
ABSTRACT
DNA double-strand breaks (DSBs) pose a major threat to the genome, so the efficient repair of such breaks is essential. DSB processing and repair is affected by 53BP1, which has been proposed to determine repair pathway choice and/or promote repair fidelity. 53BP1 and its downstream effectors, RIF1 and shieldin, control 3’ overhang length, and the mechanism has been a topic of intensive research. Here, we highlight recent evidence that 3’ overhang control by 53BP1 occurs through fill-in synthesis of resected DSBs by CST/Polα/primase. We focus on the crucial role of fill-in synthesis in BRCA1-deficient cells treated with PARPi and discuss the notion of fill-in synthesis in other specialized settings and in the repair of random DSBs. We argue that – in addition to other determinants – repair pathway choice may be influenced by the DNA sequence at the break which can impact CST binding and therefore the deployment of Polα/primase fill-in.
KEYWORDS:
Introduction
DSBs can be repaired by multiple pathways whose engagement is influenced by the structure of the DNA ends (). Blunt or minimally-processed DNA ends can be repaired throughout the cell cycle by classical non-homologous end joining (cNHEJ), a pathway mediated by Ku70/80, DNA-PKcs, DNA Ligase 4 (Lig4), and several accessory factors [Citation1]. Resected DNA ends with a 3’ single-stranded (ss) overhang can undergo error-free Rad51-dependent homology-directed repair (HDR) when the sister chromatid can be used as a donor [Citation2]. However, DNA ends with long 3’ overhangs can also be processed by the Rad52-dependent single-strand annealing (SSA) pathway [Citation3]. Since SSA is prone to deletions, excessively long 3’ overhangs threaten the integrity of the genome.
Figure 1. The 53BP1 pathway controls DNA end structure at DSBs. a, Schematic of the DNA end structures acted upon by the primary DSB repair pathways. DSB: double-strand break; cNHEJ: classical non-homologous end-joining; HDR: homology-directed repair; SSA: single-strand annealing. b, Top, overview of 53BP1, RIF1, shieldin, and CST/Polα/primase interactions. Bottom, Two models for how the 53BP1 pathway limits 3’ overhang length. c, Three context-dependent repair reactions where 53BP1 activity has been studied. Check marks indicate a requirement for a 53BP1 downstream effector or function. Question marks and reduced opacity cartoons denote uncertainties based on the current data.
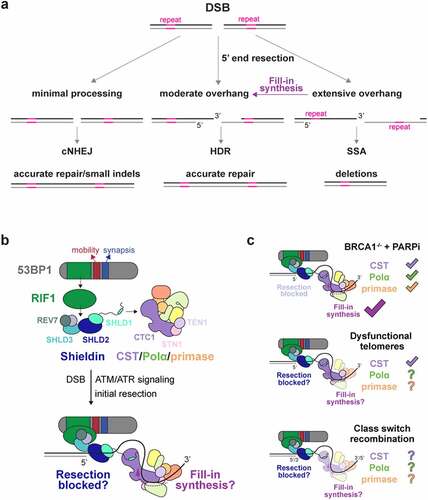
The terminal structure of DSBs is affected by 53BP1, which localizes to damaged chromatin in response to ATM or ATR signaling [Citation4]. 53BP1 limits the formation of long 3’ overhangs through its downstream effector RIF1 and its associated shieldin complex (SHLD1–3 and REV7) (see Box 1; ). Shieldin can bind ssDNA in vitro and has been proposed to block 5’ end resection [Citation21]. However, there is currently no data on the ability of shieldin to bind DNA templates with recessed 5’ ends, nor is there data showing that shieldin prevents nucleases from degrading 5’ ends in vitro. Importantly, shieldin also recruits a second ssDNA-binding complex, CST (CTC1, STN1, TEN1) and its associated DNA Polymerase alpha/primase (hereafter Polα/primase) (see Box 2; ; ), which can limit 3’ overhangs through fill-in synthesis [Citation8,Citation55].
Figure 2. CST and CST/Polα/primase structure and function. a, Domain schematics for CST and Polα/primase subunits. OB: oligosaccharide/oligonucleotide-binding fold; wH: winged helix-turn-helix; 3 H: 3-helix bundle; N: POLA1 flexible N-terminal region; NTD: N-terminal domain; EXO: exonuclease domain, inactive; CTD: C-terminal domain; CRL: CTC1-Recognition Loop; PDE: phosphodiesterase domain. Colors from domain schematics are consistent in all figures. CP mutations in fill-in components are indicated with markers. Colors indicate mutants reported to disrupt Polα/primase association (red), ssDNA-binding (cyan), or CST complex formation (orange). Black markers represent mutants with other or uncharacterized defects. b, Summary of CST substrates and reported affinities (order of magnitude). c, Cartoon summary of CST and CST/Polα/primase cellular functions. RC: recruitment complex; PIC: pre-initiation complex. d, Structural models of CST in ssDNA-bound (top) and apo (bottom) conformations. Composite models were generated from cryo-EM structures (PDB-6W6W [Citation25], maps (EMD -21,56346), and AlphaFold models of individual subunits (AF- Q54WQ3/Q9H668/Q86WV5 [Citation53,Citation54]. e, Cryo-EM structure of CST/Polα/primase in the recruitment complex conformation [Citation48]. The model is scaled and rotated about CST relative to (d) as indicated. f, Cryo-EM structure of CST/Polα/primase in the pre-initiation complex conformation [Citation49]. The model is rotated about CST relative to (e) as indicated.
![Figure 2. CST and CST/Polα/primase structure and function. a, Domain schematics for CST and Polα/primase subunits. OB: oligosaccharide/oligonucleotide-binding fold; wH: winged helix-turn-helix; 3 H: 3-helix bundle; N: POLA1 flexible N-terminal region; NTD: N-terminal domain; EXO: exonuclease domain, inactive; CTD: C-terminal domain; CRL: CTC1-Recognition Loop; PDE: phosphodiesterase domain. Colors from domain schematics are consistent in all figures. CP mutations in fill-in components are indicated with markers. Colors indicate mutants reported to disrupt Polα/primase association (red), ssDNA-binding (cyan), or CST complex formation (orange). Black markers represent mutants with other or uncharacterized defects. b, Summary of CST substrates and reported affinities (order of magnitude). c, Cartoon summary of CST and CST/Polα/primase cellular functions. RC: recruitment complex; PIC: pre-initiation complex. d, Structural models of CST in ssDNA-bound (top) and apo (bottom) conformations. Composite models were generated from cryo-EM structures (PDB-6W6W [Citation25], maps (EMD -21,56346), and AlphaFold models of individual subunits (AF- Q54WQ3/Q9H668/Q86WV5 [Citation53,Citation54]. e, Cryo-EM structure of CST/Polα/primase in the recruitment complex conformation [Citation48]. The model is scaled and rotated about CST relative to (d) as indicated. f, Cryo-EM structure of CST/Polα/primase in the pre-initiation complex conformation [Citation49]. The model is rotated about CST relative to (e) as indicated.](/cms/asset/8dc39a79-7237-40a6-b866-58d0858b9072/kccy_a_2123886_f0002_oc.jpg)
Figure 3. CST-shieldin interactions. a, Domain schematics for CST and shieldin subunits, with interacting regions mapped in gray. Abbreviations as in Figure 2A. EIF4E-l: eukaryotic translation initiation factor 4E-like. HORMA: HOP1, REV7, MAD2-like. SHLD3 interacts with two copies of REV7 in a 1:2 stoichiometry [Citation67]. b, Composite structural model of CST-shieldin complex generated from the cryo-EM structure of ssDNA-bound CST (PDB-6W6W [Citation46]) superposed with AlphaFold-multimer models [Citation61,Citation62] (default settings, max_template_date=2022-07-01, AMBER relaxed) of CTC1-SHLD1, SHLD1-SHLD2-SHLD3, and the crystal structure of SHLD2-C-REV7-O-REV7-SHLD3 (O-REV7: open conformation; C-REV7: closed conformation; PDB-6KTO [Citation67]). Flexible, unstructured regions predicted with low confidence (pLDDT < 30) are shown with dashed lines. Models were aligned in PyMOL (Schrödinger) and visualized in ChimeraX [Citation68]. c, Top, zoom-in of the CTC1-SHLD1 interface with interacting residues labeled. Residues highlighted in bold face (L18, D19, L20, and P21) are deleted in the SHLD1D mutant. Bottom, SHLD1 N-terminus sequence, adapted from [Citation7]. Asterisks indicate residues which, upon mutation, weaken the interaction between CTC1 and SHLD1. Red asterisks indicate loss of interaction due to a single amino acid substitution or deletion. Residues deleted in SHLD1D and the alpha helix (residues 23-32) are indicated. d, Predicted aligned error (PAE) plots of the top six ranked CTC1-SHLD1 Alphafold-multimer models. Domain schematics the same as in a. Green arrows indicate high-confidence in the position prediction of SHLD1 N-terminus relative to CTC1.
![Figure 3. CST-shieldin interactions. a, Domain schematics for CST and shieldin subunits, with interacting regions mapped in gray. Abbreviations as in Figure 2A. EIF4E-l: eukaryotic translation initiation factor 4E-like. HORMA: HOP1, REV7, MAD2-like. SHLD3 interacts with two copies of REV7 in a 1:2 stoichiometry [Citation67]. b, Composite structural model of CST-shieldin complex generated from the cryo-EM structure of ssDNA-bound CST (PDB-6W6W [Citation46]) superposed with AlphaFold-multimer models [Citation61,Citation62] (default settings, max_template_date=2022-07-01, AMBER relaxed) of CTC1-SHLD1, SHLD1-SHLD2-SHLD3, and the crystal structure of SHLD2-C-REV7-O-REV7-SHLD3 (O-REV7: open conformation; C-REV7: closed conformation; PDB-6KTO [Citation67]). Flexible, unstructured regions predicted with low confidence (pLDDT < 30) are shown with dashed lines. Models were aligned in PyMOL (Schrödinger) and visualized in ChimeraX [Citation68]. c, Top, zoom-in of the CTC1-SHLD1 interface with interacting residues labeled. Residues highlighted in bold face (L18, D19, L20, and P21) are deleted in the SHLD1D mutant. Bottom, SHLD1 N-terminus sequence, adapted from [Citation7]. Asterisks indicate residues which, upon mutation, weaken the interaction between CTC1 and SHLD1. Red asterisks indicate loss of interaction due to a single amino acid substitution or deletion. Residues deleted in SHLD1D and the alpha helix (residues 23-32) are indicated. d, Predicted aligned error (PAE) plots of the top six ranked CTC1-SHLD1 Alphafold-multimer models. Domain schematics the same as in a. Green arrows indicate high-confidence in the position prediction of SHLD1 N-terminus relative to CTC1.](/cms/asset/3878252c-f352-4b48-87ab-92e3cad06ab0/kccy_a_2123886_f0003_oc.jpg)
Box 1
53BP1 as a fidelity factor. 53BP1 has three main effects at DSBs. 53BP1 limits the formation of long 3’ overhangs, increases the mobility of damaged DNA ends [Citation5,Citation6], and promotes synapsis of DNA ends [Citation7] (). Overhang control is mediated by RIF1, which recruits the shieldin complex [Citation8–18]. 53BP1 activity is prominent in three contexts which have been used as surrogates for understanding how 53BP1 acts at DSBs (): 1. BRCA1-deficient cells treated with PARPi; 2. Telomeres that are recognized as DSBs when shelterin is compromised; and 3. Immunoglobulin class-switch recombination. In these three contexts, 53BP1 promotes cNHEJ, which has led to the view that 53BP1 governs DSB repair pathway choice generally, favoring cNHEJ at the expense of HDR, presumably by keeping DNA ends blunt. The binding of shieldin to ssDNA – and the discovery of fill-in synthesis at DSBs which is the subject of this review – leads to the surprising conclusion that downstream effectors of 53BP1/RIF1 most likely act on DNA substrates which have already been resected. The architecture of DNA damage foci is complex, and the spatiotemporal aspects of recruitment of 53BP1 and its downstream factors and the status and position of the DNA end within the foci are not yet known.
We recently argued that rather than controlling the “choice” between cNHEJ and HDR, 53BP1 promotes the fidelity of DSB repair by averting illegitimate recombination [Citation4]. Briefly, long 3’ overhangs containing homologous or repetitive sequences raise the danger of the mutagenic SSA pathway (). CST/Polα/primase fill-in synthesis suppresses long overhang formation but may be unable to reconstitute a blunt end. By analogy to the telomeric overhang which invades the telomeric duplex to form a t-loop [Citation19], it is plausible that at DSBs, the overhang control function of the 53BP1 pathway leaves a moderately-sized overhang competent for HDR. Its mobility and synapsis functionalities also make 53BP1 well-suited to favoring high fidelity repair over illegitimate recombination (for in-depth discussion see Mirman and de Lange [Citation4]).
The notion that 53BP1 evolved simply to promote cNHEJ is also doubtful because shieldin recruits the XPG-related nuclease, ASTE1, which contributes to CSR and PARPi efficacy in BRCA1-deficient cells [Citation20]. As ASTE1 can remove part of the 3’ overhang, its 53BP1- and shieldin-dependent recruitment is predicted to create deletions at DSBs that are shuttled into cNHEJ. It is difficult to imagine how such a mutagenic pathway could have been selected for as a mechanism to promote cNHEJ. Perhaps the actual utility of ASTE1 in general DSB repair is to remove crosslinked proteins or other blocks from 3’ ends, allowing high-fidelity repair through HDR. In summary, although 53BP1 and its downstream factors can tilt the balance toward cNHEJ in the specialized scenarios favored by current research, their role in general DSB repair is likely to be more complex (and more interesting) than promoting cNHEJ.
Box 2
CST and CST/Polα/primase. synthesis is executed by Polα/primase in complex with the trimeric CST (), which was first observed as a Polα/primase accessory factor that stimulates both the polymerase and primase activities of the enzyme [Citation22,Citation23]. CST is related to the abundant ssDNA-binding factor RPA [Citation24,Citation25]. It binds a range of ssDNA and junction substrates with a preference for ssDNA with G-runs [Citation26–28] () and these broad DNA-binding activities appear to underlie CST’s diverse functions in genome maintenance (). CST binds numerous loci containing G-rich repeats, at telomeres and genome wide, where it can facilitate replication and fork restart in part by unwinding G-quadruplex (G4) structures [Citation29–31]. Whether Polα/primase participates in this non-canonical replication function is unknown. At telomeres, CST contributes to the regulation of telomerase [Citation32] but its primary role (for which CST is thought to have evolved with Polα/primase [Citation33]) lies in the maintenance of the C-rich telomeric repeat strand [Citation29,Citation34–40].
The telomere-specific functions of CST rely on its recruitment by the TPP1 and POT1 subunits of shelterin (), which, like CST, are needed for reconstituting the 5’-ended C-strand after DNA replication [Citation32,Citation34,Citation38,Citation41–47]. Cryo-EM studies showed that CST/Polα/primase is recruited to telomeres in a compact, auto-inhibited state (recruitment complex (RC); ), where the POLA1 active site is occluded [Citation48]. In ways that are not understood, the complex then transitions into an extended, active state, where the POLA1 catalytic core is accessible for catalysis [Citation49] (pre-initiation complex (PIC); ).
Deficient telomeric fill-in synthesis results in the severe developmental disorder Coats plus syndrome (CP [Citation50]) which is proposed to involve stochastic telomere truncations resulting from unmitigated C-strand resection [Citation38]. CP patients generally are compound heterozygous, bearing one allele of CTC1 that is nonfunctional with the second allele carrying a hypomorphic point mutation or small deletion [Citation50] (). Mutations in STN1, TEN1, and POT1 have also been reported [Citation38,Citation51,Citation52]. Some of the CP mutations map to interfaces between CST subunits [Citation25] or to the interface between CST and Polα/primase in the recruitment state [Citation38] but for most CP mutations the molecular basis of their effect is not yet clear.
This review summarizes recent evidence that fill-in synthesis by CST/Polα/primase counteracts 5’ end resection at DSBs, thereby avoiding the long 3’ overhangs that could engender SSA-mediated deletions [Citation4,Citation9,Citation56]. This fill-in synthesis plays a crucial role in the efficacy of PARPi in BRCA1-deficient cells and, as we argue here, is likely to contribute to the structure of DSBs, and thus DSB repair, in other settings.
The role of CST/Polα/primase in limiting 3’ overhangs at DNA ends
53BP1, together with RIF1 and shieldin, determines PARPi efficacy in BRCA1-deficient cells, affects the processing of dysfunctional telomere ends, and promotes immunoglobulin class switch recombination (CSR) (). In each context, 53BP1 limits the formation of ssDNA at DNA ends, but whether 53BP1 acts by blocking 5’ end resection through the agency of shieldin or by mediating CST/Polα/primase-dependent fill-in of resected ends has been a matter of debate. Importantly, inhibition of CST, like 53BP1, RIF1, and shieldin, improves the survival of BRCA1-deficient cells after treatment with PARPi [Citation8]. Assuming that CST functions with Polα/primase, this finding implicates fill-in synthesis as a determinant of PARPi efficacy.
CST/Polα/primase has diverse roles in genome maintenance, including its critical role in maintaining the C-rich telomeric repeat strand at chromosome ends [Citation29,Citation34–40], resolving G4 structures that hinder replication at telomeres and other sites in the genome [Citation26,Citation29–31,Citation57], and was proposed to mediate fill-in synthesis at DSBs [Citation8] (see Box 2 and ). Data published in the past year now provide strong support for CST/Polα/primase-mediated fill-in synthesis at resected DSBs.
Fill-in synthesis was directly visualized based on the incorporation of BrdU at DSBs generated by FOKI cutting of a LacO array [Citation55]. 53BP1, shieldin, CST, Polα, and primase are required for most of the observed BrdU incorporation. (In the absence of these factors, some residual BrdU signal is expected from DNA synthesis accompanying other repair pathways.) Similarly, shieldin- and Polα/primase-dependent fill-in synthesis was documented using a proximity ligation assay for coincidence of BrdU with γH2AX at PARPi-induced chromosome breaks in BRCA1-deficient cells [Citation55].
Further evidence for fill-in synthesis at DSBs came from a combination of sequencing methods: END-seq which captures resection endpoints after removal of the 3’ overhang prior to adaptor ligation and sequencing [Citation58]; RPA ssDNA sequencing; and SAR-seq which maps nascent DNA synthesis through EdU incorporation [Citation59]. These methods were used to monitor hundreds of AsiSI-induced DSBs in v-abl-transformed murine B cells. Most resection endpoints captured by END-seq in wild-type cells map close to the AsiSI site, yet SAR-seq detected nascent DNA incorporation around these sites, indicative of post-resection fill-in synthesis [Citation60]. In cNHEJ (Lig4)-deficient cells, both END-seq and SAR-seq reads span kilobases surrounding the DSB. This fill-in synthesis depends on Polα/primase but is only mildly affected by 53BP1 or shieldin loss [Citation60], suggesting that some Polα/primase recruitment is independent of 53BP1 and shieldin. This is potentially due to the ability of CST to bind to G-rich DNA without the aid of 53BP1 (Box 2; ; and see discussion below). When Polα is inhibited with Aphidicolin (which does not affect primase), the 3’ overhangs generated at AsiSI sites could template in vitro DNA synthesis by prokaryotic or phage DNA polymerases. In contrast, 3’ overhangs generated in cells where both Polα and primase were inhibited did not support in vitro DNA synthesis, suggesting that the added DNA polymerases used primers made by primase. The involvement of primase is consistent with the finding that degradation of primase (like loss of 53BP1, shieldin, or CST) reversed hallmarks of BRCA1-deficiency [Citation55].
CST/Polα/primase fill-in synthesis is also implicated in the generation of short tandem duplications at Cas9 nickase-induced DSBs bearing 43–50 nt 3’ overhangs [Citation61]. These duplications were less frequent when Cas9 was directed to cut such that the 3’ overhang would constitute a “primase desert”, where the lack of pyrimidines in the ss template curbs the ability of primase to initiate RNA synthesis [Citation62]. Similarly, inhibition of Polα or loss of 53BP1, shieldin, or CST led to reduced incidence of the short tandem duplications [Citation61].
Testing the role of shieldin in BRCA1-deficient cells
A key question was whether CST/Polα/primase-mediated fill-in synthesis and shieldin function (presumably blocking 5’ end resection) contributed separately to PARPi efficacy, or whether shieldin primarily serves to recruit CST. This question was addressed with a SHLD1 separation of function mutant (SHLD1∆ [Citation55] see Box 3) that retains the ability to interact with other shieldin subunits but lacks its CTC1 binding site and is deficient in CST recruitment to DSBs. BRCA1-deficient cells that express SHLD1∆ resemble cells lacking SHLD1 indicating that SHLD1∆ behaves as a null mutant. Importantly, when the link between SHLD1∆ and CTC1 was restored (using chemically-induced SNAP-HALO interaction) SHLD1∆ behaved like wild-type SHLD1 [Citation55]. These experiments argue that shieldin depends on its interaction with CST for its function. Conversely, tethering an FHA-tagged version of STN1 to sites of DNA damage allows CST to function at DSBs in a 53BP1- and shieldin-independent manner [Citation55].
Box 3.
SHLD1∆. A yeast two-hybrid screen identified residues important for the interaction between SHLD1 and CTC1 [Citation55]. This allowed generation of a mutant version of SHLD1 (SHLD1∆; deletion of amino acids 18–21), which shows diminished interaction with CTC1 and inability to recruit CST to DNA damage foci [Citation55]. Remarkably, the AlphaFold-multimer protein structure prediction tool [Citation53,Citation63,Citation65] identified the same CTC1 interaction site in SHLD1 with high-confidence (). According to AlphaFold-multimer, SHLD1 uses a helix and an ordered loop (containing aa 18–21) in its flexible N-terminal extension to bind CTC1. The CTC1-SHLD1 interface is primarily hydrophobic and supplemented with solvent-exposed polar interactions, exemplified by the amphipathic helix in SHLD1 (aa 22–31) that buries a hydrophobic cleft in CTC1 (). Strikingly, this helix is not predicted in the AlphaFold model of SHLD1 alone (AF-Q8IYI0 [Citation53,Citation54,Citation64]); in silico ordering of the helix is only observed upon binding CTC1. Of the residues deleted in SHLD1Δ, L18, L20, and P21 are involved in hydrophobic interactions with a core formed by CTC1 residues Y244, V237, I246, and I291, and SHLD1 residue D19 forms salt bridges with CTC1 residues K242 and R292. This interaction was robust to 25 models predicted using five random seeds for each of five AlphaFold model parameters (). Although interactions between other subunits of CST and shieldin have been detected in yeast two-hybrid assays [8], the CTC1-SHLD1 interaction was the only one detected with high confidence by AlphaFold-multimer. However, shieldin contains many long, flexible linkers that may also become ordered upon binding other factors and/or post-translational modifications.
Is there any possibility, then, that shieldin blocks resection in BRCA1-deficient cells? The data argue against a separate function for shieldin blocking resection, as the entire effect of shieldin loss on radial chromosome formation and RAD51 loading can be reinstated by restoring CST recruitment. These data do not exclude the possibility that CST itself helps to block resection when bound to shieldin. In vitro, CST can bind substrates with a recessed 5’ end [Citation26] and therefore could potentially occlude the 5’ end from nucleases in vivo. However, such a block to resection by shieldin-bound CST is not consistent with the major effects of Polα and primase inhibition on DSB processing and PARPi-induced radial formation [Citation8,Citation55].
The role of the shieldin-CST link in CSR and at dysfunctional telomeres
Whereas the work with the SHLD1∆ mutant established that shieldin requires CST in the context of BRCA1-deficient cells, definitive data is lacking for how shieldin functions in CSR or at dysfunctional telomeres. Furthermore, evidence for Polα/primase-dependent DNA synthesis is hard to obtain for either CSR or the control of ssDNA at telomeres. In general, shieldin appears to function similarly downstream of 53BP1 in all three settings despite differences in cell type, chromatin context, DDR signaling, and DNA break substrate. Like shieldin, CST minimizes the length of the 3’ overhang at dysfunctional telomeres [Citation8], and there is some evidence that CST contributes to CSR [Citation66]. Unexpectedly, however, SHLD1∆ behaves like a wild-type allele in CSR and 3’ overhang control at dysfunctional telomeres [Citation55].
This context-dependent behavior of SHLD1∆ is not easily explained from differences such as ATM versus ATR signaling, cell cycle phase of the repair events, cell type differences, or the presence or absence of BRCA1 (see Mirman et al [Citation55] for details). What might account for the discordant behavior of the SHLD1∆ mutant in CSR and at telomeres versus at random DSBs in BRCA1-deficient cells? A notable shared feature of CSR and telomeres is the unusual DNA sequence of the processed DNA ends. At dysfunctional telomeres, the fill-in reaction copies the [TTAGGG]n sequence of the 3’ overhang. Similarly, tandem G-rich repeats are present near the AID-induced breaks in the CSR switch regions. For instance, the mouse IgA locus used in standard CSR assays contains over 60 copies of tandem 5 nucleotide repeats containing GG or GGG [Citation67]. These sequences are predicted to be good substrates for CST, as are the telomeric repeats [Citation27]. We therefore speculated [Citation55] that at genomic regions with optimal binding sites such as telomeres or IgA switch regions, CST is less dependent on shieldin for its recruitment and/or persistence at such sites, explaining the lack of effect of the SHLD1∆ mutant.
To test this idea, we performed electrophoretic mobility shift assays (EMSA) with purified CST [Citation48] and 5’ end-labeled 18 nt ssDNA substrates (). Consistent with previously reported results, CST binds telomeric repeats (GGTTAG)3 with a KD,app of 15 nM and does not detectably bind a control substrate (TATATA)3. We then measured the affinity of CST for either a G-rich (AGAGGAGGAGAGGAGAGG) or the complementary C-rich (CCTCTCCTCTCCTCCTCT) sequence from the core of the IgA locus. As predicted, the KD,app of CST on the IgA G-rich strand was 16 nM, similar to the telomeric repeats whereas the KD,app of CST on the IgA C-rich strand is moderately lower (about 4-fold; 63 nM) (). The 53BP1/shieldin-independent fill-in synthesis [Citation60] may therefore be explained by the ability of CST to stably bind G-rich regions and initiate fill-in synthesis. Similarly, CST can help facilitate replication of genome-wide G-rich sites [Citation30,Citation31], presumably localizing to such sites because of its innate preference for G-rich DNA (Box 2; ). In contrast to the CSR or telomeric sites with G runs in the ssDNA, CST may be more reliant on its SHLD1 binding at random PARPi- or IR-induced breaks (). Structural and biochemical studies of shieldin/CST complexes with various DNA substrates will be needed to test these ideas.
Figure 4. CST binding to telomeric and CSR sequences. Representative gels from EMSAs measuring CST binding to 0.1 nM radioactive 32P 5’ end-labeled substrates: AT rich (TATATA)3, Telomeric (GGTTAG)3, mouse IgA locus G-rich sequence (AGAGGAGGAGAGGAGAGG), and mouse IgA locus C-rich sequence (CCTCTCCTCTCCTCCTCT). A serial dilution of purified CST [Citation48] (shown left) was incubated at room temperature for 30 min with labeled substrate in binding buffer (20 mM HEPES-KOH pH 7.5, 150 mM KCl, 1 mM MgCl2, 0.1 mM TCEP, 0.05 mg/ml BSA, and 6% v/v glycerol) in 10 μl reactions. 0.1 nM (TATATA)3 was mixed in as a non-binding loading control for quantification. Samples were electrophoresed on 4–20% TBE gels (Invitrogen) at 250 V for 30 min in cold 0.5x TB buffer, exposed to phosphor screens, and imaged with an Amersham Typhoon scanner (GE Life Sciences). Right, quantification of three independent experiments. Signal intensity was measured with ImageJ (NIH) and normalized to intensity of the loading control. Because the intensity of the bound species was lost due to trapping in the sample well, binding was quantified using depletion of the free probe. KD,app values were calculated using the “One site – Specific binding” model in Prism 9 (GraphPad). Error bars represent standard error of the mean.
![Figure 4. CST binding to telomeric and CSR sequences. Representative gels from EMSAs measuring CST binding to 0.1 nM radioactive 32P 5’ end-labeled substrates: AT rich (TATATA)3, Telomeric (GGTTAG)3, mouse IgA locus G-rich sequence (AGAGGAGGAGAGGAGAGG), and mouse IgA locus C-rich sequence (CCTCTCCTCTCCTCCTCT). A serial dilution of purified CST [Citation48] (shown left) was incubated at room temperature for 30 min with labeled substrate in binding buffer (20 mM HEPES-KOH pH 7.5, 150 mM KCl, 1 mM MgCl2, 0.1 mM TCEP, 0.05 mg/ml BSA, and 6% v/v glycerol) in 10 μl reactions. 0.1 nM (TATATA)3 was mixed in as a non-binding loading control for quantification. Samples were electrophoresed on 4–20% TBE gels (Invitrogen) at 250 V for 30 min in cold 0.5x TB buffer, exposed to phosphor screens, and imaged with an Amersham Typhoon scanner (GE Life Sciences). Right, quantification of three independent experiments. Signal intensity was measured with ImageJ (NIH) and normalized to intensity of the loading control. Because the intensity of the bound species was lost due to trapping in the sample well, binding was quantified using depletion of the free probe. KD,app values were calculated using the “One site – Specific binding” model in Prism 9 (GraphPad). Error bars represent standard error of the mean.](/cms/asset/e6bc0955-b8f9-4276-8665-3e42b77d5357/kccy_a_2123886_f0004_oc.jpg)
In summary, the available data are consistent with a role for CST/Polα/primase fill-in at all DSBs, including those in BRCA1-proficient cells. Although the contribution of the shieldin-dependent recruitment of CST may vary depending on the sequence of the DNA ends, there is (as yet) no instance where the regulation of ssDNA at DSBs is shown to be independent of fill-in synthesis.
Conclusions and perspective
The recent data discussed here provide direct evidence of CST/Polα/primase-mediated fill-in synthesis at DSBs. Downstream of 53BP1/shieldin, fill-in synthesis plays a crucial role in promoting the chromosomal aberrations that underlie PARPi efficacy in BRCA1-deficient cells. Additionally, fill-in synthesis likely plays a role in counteracting excessive resection at dysfunctional telomeres and in CSR, although further work is needed in these areas. Fill-in synthesis also occurs in various experimental settings where DSBs are induced outside of these elaborate and specialized DSB repair reactions. Yet several outstanding questions remain to be resolved.
First, we propose CST recruitment may be affected by the availability of optimal CST binding sites at resected DSBs, explaining the 53BP1/shieldin-independent fill-in synthesis by Polα [Citation60] and the wild-type behavior of SHLD1∆ in some contexts [Citation55]. Site-specific breaks generated by Cas9 and nt-resolution sequencing could be used to evaluate the hypothesis that the sequence context around the break affects shieldin-mediated recruitment of CST. Second, recent structural studies have uncovered major conformational differences in the recruitment and active states of CST/Polα/Primase presumed to be present at telomeres [Citation48,Citation49], but the state in which CST or CST/Polα/Primase is recruited (by shieldin or on its own) to DSBs remains unknown. Third, it remains to be determined how the state of CST/Polα/Primase, with or without associated shieldin, dictates where fill-in synthesis begins. An important question to resolve is whether fill-in can begin at the very 3’ end as has been proposed [Citation61], or whether a moderate 3’ overhang remains uncopied, as in the case of fill-in synthesis at telomeres. The issue of where fill-in synthesis starts is pertinent to the question of whether CST/Polα/Primase can promote cNHEJ or is more likely to generate DNA ends poised for HDR.
There are also important questions regarding the fate of the CST/Polα/Primase fill-in product. In canonical DNA replication, Polα/Primase only generates a short DNA oligo that is elongated into the mature Okazaki fragment by Polα. Whether the same hand-off occurs at DSBs is not known. It is possible that there is no hand-off and that CST provides sufficient processivity to Polα/primase for extensive fill-in synthesis. It is also unclear how the product of fill-in synthesis becomes ligated to the 5’ end of the original resected DNA. Does this ligation involve the canonical Okazaki fragment processing machinery or does it use a specialized mechanism such as the PARP1-dependent maturation pathway [Citation68]? CST has emerged from the far ends of the genome, but no longer remains on the sidelines of the field. Having joined the 53BP1 team as an exciting new recruit, CST/Polα/primase is fast becoming a key playmaker.
Author contributions
All authors contributed equally to the writing of this review. S.W.C. performed the gel-shift experiments and generated the AlphaFold-multimer models.
Disclosure statement
T.d.L. is a member of the SAB of Calico, LLC, San Francisco, USA. The other authors have no competing interests.
Data availability statement
The data of the present study are available from the corresponding author on reasonable request.
Additional information
Funding
References
- Stinson BM, Loparo JJ. Repair of DNA double-strand breaks by the nonhomologous end joining pathway. Annu Rev Biochem. 2021;90:137–164.
- Wright WD, Shah SS, Heyer WD. Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem. 2018;293:10524–10535.
- Bhargava R, Onyango DO, Stark JM. Regulation of single-strand annealing and its role in genome maintenance. Trends Genet. 2016;32:566–575.
- Mirman Z, de Lange T. 53BP1: a DSB escort. Genes Dev. 2020;34:7–23.
- Dimitrova N, Chen YC, Spector DL, et al. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528.
- Lottersberger F, Karssemeijer RA, Dimitrova N, et al. 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair. Cell. 2015;163:880–893.
- Difilippantonio S, Gapud E, Wong N, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–533.
- Mirman Z, Lottersberger F, Takai H, et al. 53BP1-RIF1-Shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature. 2018;560:112–116.
- Escribano-Díaz C, Orthwein A, Fradet-Turcotte A, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49:872–883.
- Zimmermann M, Lottersberger F, Buonomo SB, et al. 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science. 2013;339:700–704.
- Di Virgilio M, Callen E, Yamane A, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339:711–715.
- Chapman JR, Barral P, Vannier JB, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49:858–871.
- Boersma V, Moatti N, Segura-Bayona S, et al. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5’ end resection. Nature. 2015;521:537–540.
- Xu G, Chapman JR, Brandsma I, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521:541–544.
- Dev H, Chiang TW, Lescale C, et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol. 2018;20:954–965.
- Ghezraoui H, Oliveira C, Becker JR, et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature. 2018;560:122–127.
- Gupta R, Somyajit K, Narita T, et al. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell. 2018;173:972–988.e23.
- Noordermeer SM, Adam S, Setiaputra D, et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature. 2018;560:117–121.
- de Lange T. Shelterin-Mediated telomere protection. Annu Rev Genet. 2018;52:223–247.
- Zhao F, Kim W, Gao H, et al. ASTE1 promotes shieldin-complex-mediated DNA repair by attenuating end resection. Nat Cell Biol. 2021;23:894–904.
- Setiaputra D, Durocher D. Shieldin - the protector of DNA ends. EMBO Rep. 2019;20:5.
- Goulian M, Heard CJ, Grimm SL. Purification and properties of an accessory protein for DNA polymerase alpha/primase. J Biol Chem. 1990;265:13221–13230.
- Goulian M, Heard CJ. The mechanism of action of an accessory protein for DNA polymerase alpha/primase. J Biol Chem. 1990;265:13231–13239.
- Gao H, Cervantes RB, Mandell EK, et al. RPA-Like proteins mediate yeast telomere function. Nat Struct Mol Biol. 2007;14:208–214.
- Lim CJ, Barbour AT, Zaug AJ, et al. The structure of human CST reveals a decameric assembly bound to telomeric DNA. Science. 2020;368:1081–1085.
- Bhattacharjee A, Wang Y, Diao J, et al. Dynamic DNA binding, junction recognition and G4 melting activity underlie the telomeric and genome-wide roles of human CST. Nucleic Acids Res. 2017;45:12311–12324.
- Hom RA, Wuttke DS. Human CST prefers G-rich but not necessarily telomeric sequences. Biochemistry. 2017;56:4210–4218.
- Miyake Y, Nakamura M, Nabetani A, et al. RPA-Like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol Cell. 2009;36:193–206.
- Stewart JA, Wang F, Chaiken MF, et al. Human CST promotes telomere duplex replication and general replication restart after fork stalling. Embo J. 2012;31:3537–3549.
- Chastain M, Zhou Q, Shiva O, et al. Human CST Facilitates genome-wide RAD51 recruitment to GC-rich repetitive sequences in response to replication stress. Cell Rep. 2016;16:1300–1314.
- Zhang M, Wang B, Li T, et al. Mammalian CST averts replication failure by preventing G-quadruplex accumulation. Nucleic Acids Res. 2019;47:5243–5259.
- Chen LY, Redon S, Lingner J. The human CST complex is a terminator of telomerase activity. Nature. 2012;488:540–544.
- Lue NF. Evolving linear chromosomes and telomeres: a C-strand-centric view. Trends Biochem Sci. 2018;43:314–326.
- Wu P, Takai H, de Lange T. Telomeric 3’ overhangs derive from resection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell. 2012;150:39–52.
- Gu P, Min JN, Wang Y, et al. CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. Embo J. 2012;31:2309–2321.
- Wang F, Stewart JA, Kasbek C, et al. Human CST has independent functions during telomere duplex replication and C-strand fill-in. Cell Rep. 2012;2:1096–1103.
- Chen LY, Majerská J, Lingner J. Molecular basis of telomere syndrome caused by CTC1 mutations. Genes Dev. 2013;27:2099–2108.
- Takai H, Jenkinson E, Kabir S, et al. A POT1 mutation implicates defective telomere end fill-in and telomere truncations in coats plus. Genes Dev. 2016;30:812–826.
- Feng X, Hsu SJ, Kasbek C, et al. CTC1-Mediated C-strand fill-in is an essential step in telomere length maintenance. Nucleic Acids Res. 2017;45:4281–4293.
- Gu P, Jia S, Takasugi T, et al. CTC1-STN1 coordinates G- and C-strand synthesis to regulate telomere length. Aging Cell. 2018;17:e12783.
- Wan M, Qin J, Songyang Z, et al. OB fold-containing protein 1 (OBFC1), a human homolog of yeast Stn1, associates with TPP1 and is implicated in telomere length regulation. J Biol Chem. 2009;284:26725–26731.
- Pinzaru AM, Hom RA, Beal A, et al. Telomere replication stress induced by POT1 inactivation accelerates tumorigenesis. Cell Rep. 2016;15:2170–2184.
- Hockemeyer D, Daniels JP, Takai H, et al. Recent expansion of the telomeric complex in rodents: two distinct POT1 proteins protect mouse telomeres. Cell. 2006;126:63–77.
- Hockemeyer D, Palm W, Wang RC, et al. Engineered telomere degradation models dyskeratosis congenita. Genes Dev. 2008;22:1773–1785.
- Hockemeyer D, Palm W, Else T, et al. Telomere protection by mammalian Pot1 requires interaction with Tpp1. Nat Struct Mol Biol. 2007;14:754–761.
- Kibe T, Osawa GA, Keegan CE, et al. Telomere protection by TPP1 is mediated by POT1a and POT1b. Mol Cell Biol. 2010;30:1059–1066.
- Kibe T, Zimmermann M, de Lange T. TPP1 blocks an ATR-mediated resection mechanism at telomeres. Mol Cell. 2016;61:236–246.
- Cai SW, Zinder JC, Svetlov V, et al. Cryo-EM structure of the human CST–Polα/primase complex in a recruitment state. Nat Struct Mol Biol. 2022;29:813–819.
- He Q, Lin X, Chavez BL, et al. Structures of the human CST-Polα-primase complex bound to telomere templates. Nature. 2022;608(7924).
- Anderson BH, Kasher PR, Mayer J, et al. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat Genet. 2012;44:338–342.
- Simon AJ, Lev A, Zhang Y, et al. Mutations in STN1 cause coats plus syndrome and are associated with genomic and telomere defects. J Exp Med. 2016;213:1429–1440.
- Passi GR, Shamim U, Rathore S, et al. An Indian child with coats plus syndrome due to mutations in STN1. Am J Med Genet a. 2020;182:2139–2144.
- Jumper J, Evans R, Pritzel A, et al. Applying and improving AlphaFold at CASP14. Proteins. 2021;89:1711–1721.
- Varadi M, Anyango S, Deshpande M, et al. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2021;50(D1):D439–D444.
- Mirman Z, Sasi NK, King A, et al. 53BP1-Shieldin-Dependent DSB processing in BRCA1-deficient cells requires CST-Polα-primase fill-in synthesis. Nat Cell Biol. 2022;24:51–61.
- Ochs F, Somyajit K, Altmeyer M, et al. 53BP1 fosters fidelity of homology-directed DNA repair. Nat Struct Mol Biol. 2016;23:714–721.
- Zaug AJ, Goodrich KJ, Song JJ, et al. Reconstitution of a telomeric replicon organized by CST. Nature 2022;
- Canela A, Sridharan S, Sciascia N, et al. DNA breaks and end resection measured genome-wide by end sequencing. Mol Cell. 2016;63:898–911.
- Wu W, Hill SE, Nathan WJ, et al. Neuronal enhancers are hotspots for DNA single-strand break repair. Nature. 2021;593:440–444.
- Paiano J, Zolnerowich N, Wu W, et al. Role of 53BP1 in end protection and DNA synthesis at DNA breaks. Genes Dev. 2021;35:1356–1367.
- Schimmel J, Muñoz-Subirana N, Kool H, et al. Small tandem DNA duplications result from CST-guided Pol α-primase action at DNA break termini. Nat Commun. 2021;12:4843.
- Kuchta RD, Stengel G. Mechanism and evolution of DNA primases. Biochim Biophys Acta. 2010;1804:1180–1189.
- Evans R, O’Neill M, Pritzel A, et al. Protein complex prediction with Alphafold-multimer. bioRxiv. 2021
- Liang L, Feng J, Zuo P, et al. Molecular basis for assembly of the shieldin complex and its implications for NHEJ. Nat Commun. 2020;11:1972.
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021;30:70–82.
- Barazas M, Annunziato S, Pettitt SJ, et al. The CST complex mediates end protection at double-strand breaks and promotes PARP inhibitor sensitivity in BRCA1-deficient cells. Cell Rep. 2018;23:2107–2118.
- Chaudhuri J, Alt FW. Class-Switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat Rev Immunol. 2004;4:541–552.
- Hanzlikova H, Kalasova I, Demin AA, et al. The importance of poly(ADP-ribose) polymerase as a sensor of unligated okazaki fragments during DNA replication. Mol Cell. 2018;71:319–331.e3.