
ABSTRACT
MicroRNA-141-3p (miR-141-3p) has been found to be altered in the brain following a stroke. Herein, we investigate the impact of miR-141-3p on the apoptosis of neural stem cells (NSCs) in mice with middle cerebral artery occlusion (MCAO) and the potential mechanisms involved. Eight-week-old mice were injected intracerebroventricularly with miR-141-3p, antagomir-141-3p, or agomir negative control 2 h before MCAO, and animal behavior tests and infraction volume measurements were performed 24 h later. MCAO-mediated brain injury and NSCs apoptosis were observed by H&E, TTC, and TUNEL staining. The expression of cleaved caspase-3 and Ki67 was detected by western blotting. The luciferase reporter assay proved that miR-141-3p in combination with its target gene PBX homeobox 1 (PBX1). Exogenous miR-141-3p (agomir-141-3p) treatment increased infraction volume and brain edema and damaged neurological functions compared to control mice. Agomir-141-3p increased miR-141-3p expression in brain tissue of mice with MCAO and suppressed PBX1 expression. The effects of the agomir-141-3p-induced apoptosis in NSCs treated with oxygen–glucose deprivation (OGD)/reoxygenation (R) were abolished by PBX1 overexpression. The results from UCSC and JASPAR database showed that prokineticin 2 (PROK2) was a transcription factor of PBX1. The expression of PROK2 was transcriptionally regulated by PBX1 using RT-PCR and western blot assays. The effects of the apoptosis-promoting caused by PBX1 inhibition in NSCs treated with OGD/R were reversed by PROK2 inhibition. In conclusion, the miR-141-3p/PBX1/PROK2 axis might be a novel therapeutic target for the apoptosis of NSCs in MCAO.
KEYWORDS:
Introduction
Ischemic stroke is one of the most common diseases in emergency and critical medical departments, which is a necrotic disease of brain tissue caused by stenosis or occlusion of cerebral feeding arteries (carotid artery and vertebral artery) and insufficient cerebral blood supply [Citation1,Citation2]. Neural stem cells (NSCs) are a kind of pluripotent undifferentiated pluripotent stem cells that can self-renewal and differentiate into all major types of cells in the central nervous system, including neurons, astrocytes, and oligodendrocytes [Citation3]. NSCs locating widely in the ventricular area of embryonic nervous system maintains the steady-state of stem cell bank by symmetrical and asymmetric division and can fuse well with the nerve tissue of the host [Citation4,Citation5]. NSCs were considered as related to nervous system developmental diseases and the occurrence and progression of stroke, attributing to the production of a large number of neurons and glial cells induced by NSCs [Citation3,Citation5]. It was reported that NSCs apoptosis is one of the main causes of ischemic stroke [Citation3,Citation6].
MicroRNAs (miRNAs) are the kinds of highly conserved non-coding RNAs with a length of about 22 nt [Citation3,Citation7], which mainly bind to the 3’-UTR of the downstream target gene to degrade mRNA or inhibit its translation process, thereby regulating the expression of downstream target genes [Citation3]. According to research, miRNAs play important roles in the occurrence and development of ischemic stroke. MiR-145 expression is downregulated after ischemic stroke, and its high expression inhibits the apoptosis of NSCs, alleviating the progress of ischemic stroke [Citation8]. MiR-141-3p acts as an important role in a variety of diseases. MiR-141-3p is a key negative regulator of the EGFR pathway in osteosarcoma [Citation9], and it promotes proliferation and metastasis of nasopharyngeal carcinoma by targeting NME1 [Citation10]. MiR-141-3p upregulation of neural cell adhesion molecule 1 suppresses ameloblastoma cell migration [Citation11]. Inhibition of miR-141-3p promotes neurogenesis in rat NSCs and ameliorates stroke in aged mice [Citation12]. What’s more, inhibition of miR-141-3p improved mortality, neurological deficits, and decreased infarct volumes. The above studies fully indicate that miR-141-3p, a miRNA closely related to cell proliferation and apoptosis, is linked to the occurrence and development of brain diseases. However, the role of miR-141-3p in MCAO and its mechanism are unclear, and whether regulating neuronal cell apoptosis can affect MCAO-mediated brain damage needs to be investigated further.
Materials and methods
Animals and experimental design
The study had been approved by Animal Care and Use Committee of Nanjing University. All animal protocols were performed in accordance with the guidelines of the Committee. A total of 40 C57BL/6 mice (male, weight 20–25 g) was purchased from Shanghai Slac Laboratory Animal Company (China). Mice were randomly divided into eight groups: sham operation (n = 10), MCAO + miR-141-3p group (MCAO+agomir-141-3p; n = 5), MCAO+miR-141-3p antagonist group (MCAO+antagomiR-141-3p; n = 5), MCAO + miR-141-3p negative control group (MCAO+agomir-NC; n = 5), MCAO + NC group (n = 5), MCAO + sh-PBX1 group (n = 5), MCAO + oe-PBX1 group (n = 5). Agomir-141-3p, antagomiR-141-3p, and agomir-NC were synthesized by Sangon Biotech, Shanghai, China.
MCAO model
Transient MCAO was induced as previously described [Citation13]. The mice were deeply anesthetized with 2% chloralhydrate (1 ml/100 g, intraperitoneal injection). A middle line incision was made in the neck, and the left carotid artery was carefully separated. The left internal carotid artery was then sutured with a monofilament suture (Doccol, Sharon, MA, USA). Using a temperature control heating pad, the rectal temperature was maintained at 37°C during and after surgery. Transcranial laser Doppler (Moor Instruments, UK) was used to monitor animal regional cerebral blood flow (rCBF), and animals with CBF greater than 20% of the original baseline value were excluded from the study. The monofilament suture was removed slowly 1 h after occlusion. Mice were housed under controlled temperature and photoperiod conditions [12 h light/dark], with food and water freely available. Neurological scores, foot-fault test, and brain water content were evaluated as previously described [Citation13].
Intracerebroventricular injection
Agomir-141-3p, antagomir-141-3p, and agomir-NC (0.8 nmol dissolved in 4 μl PBS) were administered 2 h before MCAO via intraventricular injection (ICV). The injections were performed as previously described [Citation13]. Mice were anesthetized and positioned prone in a stereotactic head frame (RWD Life Science, China). A scalp incision was made along the midline and a burr hole was drilled into the right side of the skull (0.5 mm posterior and 1.0 mm lateral to the bregma). miR-141-3p, antagomiR-141-3p, miR-NC (4 μl), lentivirus expressing shRNA against PBX (sh-PBX1), pcDNA3.1 vector expressing PBX1 plasmid (pcDNA-PBX1), or pcDNA3.1 vector expression plasmid (pcDNA-vector) (20 μM) were microinfused into right lateral ventricles through a Hamilton syringe (2.5 mm vertically), which was driven by a microinfusion pump (KD Scientific) with 0.2 μl/min. To prevent possible leakage, the needle was left in place for an additional 5 min after injection before being slowly withdrawn within 4 min. The burr hole was sealed with bone wax after the needle was removed; the incision was closed with sutures and the mice were allowed to recover. After 6 h of modeling, the mice in each group were immediately executed with a severed head. To be used in the following experiments, brain tissue is carefully dissected and washed twice with Hank’s buffer.
2,3,5-Triphenyltetrazolium hydrochloride (TTC) staining
The brain was collected after 24 h of reperfusion, sectioned coronally at 1 mm interval, and stained with TTC at 37°C for 20 min. The infracted volume was expressed as a ratio of the whole contralateral hemisphere.
Luciferase reporter assay
A fragment of PBX1 gene 3’-UTR (wild-3’-UTR) containing a miR-141-3p targeting site was identified using the TargetScan database, and its respective mutant was amplified by RT-PCR and cloned into the psiCHECK-2 vector (Promega, USA). In 24-well plates, cells were co-transfected with the dual-luciferase reporter vector and agomir-141-3p, agomir-NC or antagomir-141-3p using HilyMax transfection reagent (Dojindo, Japan). Luciferase assays were performed 48 h after transfection with a Dual-Luciferase Reporter Assay System (Promega, USA) according to the manufacturer’s protocol. Renilla luciferase activity was normalized to that of firefly luciferase.
Real-Time PCR (RT-PCR)
The total RNA was extracted from the brain using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s instructions and then reverse-transcribed into cDNA using miRcute miRNA First-Strand cDNA (Tiangen, Beijing, China). The following primers were used (Tiangen, Beijing, China): U6, GCTCACTCGCAGGCATC (forward) and TTACCTTGGCTGTGGTAATC (reverse); miR-141-3p, GCTTACACTGTCCCTAAACGTGG (forward) and TTCTGCAACCAACACTGGGCT (reverse). The miRNA was detected by Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, Hemel Hempstead, UK) using the miRcute miRNA qPCR Detection Kit (Tiangen, Beijing, China), and U6 served as an internal reference.
The first-strand cDNA synthesis kit (Roche) was used to synthesize cDNA. The FastStart Universal SYBR Green Master (Roche) was then used to carry out RT-PCR to detect mRNA expression. GAPDH served as an internal reference. The following primers were used (Tiangen, Beijing, China): GAPDH, ATCCGTGTCAATGTGCCATTCGCG (forward) and TACCGGCTGCATGGCACAATGGG (reverse); PBX1, GCGTGGCAACGTCATGCTCCAA (forward) and ACTGTGCGTGCAACAATGTTCA (reverse); PROK2, AATCTGTGCCCTGCCTGCACTTGGC (forward) and CGCCGTGCTGGCAAAGCTCGT (reverse). The relative miRNA or mRNA level was calculated according to the 2−ΔΔCT approach.
Western blot assay
The whole protein was homogenized in RIPA buffer (Beyotime) containing protease inhibitor cocktail (Sigma, St. Louis, MO, USA) before being sonicated on ice. The supernatant was collected after centrifugation for western blot assays. The membrane was blocked with 5% nonfat milk in TBST for 2 h and then incubated with primary antibodies against cleaved caspase-3 (1:1000), Ki67 (1:1000), PBX1 (1:800), PROK2 (1:1000) or GAPDH (1:1000) at 4°C overnight. After 2 h of incubation with secondary antibody at 37°C, the cells were visualized using a chemiluminescence apparatus (ImageQuant LAS 4000 mini, GE Healthcare, USA). As the relative protein expression, the gray value of proteins was measured using Image-J software and normalized to that of GAPDH.
NSCs culture
The NSCs were harvested from embryonic C57BL/6 mouse brain as described previously [Citation14]. The neurosphere culture was kept in T25 tissue culture flasks (Nunc, Roskilde) containing DMEM/12 serum-free neurobasal medium supplemented with 2% B27, 20 ng/mL human recombinant epidermal growth factor (EGF; Gibco, USA), 20 ng/mL basic fibroblast growth factor (bFGF; Gbico, USA) and 5 µg/mL heparin (Sigma-Aldrich, USA). Cells were grown as free-floating neurospheres in a humidified 5% CO2/95% air incubator at 37°C. The medium was changed every three days. The passage was carried out when the neurospheres grew to 50–100 μm in diameter. Depending on the further tests, the neurospheres were digested to a single-cell suspension and plated on a PDL-precoated surface.
NSC OGD/Reoxygenation induction
With slight modifications, we administrated OGD/reoxygenation to NSCs as previously reported [Citation6]. Following starvation, the monolayer NSCs were transferred into a deoxygenated, glucose-free HBSS buffer. The culture was then incubated for 2 h at 37°C in an oxygen-deprived box aerated with 5% CO2 and 95% N2 to simulate OGD-injured NSCs. Ischemic NSCs were then transferred back to the DMEM/F12 medium and incubated at 37°C with 5% CO2 for 2, 4, 8, 12, and 24 h to induce further reoxygenation. As a negative control, the starved NSCs were incubated in HBSS with glucose for 2 h at 37°C, 5% CO2, followed by oxygen and glucose re-treatment.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assays were performed using the ChIP Assay Kit (Beyotime, Shanghai, China) according to the manufacturer’s protocol with slight modifications. Cells were cross-linked with 1% formaldehyde and terminated after 10 min by the addition of glycine at a final concentration of 0.125 M. DNA was immunoprecipitated from the sonicated cell lysates using a PBX1 antibody (Cell Signaling Technology, Beverly, MA, USA); IgG (BD Biosciences, San Diego, CA, USA) served as the negative control. The DNA was subjected to PCR to amplify the PBX1 binding sites. The amplified fragments were then run through an agarose gel for analysis. Chromatin (10%) was used as an input control before immunoprecipitation. The primer sequence was as follows: 5’-CGCGTTCGTGTTTGCATC-3’ and 5’-CTTCCTCACCAACCGAGCT-3’.
Caspase-3 activity assay
The activity of caspase-3 was quantified using the commercially available caspase-3 colorimetric assay kit (Beyotime). Caspase-3 activity was detected at 405 nm by a spectrophotometer on an ELISA reader (EMax, Molecular Devices LLC, San Jose, CA, USA).
Cell viability assay
Cell viability was determined using the CCK-8 reagent (Beyotime) according to the manufacturer’s instructions. The absorbance was detected at 450 nm using a Microplate Reader (Bio-Rad, Hercules, CA, USA).
Apoptosis assay using Annexin V-FITC
Apoptosis was measured using an Annexin V-FITC apoptosis detection kit (Calbiochem, CA, USA) according to manufacturer’s instructions. In brief, cells were seeded at a low density (3 × 105) in 12-well plates and allowed to attach overnight in DMEM medium. After 48 h, the cells were stained with FITC-Annexin V and propidium iodide and subjected to flow cytometry.
Immunofluorescence staining
NSCs were routinely inoculated and cultured in 48-well plates at a density of 2 × 104 cells/cm2. NSCs were fixed in 4% paraformaldehyde at room temperature for 25 min and then blocked in 10% donkey serum in phosphate-buffered saline containing 1% BSA and 0.2% Triton X-100 for 50 min. We identified cells via immunofluorescence staining with Nestin antibody (1:1000) and anti-rabbit IgG (1:1200) overnight at 4°C at room temperature in the dark. The nuclei were then stained for 1 min with 5 μg/ml DAPI. The fluorescence intensity in images was analyzed using Volocity software.
Agomir-141-3p, agomir-NC or antagomir-141-3p transfection
The neurosphere culture was kept at 37°C with 5% CO2 in DMEM/F12 medium containing B27, EGF, and bFGF. Agomir-141-3p, agomir-NC or antagomir-141-3p (100 μM) were transfected into NSCs for 48 h using Lipofectamine RNAiMAX Transfection Reagent (Invitorgen, USA) according to the manufacturer’s protocol. To model the OGD-injured NSCs, the cells were then incubated for 2 h in an oxygen-deprived box aerated with 5% CO2 and 95% N2 at 37°C. Ischemic NSCs were then transferred back into the DMEM/F12 medium and incubated for 2 h at 37°C with 5% CO2 for further reoxygenation induction.
Cell transfection
NSCs were seeded in a 6-well plate at a density of 5 × 105 cells/well. Then, lentivirus expressing shRNA against either PBX1 or PROK2 (sh-PBX1/or sh-PROK2 group) or blank control lentivirus (sh-scramble group) was transfected into NSCs at the indicated concentrations (MOI = 10). Next, Lipofectamine 2000 (Invitrogen) was used to carry out the transfection of pcDNA3.1 vector expressing PBX1 plasmid (pcDNA-PBX1 group) or pcDNA3.1 vector expression plasmid (pcDNA-vector group) (GeneChem, Shanghai, China) following the manufacturer’s protocol. After 12 h, the cells were washed twice with PBS and a fresh medium was added. After 24 h, the cells were collected, the transfection efficiency of PBX1 or PROK2 was evaluated using immunocytochemistry, RT-PCR, and western blot assays.
TUNEL staining
The sections were further incubated for 1 h in the dark with 50 μl TUNEL test solution (Beyotime) at 37°C. After incubation, they were observed under a fluorescence microscope.
H&E staining
The frozen sections of brain tissue were washed after being placed in an oven at 37°C for 10 min to dissolve the tissue embedding agent. The H&E staining was performed as previously described [Citation7].
Statistical analysis
All data were from three independent experiments and represented mean ± standard derivations (SD). Statistical significance between different groups was analyzed by One-way Analysis of Variance (ANOVA) with Duncan’s multiple range tests in SPSS 20 (IBM, NY). P < 0.05 was considered to indicate statistical significance.
Results
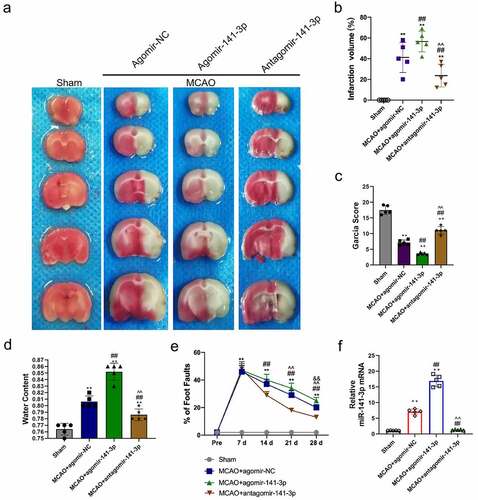
Administration of agomir-141-3p aggravated MCAO-mediated infarction volume and neurological deficits
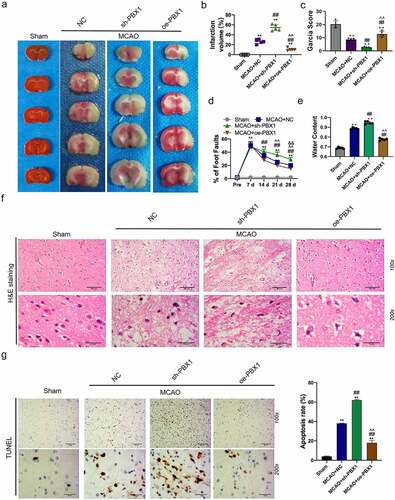
TTC staining was used to reveal cerebral infarct, whereas normal brain tissue is stained red, and the infarct lesion remains unstained (white color) (). MCAO for one hour caused a significant brain infarction and deteriorated neurobehavior at 24 h after surgery, and administration of antagomir-141-3p reduced the infarct volume and improved the neurological scores, and the synthesized agomir-141-3p that was specific to increasing endogenous miR-141-3p, afterward abolished the protective effect of antagomir-141-3p (). In the long term, administration of antagomir-141-3p greatly improved the performance of the foot fault test at 28 days after MCAO, which was reversed by agomir-141-3p (). Meanwhile, antagomir-141-3p largely reduced the brain water content in the ischemic hemisphere, and agomir-141-3p increased it at 24 h after MCAO (). Next, we found that miR-141-3p was significantly increased in the MACO group. The administration of agomir-141-3p further increased miR-141-3p expression in the MACO group, while treatment of antagomir-141-3p inhibited the effects ().
Figure 1. Administration of exogenous agomir-141-3p aggravated MCAO-mediated infarction volume, neurological deficits.
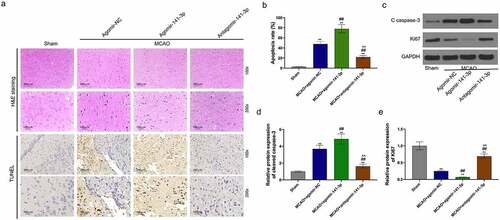
Administration of agomir-141-3p aggravated MCAO-mediated brain injury and apoptosis in vivo
H&E staining and TUNEL analysis showed that MCAO for one hour caused a significant brain injury and cell apoptosis 24 h later (), and that administration of antagomir-141-3p reduced the brain injury and cell apoptosis in MCAO mice. The apoptosis-inhibiting effect of antagomir-141-3p was abolished by agomir-141-3p of. Western blot analysis showed that agomir-141-3p increased MCAO-medicated cleaved caspase-3 expression and decreased Ki67 expression, however, antagomir-141-3p reversed these effects ().
Figure 2. Administration of exogenous agomir-141-3p aggravated MCAO-mediated brain injury and apoptosis in vivo.
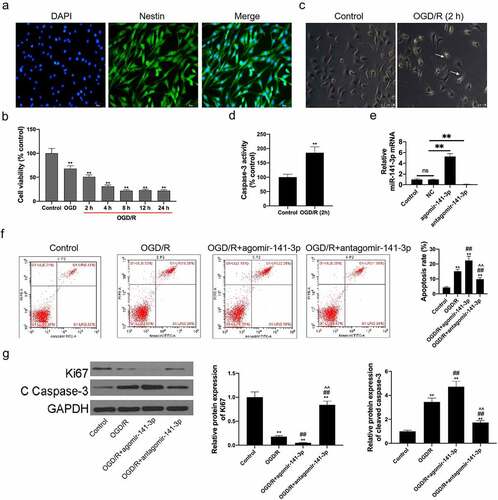
Agomir-141-3p promoted OGD/reoxygenation-induced NSCs apoptosis
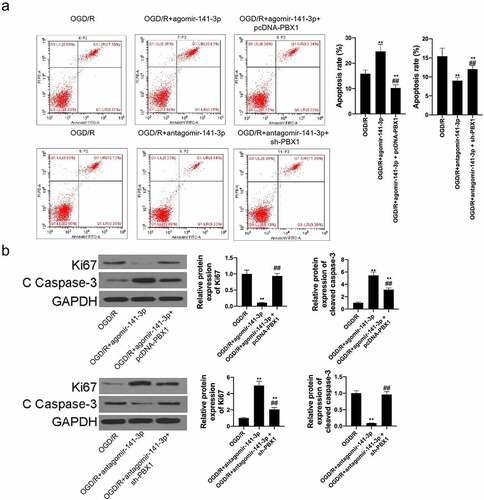
In this study, NSCs were isolated from mouse brain tissues and identified with up to 95% of Nestin (the NSC marker protein) positive cells (). The starved NSCs were culture in monolayers for OGD/reoxygenation and administration. Furthermore, CCK-8 assay results demonstrated an exact NSC injury by OGD for 4 h, with cell viability dropping from 99.5 ± 1.2% to 68.0 ± 5.8%; further injury was promoted by 2–24 h reoxygenation, with cell viability dropping to 51.0 ± 3.9%, 31.0 ± 2.7%, 22.0 ± 1.8%, 23.0 ± 2.1%, and 22.0 ± 1.9% (). OGD for 4 h followed by reoxygenation for 2 h was used for experimental administration with moderate cell injury (cell viability dropped to 51.0 ± 3.9%). The results of morphology () showed that the OGD/reoxygenation-treated NSCs were detected with broken cytomembrane and axons and a wizened cell body, compared to healthy control NSCs with a bright and clear cell body and smooth and extended axons. Caspase-3 activity in OGD/reoxygenation-treated NSCs increased significantly (). Transfection of agomir-141-3p could upregulate miR-141-3p level and transfection of antagomir-141-3p could downregulate its level in NSCs (). Agomir-141-3p increased OGD/reoxygenation-induced apoptosis of the NSCs, and antagomir-141-3p abolished the apoptosis-promoting effect caused by OGD/reoxygenation (). Ki67 expression in OGD/reoxygenation-treated NSCs carrying with agmir-141-3p was lower than that in OGD/reoxygenation-treated NSCs, and cleaved caspase-3 expression in OGD/reoxygenation-treated NSCs carrying with agmir-141-3p was higher than that in OGD/reoxygenation-treated NSCs (). Antagomir-141-3p reversed the functions of OGD/reoxygenation inhibiting Ki67 expression and increasing cleaved caspase-3 expression.
Figure 3. Administration of exogenous agomir-141-3p aggravated MCAO-mediated brain injury and apoptosis in vitro.
miR-141-3p regulated MCAO-induced brain injury and NSCs apoptosis via targeting PBX1 in vivo and in vitro
TargetScan database was used to predict the possible target of miR-141-3p and PBX1 was predicted as its target (). Co-transfection of the dual-luciferase reporter plasmid containing PBX1 WT- 3’-UTR with agomir-141-3p decreased the reporter activity in cells, and co-transfection of the plasmid containing PBX1 MUT- 3’-UTR with antagomir-141-3p increased the activity in cells (). Brain tissue miR-141-3p was detected by RT-PCR and significantly increased in agomir-141-3p group, on the contrary, intracerebroventricular injection of antagomir-141-3p reduced the miR-141-3p level, otherwise, brain tissue PBX1 was detected by RT-PCR and significantly decreased in agomir-141-3p group, and intracerebroventricular injection of antagomir-141-3p increased PBX1 level (). PBX1 expression was inhibited by agomir-141-3p treatment and was increased by antagomir-141-3p treatment (). Treatment of agomir-141-3p increased miR-141-3p expression () and inhibited PBX1 expression (). Next, miR-141-3p expression was reduced in NSCs treatment with antagomir-141-3p and PBX1 expression was increased in NSCs treatment with antagomir-141-3p. PBX1 expression in sh-PBX1 transfected NSCs decreased significantly and in pcDNA-PBX1 transfected NSCs increased significantly (). Next, TTC staining analysis of the cerebral infarct in mice infection with sh-PBX1 and pcDNA3.1-PBX1 was shown in . MCAO caused a significant brain infarction and deteriorated neurobehavior at 24 h after surgery, and administration of PBX1 overexpression reduced the infarct volume and improved the neurological scores, and the PBX1 silencing that specific to inhibit PBX1 expression, afterward abolished the protective effect of PBX1 overexpression (). After administration for 21 days in MCAO mice, PBX1 overexpression greatly ameliorated the performance of the foot fault test, which was reversed by PBX1 inhibition (). PBX1 inhibition significantly reduced the brain water content in the ischemic hemisphere, and PBX1 overexpression increased it in the MCAO mice (). The results of H&E staining and TUNEL analysis showed that MCAO induced a significant brain injury and cell apoptosis at 24 h after surgery (), and administration of PBX1 overexpression reduced the brain injury and cell apoptosis in MCAO mice. However, PBX1 silencing aggravated MCAO-induced brain injury and cell apoptosis ().
Figure 4. miR-141-3p regulated MCAO-induced brain injury and NSCs apoptosis via targeting PBX1 in vitro .
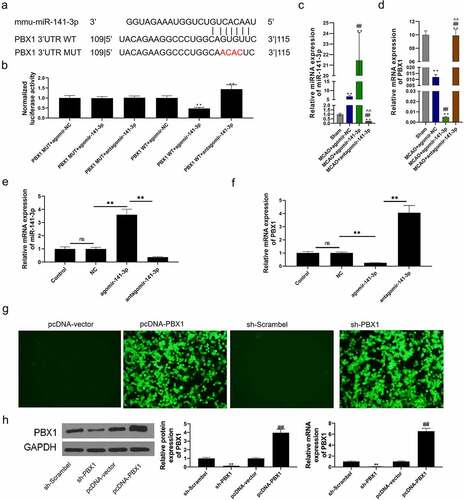
Figure 5. miR-141-3p regulated MCAO-induced brain injury and NSCs apoptosis via targeting PBX1 in vivo.
Whereafter, we found that PBX1-overexpression inhibited the apoptosis-promoting effects of agomir-141-3p on OGD/reoxygenation-treated NSCs, likewise, suppression of PBX1 inhibited the apoptosis-inhibiting effects of antagomir-141-3p on OGD/reoxygenation-treated NSCs (). In addition, overexpression of PBX1 abolished the Ki67-decreasing and cleaved caspase-3-increasing effects of agomir-141-3p on OGD/reoxygenation-treated NSCs. Suppression of PBX1 reversed the Ki67-increasing and cleaved caspase-3-decreasing effects of antagomir-141-3p on OGD/reoxygenation-treated NSCs ().
Figure 6. miR-141-3p regulated MCAO-induced brain injury and NSCs apoptosis via targeting PBX1 in vitro.
PBX1 downregulated PROK2 expression transcriptionally in NSCs and affected OGD/reoxygenation-induced cell apoptosis
With the aid of two online bioinformatics tools (UCSC and JASPAR), PBX1 was predicted to have binding sites in the region of the PROK2 promoter (). Brain tissue PROK2 expression was significantly decreased in the MACO and agomir-141-3p group, on the contrary, intracerebroventricular injection of antagomir-141-3p upregulated the PROK2 level (). Next, PROK2 expression was inhibited by agomir-141-3p treatment and increased by antagomir-141-3p treatment (). Thereafter, we discovered that PROK2 expression was apparently increased under PBX1-inhibition and decreased under PBX1 overexpression (). PROK2 expression in sh-PROK2 transfected NSCs decreased significantly (). ChIP assays further confirmed that PBX1was efficiently bound to the PROK2 promoter-specific region in NSCs (). Moreover, PROK2-inhibition reversed the apoptosis-promoting effects of PBX1-inhibition on OGD/reoxygenation-treated NSCs (). The functions of PBX1-inhibition inhibiting Ki67 expression and increasing cleaved caspase-3 expression in OGD/reoxygenation-treated NSCs were reversed by PROK2-inhibition ().
Figure 7. PBX1 downregulated PROK2 expression transcriptionally in NSCs and affected OGD/reoxygenation-induced cell apoptosis.
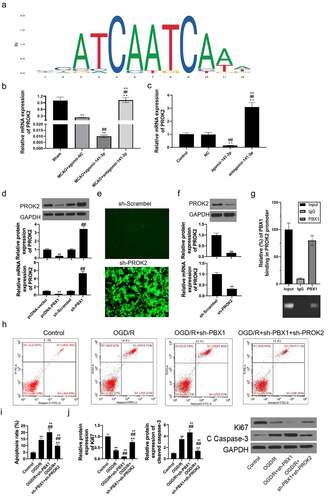
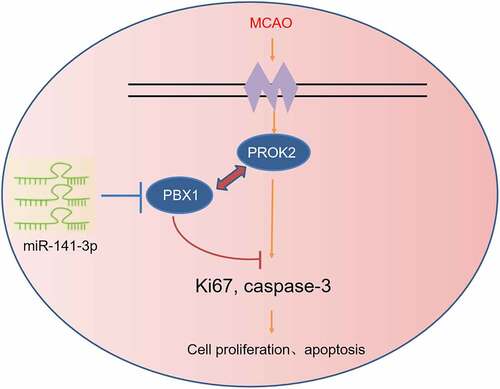
Figure 8. The schematic diagram depicts the regulatory mechanism of miR-141-3p in MCAO.
Discussion
In the present study, we found that miR-141-3p expression was significantly increased. The administration of miR-141-3p agonist (agomir-141-3p) aggravated MCAO-mediated infarction volume, neurological deficits, and apoptosis. Next, the damage effects of agomir-141-3p antagonist were abolished by antiagomir-141-3p. Therefore, we demonstrated that the suppression of miR-141-3p has a protective effect on MCAO mice. It was reported that miR-141-3p involved in the neurogenesis of NSCs. Therefore, we speculated that miR-141-3p was served as a therapeutic target in ischemic stroke patients. Subsequently, we explored their molecular mechanism. PBX1,a target gene of miR-141-5p, belongs to the family of transcription factors in pre-B cell leukemia (pre-B cell leukemia, PBC), which contributes to the formation of embryonic proximal-distal axis and participates in the regulation of proliferation, apoptosis, and differentiation during embryonic development [Citation15]. The deletion of PBX1 gene in mouse embryos will lead to the death of embryos during development, accompanied by impaired hematopoiesis, and organ regeneration disorders, as well as developmental deformities of cervical vertebrae, ribs, proximal limbs, and heart [Citation16,Citation17]. PBX1 protein can directly regulate the growth of the nervous system, hematopoietic system, and organs through its transcriptional activation. In addition, PBX1 regulated stem cells’ self-renewal, proliferation, and differentiation. It was reported that PBX1 expression downregulated significantly in nigral dopaminergic neurons of the patients with Parkinson’s disease (PD) [Citation15]. Therefore, PBX1 protein may be directly involved in neurons proliferation and apoptosis. Results indicated that overexpression of PBX1 played an important protective role in the development of the nervous system. Coincidentally, we further demonstrated that agomir-141-3p targeting PBX1 to suppress its expression, consequently regulating Ki67, and cleaved caspase-3 expression in brain tissues of mice after MCAO and NSCs after OGD/reoxygenation and promoting its apoptosis. These suggested that overexpression of miR-141-3p aggravated MCAO-mediated infarction volume, neurological deficits, and NSCs apoptosis via inhibiting PBX1 expression.
However, there are interactions among genes. PBX1 cannot regulate cell proliferation and apoptosis alone, and the genes that interact with it need to be further screened. Herein, we demonstrated for the first time that PROK2 was transcriptionally modulated by PBX1 in NSCs. It has been reported that PROK2 is a cysteine-rich secreted protein that contains a conserved N-terminal sequence of AVITGA and 10 cysteines [Citation18]. Some studies showed that PROK2 was an oncogene in human cancer and an insult-inducible endangering factor for cerebral ischemic injury [Citation18,Citation19]. The pharmacological blockade of the prokineticin system can exert neuroprotective effects by preventing PROK2 upregulation, thus suggesting that it is a new pathological hallmark for AD at least in animal models [Citation18]. These studies show that inhibition of PROK2 protected the nervous system. Next, we found that suppression of PBX1 promoted the apoptosis-promoting effect of OGD/reoxygenation-treated NSCs, which were abolished by inhibition of PROK2. It showed that overexpression of PBX1 inhibited OGD/reoxygenation-induced apoptosis in NSCs via downregulated PROK2 transcription. To sum up, miR-141-3p/PBX1/PROK2 axis plays an important role in MCAO ().
In this study, the high expression of miR-141-3p could aggravate MCAO-mediated brain injury and NSCs apoptosis, which was further confirmed at the MCAO animal model and cellular level. Whereafter, its target gene-PBX1 silencing could also aggravate MCAO-mediated brain injury and NSCs apoptosis. The above studies showed that the high expression of miR-141-3p was consistent with the role of PBX1 silencing in MCAO. It proved that miR-141-3p targeting PBX1 accelerated the progression of MCAO. Furthermore, PROK2, a downstream transcription factor of PBX1, also participated in OGD/reoxygenation-induced cell apoptosis through interaction with PBX1. Because the constraints of time and budget, we did not continue to explore the role of the co-interaction of PROK2 and PBX1 in the progression of MCAO and the signaling pathways involved in vivo. Therefore, we hope to collect relevant clinical samples at future to continually study the expression levels of miR-141-3p, PBX1, and PROK2 in patients with MCAO and analyze their correlation with clinical phenotypes. Finally, the significance of the miR-141-3p/Pbx1/Prok2 axis for the occurrence and development of MCAO was discussed from the animal, cell, and clinical levels.
Conclusion
miR-141-3p increased MCAO-mediated infarction volume, neurological deficits, and NSCs apoptosis via inhibiting PBX1 expression to downregulate PROK2 transcription.
Ethics approval
The study had been approved by Animal Care and Use, Committee of Nanjing University. All animal protocols were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee.
Author contributions
Tingting Liu and Jinzhou Feng wrote the main manuscript, performed all the experiments and analyzed the data. Zhenjie Sun, Linlin Sun, Shuangshuang Dong, and Zhenwei Guo collected the data. Mingli He and Guanghui Zhang designed the study. All authors read and approved the final manuscript and agree to be accountable for all aspects of the research in ensuring that the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Additional information
Funding
References
- Randolph SA. Ischemic stroke. Workplace Health Saf. 2016;64(9):444.
- Larsson A, Karlsson C, Rentzos A, et al. Do patients with large vessel occlusion ischemic stroke harboring prestroke disability benefit from thrombectomy? J Neurol. 2020;267(9):2667–2674. DOI:10.1007/s00415-020-09882-5
- Ma J, Shui S, Han X, et al. microRNA-200a silencing protects neural stem cells against cerebral ischemia/reperfusion injury. PLoS One. 2017;12:e0172178.
- Buckley A, Hagler SB, Lettry V, et al. Generation and profiling of tumor-homing induced neural stem cells from the skin of cancer patients. Mol Ther. 2020:28(7):1614–1627. DOI:10.1016/j.ymthe.2020.04.022
- Lin CQ, Chen LK. Cerebral dopamine neurotrophic factor promotes the proliferation and differentiation of neural stem cells in hypoxic environments. Neural Regen Res. 2020;15(11):2057–2062.
- Liu Q, Li Y, Zhou L, et al. GRP78 promotes neural stem cell antiapoptosis and survival in response to oxygen-glucose deprivation (Ogd)/reoxygenation through PI3K/Akt, ERK1/2, and NF-kappaB/p65 pathways. Oxid Med Cell Longev. 2018;3541807. DOI:10.1155/2018/3541807
- Wang J, Hu Y, Ye C, et al. miR-1224-5p inhibits the proliferation and invasion of ovarian cancer via targeting SND1. Hum Cell. 2020;33(3):780–789. DOI:10.1007/s13577-020-00364-4.
- Xue WS, Wang N, Wang NY, et al. miR-145 protects the function of neuronal stem cells through targeting MAPK pathway in the treatment of cerebral ischemic stroke rat. Brain Res Bull. 2019;144:28–38.
- Wang J, Wang G, Li B, et al. miR-141-3p is a key negative regulator of the EGFR pathway in osteosarcoma. Onco Targets Ther. 2018;11:4461–4478.
- Li M, Huang H, Cheng F, et al. Promotes proliferation and metastasis of nasopharyngeal carcinoma by targeting NME1. Adv Med Sci. 2020;65(2):252–258. miR-141-3p. DOI:10.1016/j.advms.2020.03.005.
- Guan G, Niu X, Qiao X, et al. Upregulation of Neural Cell Adhesion Molecule 1 (NCAM1) by hsa-miR-141-3p suppresses ameloblastoma cell migration. Med Sci Monit. 2020;26:e923491.
- Verma R, Ritzel RM, Harris NM, et al. Inhibition of miR-141-3p ameliorates the negative effects of poststroke social isolation in aged mice. Stroke. 2018;49:1701–1707.
- Zuo X, Lu J, Manaenko A, et al. MicroRNA-132 attenuates cerebral injury by protecting blood-brain-barrier in MCAO mice. Exp Neurol. 2019;316:12–19.
- Zhang W, Gu GJ, Zhang Q, et al. NSCS promote hippocampal neurogenesis, metabolic changes and synaptogenesis in APP/PS1 transgenic mice. Hippocampus. 2017;27(12):1250–1263. DOI:10.1002/hipo.22794
- Villaescusa JC, Li B, Toledo EM, et al. A PBX1 transcriptional network controls dopaminergic neuron development and is impaired in Parkinson’s disease. Embo J. 2016;35(18):1963–1978. DOI:10.15252/embj.201593725
- Xu X, Zhou Y, Fu B, et al. PBX1 promotes development of natural killer cells by binding directly to the Nfil3 promoter. Faseb J. 2020;34(5):6479–6492. DOI:10.1096/fj.202000121R
- Zhou Y, Fu B, Xu X, et al. PBX1 expression in uterine natural killer cells drives fetal growth. Sci Transl Med. 2020;12(537):eaax1798. DOI:10.1126/scitranslmed.aax1798
- Lattanzi R, Maftei D, Petrella C, et al. Involvement of the chemokine prokineticin-2 (PROK2) in Alzheimer’s disease: from animal models to the human pathology. Cells. 2019;8(11):1430. DOI:10.3390/cells8111430
- Kurebayashi H, Goi T, Shimada M, et al. Prokineticin 2 (PROK2) is an important factor for angiogenesis in colorectal cancer. Oncotarget. 2015;6:26242–26251.