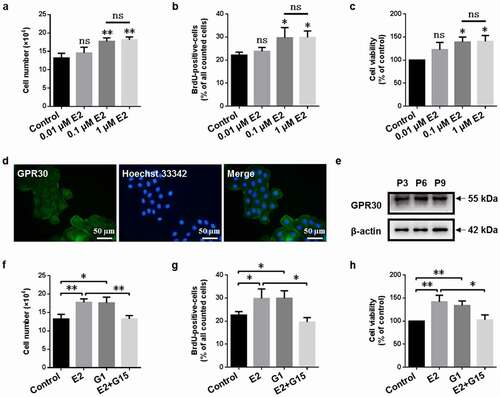
3.3. GPR30 mediates 17β-estradiol-induced cell proliferation of gMECs
To explore whether estrogen membrane receptor GPR30 mediated the pro-proliferative effect of 17β-estradiol on gMECs, we firstly examined the expression of GPR30. The results of immunofluorescence and Western blot showed that gMECs expressed GPR30 during the experiments (P3–P9) (). Subsequently, we investigated the changes of cell proliferation of gMECs exposed to GPR30 agonist and GPR30 antagonist. GPR30 agonist G1 increased cell numbers, BrdU-positive cell counts, and cell viability of gMECs compared to control, but GPR30 antagonist G15 suppressed 17β-estradiol-induced activities (), suggesting a pro-proliferative role of GPR30 activation in gMECs.
Figure 2. GPR30 silencing suppresses cell proliferation of gMECs. (a) Western blot was used to determine the protein level of GPR30 in gMECs. Cell counting assay (b), BrdU incorporation assay (c), and CCK-8 assay (d) were performed to assess cell proliferation of gMECs with GPR30 silencing. β-Actin serves as a loading control. *p < 0.05, **p < 0.01, ***p < 0.001.
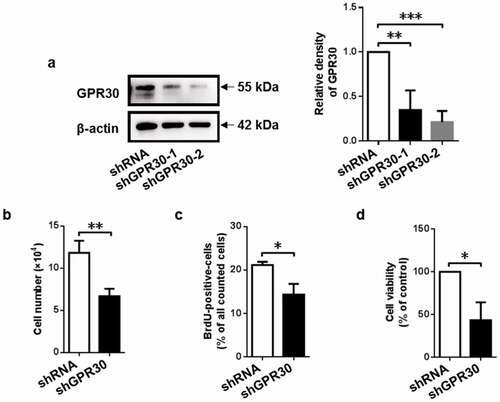
3.4. GPR30 silencing suppresses cell proliferation of gMECs
To further confirm whether GPR30 mediated 17β-estradiol-induced cell proliferation of gMECs, GPR30 silencing was performed by its specific shRNA. As shown in ), the knockdown efficiency of shGPR30-2 on GPR30 expression was higher than that of shGPR30-1 ()), thereby shGPR30-2 was utilized in the followed GPR30 silencing experiments. As expected, GPR30 silencing suppressed cell proliferation, as indicated by downregulation of cell numbers, BrdU-positive cell counts, and cell viability compared to negative control (). These data suggested that GPR30 mediated 17β-estradiol-induced cell proliferation of gMECs.
3.5. GPR30 silencing inhibits cell cycle progression
To determine the cellular mechanism by which 17β-estradiol binding to GPR30 enlarged cell proliferation, cell cycle analysis was performed. Analysis of flow cytometry showed that GPR30 silencing decreased the proportion of S phase (26.04 ± 0.78% vs. 20.44 ± 0.81%), but increased the proportion of G2/M phase (4.89 ± 1.35% vs. 8.16 ± 1.18%) compared to negative control ()).
Figure 3. GPR30 silencing arrests cell cycle progression of gMECs. (a) Flow cytometry was used to analyze the distribution of cell cycle of gMECs with GPR30 silencing. (b) The expression of cell cycle checkpoint regulators of gMECs with GPR30 silencing was detected by Western blot. β-Actin serves as a loading control. nsp > 0.05, *p < 0.05.
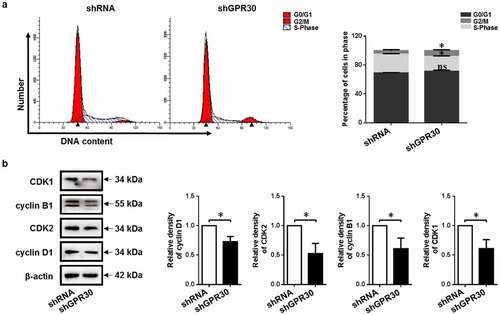
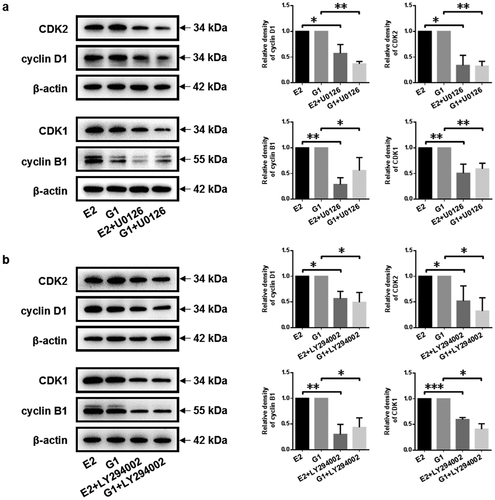
To further elucidate the underlying mechanisms, the expression of cell cycle checkpoint regulators [Citation33] was examined. GPR30 silencing decreased the expression of S-phase checkpoint regulator cyclin D1 and CDK2 compared to negative control. Meanwhile, downregulation of G2/M-phase checkpoint regulator cyclin B1 and CDK1 was determined after GPR30 silencing compared to negative control ()). These indicated that GPR30 silencing induced cell cycle arrest in the G2/M phase and suppressed cell cycle progression of gMECs.
3.6. Effect of 17β-estradiol targeting GPR30 on MEK/ERK and PI3K/AKT signaling pathway
According to the aforementioned results of GPR30 agonist and antagonist, we investigated the molecular regulation mechanisms of 17β-estradiol targeting GPR30 in cell proliferation of gMECs. As a result, an abundance of phosphorylated ERK1/2 (p-ERK1/2) was determined upon 17β-estradiol stimulation, which was reversed when exposed to GPR30 antagonist G15 simultaneously. And GPR30 agonist G1 increased the phosphorylation of ERK1/2 compared to control ()). Moreover, GPR30 silencing decreased the protein level of p-ERK1/2 compared to negative control ()), suggesting that 17β-estradiol targeting GPR30 activated the MEK/ERK signaling.
Figure 4. 17β-Estradiol binding to GPR30 activates MEK/ERK and PI3K/AKT signaling pathway. (a, c) Western blot was performed to detect the protein level of p-ERK1/2 and p-AKT of gMECs cultured with 17β-estradiol (0.1 μM), G1 (0.1 μM), and 17β-estradiol (0.1 μM) + G15 (1 μM) for 24 h, respectively. (b, d) The protein level of p-ERK1/2 and p-AKT of gMECs with GPR30 silencing were detected by Western blot, respectively. β-Actin serves as a loading control. *p < 0.05, **p < 0.01.
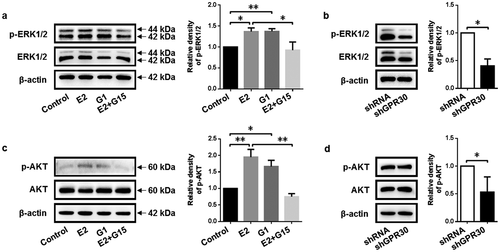
Meanwhile, 17β-estradiol increased the protein level of phosphorylated AKT (p-AKT). Of note, the activity was obviously reduced by treatment with GPR30 antagonist G15, yet GPR30 agonist G1 increased the phosphorylation of AKT compared to control ()). Further, GPR30 silencing decreased the protein level of p-AKT compared to negative control ()), suggesting that 17β-estradiol targeting GPR30 activated the PI3K/AKT signaling. These data indicated that 17β-estradiol targeting GPR30 activated the MEK/ERK and PI3K/AKT signaling pathway.
3.7. Effect of 17β-estradiol targeting GPR30 on cell proliferation through MEK/ERK and PI3K/AKT signaling pathway
To confirm whether MEK/ERK and PI3K/AKT signaling pathway mediated 17β-estradiol-induced cell proliferation of gMECs, small molecular inhibitors[Citation23] were applied. As shown in , MEK inhibitor U0126, inhibition of MEK/ERK signaling activity, decreased the expression of S-phase checkpoint regulator cyclin D1 and CDK2 and G2/M-phase checkpoint regulator cyclin B1 and CDK1 compared to negative control under 17β-estradiol and GPR30 agonist G1 treatment, respectively ()). Meanwhile, downregulation of S-phase checkpoint regulator cyclin D1 and CDK2 and G2/M-phase checkpoint regulator cyclin B1 and CDK1 was determined upon PI3K inhibitor LY294002 stimulation, when exposed to 17β-estradiol or GPR30 agonist G1 simultaneously ()). These data suggested that MEK/ERK and PI3K/AKT signaling pathway mediated cell cycle progression of gMECs.
Figure 5. Inhibition of MEK/ERK and PI3K/AKT signaling pathway arrests cell cycle progression of gMECs. (a, b) The protein expression of cell cycle checkpoint regulators was detected by Western blot after treatment with MEK inhibitor U0126 (10 μM) and PI3K inhibitor LY294002 (10 μM) in the presence of 17β-estradiol (0.1 μM) and G1 (0.1 μM) for 48 h, respectively. β-Actin serves as a loading control. *p < 0.05, **p < 0.01, ***p < 0.001.
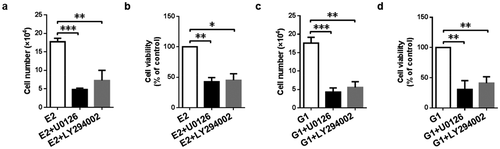
To confirm the relationship between MEK/ERK and PI3K/AKT signaling pathway and cell proliferation further, we detected the proliferation of gMECs after treatment with MEK inhibitor U0126 and PI3K inhibitor LY294002. As shown in , MEK inhibitor U0126 inhibited cell proliferation of gMECs, as evidence by downregulation of cell numbers and cell viability compared to 17β-estradiol and GPR30 agonist G1, respectively (). And inhibition of cell proliferation was detected by treatment with PI3K inhibitor LY294002 in the presence of 17β-estradiol and GPR30 agonist G1, respectively ()). These data revealed that MEK/ERK and PI3K/AKT signaling pathway was responsible for 17β-estradiol-induced and GPR30-mediated cell proliferation of gMECs.
Figure 6. 17β-Estradiol-induced and GPR30-mediated cell proliferation via MEK/ERK and PI3K/AKT signaling pathway. Cell counting assay (a, c) and CCK-8 assay (b, d) were implemented to evaluate cell proliferation after treatment with MEK inhibitor U0126 (10 μM) and PI3K inhibitor LY294002 (10 μM) in the presence of 17β-estradiol (0.1 μM) and G1 (0.1 μM) for 5 d, respectively. *p < 0.05, **p < 0.01, ***p < 0.001.
4. Discussion
Goat milk act as a pivotal role in milk consumption of human diet and is a good supplier for providing exogenous protein [Citation1,Citation2]. The mammary gland is a milk-secretory organ. Importantly, mammary epithelial cells are considered to be central to control the synthesis and secret of milk, and the number and status of mammary epithelial cells are responsible for milk yield and milk quality [Citation3,Citation4]. In addition, estrogen and its estrogen membrane receptor GPR30 play an important regulator of controlling cell proliferation and survival, positively or negatively [Citation7,Citation8]. Studies reported that estrogen significantly promoted cell proliferation [Citation26,Citation34] and GPR30 agonist enhanced cell proliferation and cell viability, whereas GPR30 antagonist and GPR30 siRNA reduced these activities [Citation23,Citation35]. This study discovered that the proliferation and viability of gMECs were driven by estrogen and GPR30 agonist but inhibited by GPR30 antagonist and GPR30 silencing, respectively (). Intriguingly, other evidence showed that the inhibitory effect of GPR30 agonist on cell proliferation could be blocked by GPR30 antagonist and GPR30 siRNA [Citation36,Citation37]. This may be because the effect of GPR30 on cell proliferation depends on different cell types and microenvironment. Nevertheless, all these studies conclude that estrogen has moderate effects on cell proliferation via GPR30.
Thereafter, our study discovered that GPR30 silencing resulted in S phase downregulation and G2/M phase arrest, inhibiting cell cycle progression (). 17β-estradiol and GPR30 agonist stimulated cell proliferation accompanied by increasing the cells in S phase, yet these effects were reversed upon GPR30 silencing treatment [Citation38]. Besides, GPR30 antagonist suppressed cell viability by inducing G2/M phase arrest [Citation39]. Further, GPR30 knockout did not increase liver growth in response to 17β-estradiol [Citation40]. On the other hand, studies reported that GPR30 agonist attenuated cell proliferation via inhibition of DNA synthesis [Citation36] and by accumulation of G2/M cells [Citation41,Citation42]. Cell cycle progression is intimately associated with cell proliferation [Citation43,Citation44].
MEK/ERK and PI3K/AKT signaling involve multiple cellular processes containing proliferation, differentiation, and apoptosis [Citation45,Citation46]. Estrogenic induction of cell proliferation was mediated by GPR30 via ERK/AKT signaling [Citation47]. GPR30 activation has been observed to induce rapid activation of ERK1/2, in turn, GPR30 silencing caused a reverse [Citation48]. Interestingly, GPR30 activation decreased the expression of phosphorylated ERK1/2 and inhibited cell proliferation, while GPR30 antagonist counteracted this effect [Citation37,Citation49]. There are two pathways that GPR30 regulates the ERK1/2 activity. Namely, Gβγ-subunit protein-dependent transactivation of epidermal growth factor increases the activity of ERK1/2. And another is cAMP-dependent signaling leading to Raf-1 inactivation and then downregulated ERK1/2 activity [Citation20]. Thus, in gMECs we speculate activation of GPR30 increasing the phosphorylation of ERK1/2 possibly via the former manner. In addition, activation of PI3K/AKT signaling contributed to the enhance of cell proliferation [Citation50]. On the contrary, inhibition of cell proliferation was mediated by antagonizing PI3K/AKT signaling activity [Citation51]. This study found that PI3K/AKT signaling was activated by estrogen binding to GPR30 (), and inhibition of these activities diminished estrogen-induced and G1-driven promotion of cell proliferation (). GPR30-mediated estrogen non-genomic signaling act as critical roles in cell fate determination.
5. Conclusion
In summary, this work determines that estrogen binding to GPR30 promotes the proliferation of goat mammary epithelial cells by driving cell cycle progression via MEK/ERK and PI3K/AKT signaling pathway. This work may provide a strong theoretical basis for researching estrogen sustained-release drugs which promote breast development and improve lactation performance.
Authors’ contributions
YZ, BHM, QW and XEZ were responsible for the study concept and design. YZ, HKL and MZF were responsible for cell culture and experiments. YZ, HKL and YYM performed data analysis. YZ finalized the manuscript. All read and approved the final manuscript.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics approval and consent to participate
This study was approved by the Institutional Animal Care and Use Committee (IACUC) of Northwest A&F University, Shaanxi, China. All animal experiments were performed under the control of the Guidelines for Animal Experiments by the Institutional Animal Care and Use Committee (IACUC) of Northwest A&F University, Shaanxi, China.
Supplemental Material
Download Zip (545.4 KB)Acknowledgments
We are grateful to Professor Yulin Chen (College of Animal Science and Technology, Northwest A&F University, China) for the vectors of synthesized lentivirus and their technical assistance.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384101.2022.2083708
Additional information
Funding
References
- Marius LN, Shipandeni MNT, Togarepi C. Review on the status of goat production, marketing, challenges and opportunities in Namibia. Trop Anim Health Prod. 2020;53(1):30.
- Tripathi MK. Comforts in quality and production of goat milk. J Adv Dairy Res. 2015;03(1):1–2.
- Inman JL, Robertson C, Mott JD, et al. Mammary gland development: cell fate specification, stem cells and the microenvironment. Development. 2015;142(6):1028–1042.
- Rezaei R, Wu Z, Hou Y, et al. Amino acids and mammary gland development: nutritional implications for milk production and neonatal growth. J Anim Sci Biotechnol. 2016;7(1):20.
- Hassiotou F, Geddes D. Anatomy of the human mammary gland: current status of knowledge. Clin Anat. 2013;26(1):29–48.
- Macias H, Hinck L. Mammary gland development. Wiley Interdiscip Rev Dev Biol. 2012;1(4):533–557.
- Alex A, Bhandary E, McGuire KP. Anatomy and physiology of the breast during pregnancy and lactation. Adv Exp Med Biol. 2020;1252:3–7.
- Hilton HN, Clarke CL, Graham JD. Estrogen and progesterone signalling in the normal breast and its implications for cancer development. Mol Cell Endocrinol. 2018;466:2–14.
- Barton M, Filardo EJ, Lolait SJ, et al. Twenty years of the G protein-coupled estrogen receptor GPER: historical and personal perspectives. J Steroid Biochem Mol Biol. 2018;176:4–15.
- Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. 2011;7(12):715–726.
- Urrego D, Tomczak AP, Zahed F, et al. Potassium channels in cell cycle and cell proliferation. Philos Trans R Soc Lond B Biol Sci. 2014;369(1638):20130094.
- Dai WT, White RR, Liu JX, et al. Seryl-tRNA synthetase-mediated essential amino acids regulate β-casein synthesis via cell proliferation and mammalian target of rapamycin (mTOR) signaling pathway in bovine mammary epithelial cells. J Dairy Sci. 2018;101(11):10456–10468.
- Jiao BL, Zhang XL, Wang SH, et al. MicroRNA-221 regulates proliferation of bovine mammary gland epithelial cells by targeting the STAT5a and IRS1 genes. J Dairy Sci. 2019;102(1):426–435.
- Zhu P, Liao LY, Zhao TT, et al. GPER/ERK&AKT/NF-κB pathway is involved in cadmium-induced proliferation, invasion and migration of GPER-positive thyroid cancer cells. Mol Cell Endocrinol. 2017;442:68–80.
- Chan QK, Lam HM, Ng CF, et al. Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of ERK1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G(2) cell-cycle arrest. Cell Death Differ. 2010;17(9):1511–1523.
- Ribeiro MPC, Santos AE, Custódio JBA. The activation of the G protein-coupled estrogen receptor (GPER) inhibits the proliferation of mouse melanoma K1735-M2 cells. Chem Biol Interact. 2017;277:176–184.
- Hernández-Silva CD, Villegas-Pineda JC, Pereira-Suárez AL. Expression and role of the g protein-coupled estrogen receptor (GPR30/GPER) in the development and immune response in female reproductive cancers. Front Endocrinol. 2020;11:544.
- Filardo EJ. Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: a novel signaling pathway with potential significance for breast cancer. J Steroid Biochem Mol Biol. 2002;80(2):231–238.
- Filardo EJ, Quinn JA, Bland KI, et al. Estrogen-induced activation of ERK-1 and ERK-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14(10):1649–1660.
- Filardo EJ, Quinn JA, Frackelton AR Jr., et al. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16(1):70–84.
- Ge LC, Chen ZJ, Liu HY, et al. Involvement of activating ERK1/2 through G protein coupled receptor 30 and estrogen receptor α/β in low doses of bisphenol a promoting growth of sertoli TM4 cells. Toxicol Lett. 2014;226(1):81–89.
- He YY, Cai B, Yang YX, et al. Estrogenic G protein-coupled receptor 30 signaling is involved in regulation of endometrial carcinoma by promoting proliferation, invasion potential, and interleukin-6 secretion via the MEK/ERK mitogen-activated protein kinase pathway. Cancer Sci. 2009;100(6):1051–1061.
- Scaling AL, Prossnitz ER, Hathaway HJ. GPER mediates estrogen-induced signaling and proliferation in human breast epithelial cells and normal and malignant breast. Horm Cancer. 2014;5(3):146–160.
- Dennis MK, Burai R, Ramesh C, et al. In vivo effects of a GPR30 antagonist. Nat Chem Biol. 2009;5(6):421–427.
- Dennis MK, Field AS, Burai R, et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J Steroid Biochem Mol Biol. 2011;127(3–5):358–366.
- Wei Y, Zhang Z, Liao H, et al. Nuclear estrogen receptor-mediated notch signaling and GPR30-mediated PI3K/AKT signaling in the regulation of endometrial cancer cell proliferation. Oncol Rep. 2012;27(2):504–510.
- Li C, Wang M, Zhang T, et al. Insulin-induced gene 1 and 2 isoforms synergistically regulate triacylglycerol accumulation, lipid droplet formation, and lipogenic gene expression in goat mammary epithelial cells. J Dairy Sci. 2019;102(2):1736–1746.
- Tong HL, Li QZ, Gao XJ, et al. Establishment and characterization of a lactating dairy goat mammary gland epithelial cell line. Vitro Cell Dev Biol Anim. 2012;48(3):149–155.
- Yang D, Jiang T, Lin P, et al. Knock-down of apoptosis inducing factor gene protects endoplasmic reticulum stress-mediated goat granulosa cell apoptosis. Theriogenology. 2017;88:89–97.
- Kaushik R, Singh KP, Kumari A, et al. Isolation, characterization, and EGFP expression in the buffalo (Bubalus bubalis) mammary gland epithelial cell line. Vitro Cell Dev Biol Anim. 2013;49(1):1–7.
- Zheng YM, He XY, Zhang Y. Characteristics and EGFP expression of goat mammary gland epithelial cells. Reprod Domest Anim. 2010;45(6):e323–331.
- Hu H, Wang J, Bu D, et al. In vitro culture and characterization of a mammary epithelial cell line from Chinese holstein dairy cow. PLoS One. 2009;4(11):e7636.
- Thu KL, Soria-Bretones I, Mak TW, et al. Targeting the cell cycle in breast cancer: towards the next phase. Cell Cycle. 2018;17(15):1871–1885.
- Zhang J, Yang Y, Zhang Z, et al. Gankyrin plays an essential role in estrogen-driven and GPR30-mediated endometrial carcinoma cell proliferation via the PTEN/PI3K/AKT signaling pathway. Cancer Lett. 2013;339(2):279–287.
- Chuang SC, Chen CH, Chou YS, et al. G protein-coupled estrogen receptor mediates cell proliferation through the cAMP/PKA/CREB pathway in murine bone marrow mesenchymal stem cells. Int J Mol Sci. 2020;21(18):6490.
- Holm A, Baldetorp B, Olde B, et al. The GPER1 agonist G-1 attenuates endothelial cell proliferation by inhibiting DNA synthesis and accumulating cells in the S and G2 phases of the cell cycle. J Vasc Res. 2011;48(4):327–335.
- Zhong J, Ge HF, Zhang C, et al. G protein-coupled estrogen receptor 1 negatively regulates the proliferation of mouse-derived neural stem/progenitor cells via extracellular signal-regulated kinase pathway. Brain Res. 2019;1714:158–165.
- Liu H, Yan Y, Wen H, et al. A novel estrogen receptor GPER mediates proliferation induced by 17β-estradiol and selective GPER agonist G-1 in estrogen receptor α (ERα)-negative ovarian cancer cells. Cell Biol Int. 2014;38(5):631–638.
- Bai LY, Weng JR, Hu JL, et al. G15, a GPR30 antagonist, induces apoptosis and autophagy in human oral squamous carcinoma cells. Chem Biol Interact. 2013;206(2):375–384.
- Chaturantabut S, Shwartz A, Evason KJ, et al. Estrogen activation of g-protein-coupled estrogen receptor 1 regulates phosphoinositide 3-kinase and mTOR signaling to promote liver growth in zebrafish and proliferation of human hepatocytes. Gastroenterology. 2019;156(6):1788–1804.e1713.
- Gui Y, Shi Z, Wang Z, et al. The GPER agonist G-1 induces mitotic arrest and apoptosis in human vascular smooth muscle cells independent of GPER. J Cell Physiol. 2015;230(4):885–895.
- Wang C, Lv X, He C, et al. The G-protein-coupled estrogen receptor agonist G-1 suppresses proliferation of ovarian cancer cells by blocking tubulin polymerization. Cell Death Dis. 2013;4(10):e869.
- Alenzi FQ. Links between apoptosis, proliferation and the cell cycle. Br J Biomed Sci. 2004;61(2):99–102.
- Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411(6835):342–348.
- Barbosa R, Acevedo LA, Marmorstein R. The MEK/ERK network as a therapeutic target in human cancer. Mol Cancer Res. 2021;19(3):361–374.
- Yu JS, Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143(17):3050–3060.
- Akimoto T, Takasawa A, Takasawa K, et al. Estrogen/GPR30 signaling contributes to the malignant potentials of ER-negative cervical adenocarcinoma via regulation of claudin-1 expression. Neoplasia. 2018;20(10):1083–1093.
- Chen Y, Li Z, He Y, et al. Estrogen and pure antiestrogen fulvestrant (ICI 182 780) augment cell-matrigel adhesion of MCF-7 breast cancer cells through a novel G protein coupled estrogen receptor (GPR30)-to-calpain signaling axis. Toxicol Appl Pharmacol. 2014;275(2):176–181.
- Grassi D, Lagunas N, Pinos H, et al. NADPH-diaphorase colocalizes with GPER and is modulated by the GPER agonist G1 in the supraoptic and paraventricular nuclei of ovariectomized female rats. Neuroendocrinology. 2017;104(1):94–104.
- Xu L, Chen J, Jia L, et al. SLC1A3 promotes gastric cancer progression via the PI3K/AKT signalling pathway. J Cell Mol Med. 2020;24(24):14392–14404.
- Ni S, Wei Q, Yang L. ADORA1 promotes hepatocellular carcinoma progression via PI3K/AKT pathway. Onco Targets Ther. 2020;13:12409–12419.