
ABSTRACT
Abdominal aortic aneurysm (AAA) is a vascular disorder greatly threatening life of the elderly population. Dexmedetomidine (DEX), an α2-adrenergic receptor agonist, has been shown to suppress AAA development. Nevertheless, the signaling pathways that might be mediated by DEX in AAA has not been clarified. Vascular smooth muscle cells (VSMCs) and endothelial cells (ECs) were treated with Angiotensin II (Ang II) to mimic AAA in vitro. BrdU, wound healing, and Transwell assays were utilized for measuring VSMC proliferation and migration. Western blotting was used for evaluating protein levels of contractile VSMC markers, collagens and matrix metalloproteinases (MMPs) in VSMCs as well as apoptosis- and HMGB1/TLR4/NF-κB signaling-related markers in ECs. Cell adhesion molecule expression and monocyte-endothelial adhesion were assessed by immunofluorescence staining and adhesion assays. Flow cytometry was implemented for analyzing EC apoptosis. Hematoxylin-eosin staining and ELISA were used to detect the effect of DEX in vivo. In this study, DEX inhibited Ang II-evoked VSMC phenotype switch and extracellular matrix degradation. DEX suppressed the inflammatory response and apoptosis of ECs induced by Ang II. DEX inhibited HMGB1/TLR4/NF-κB signaling pathway in Ang II-treated ECs. DEX attenuated Ang II-induced AAA and inflammation in mice. Overall, DEX ameliorates Ang II-induced VSMC phenotype switch, and inactivates HMGB1/TLR4/NF-κB signaling pathway to alleviate Ang II-induced EC dysfunction.
Introduction
Abdominal aortic aneurysm (AAA) is a vascular disorder characterized by permanent dilation of abdominal aorta resulting from the degeneration of abdominal aortic wall [Citation1]. AAA occurs mostly in men over the age of 65, with a high mortality rate [Citation2]. It is estimated that the mortality rate is up to 90% upon AAA rupture [Citation3]. Open and endovascular surgical repair are the major therapeutic strategies for patients with AAA; however, they are not applicable for small AAA (˂5.5 cm in diameter) [Citation4]. Hence, it is urgent to better understand AAA pathogenesis and find more effective approaches for AAA therapy.
The main pathological features of AAA include vascular inflammation, extracellular matrix (ECM) degradation, vascular smooth muscle cell (VSMC) phenotype switch and endothelial cell dysfunction [Citation5]. Vascular inflammation is considered as the primary inducement of AAA; the infiltration and accumulation of inflammatory cells especially macrophages in abdominal aortic wall cause the secretion of matrix metalloproteinases (MMPs) and inflammatory cytokines [Citation4]. MMPs lead to the degradation of ECM components, including collagen and elastin, the main factors determining the mechanical strength of aortic wall [Citation6]. Endothelial cells (ECs) on the vessel surface function as vital regulators in maintaining vascular homeostasis and dysfunction of ECs is strongly associated with AAA pathogenesis [Citation7]. VSMCs are the main inherent cells in the tunica media of abdominal aortic wall, and it was reported that the phenotype switch of VSMCs contributes to the pathogenesis of AAA [Citation8]. VSMC switching from a contractile state to a synthetic phenotype is featured with a reduction in contractile markers (SM-22α and α-SMA) and an enhancement in MMPs [Citation9]. VSMCs in a synthetic phenotype exhibit increased proliferative and migratory abilities, which contributes to the pathogenesis of many vascular disorders, including AAA [Citation10].
Dexmedetomidine (DEX), a highly selective α2-adrenergic receptor agonist, has been widely used as a sedative in surgery and intensive care units [Citation11]. DEX displays an anti-inflammatory effect in multiple disorders. For example, DEX attenuates microglia-stimulated spinal inflammatory response in neonatal rats [Citation12]. Additionally, DEX mitigates chronic constriction injury-induced neuropathic pain and inflammation by the high mobility group box 1/toll-like receptor 4/nuclear factor κB (HMGB1/TLR4/NF-κB) pathway [Citation13]. Moreover, DEX mitigates ischemia-reperfusion injury-mediated myocardial dysfunction by inactivating HMGB1/TLR4/NF-κB signaling [Citation14]. Studies have demonstrated that suppression of TLR4 signaling can protect against AAA and TRL4 can be activated by HMGB1 to promote inflammatory response [Citation15]. Furthermore, evidence showed that ADAM10 can alleviate AAA progression largely by inhibiting HMGB1/NF-κB signaling [Citation16]. Intriguingly, DEX was reported to inhibit AAA development in rats via the miR-21/ODCD4 axis [Citation17]. Nevertheless, the signaling pathways that might be mediated by DEX in AAA have not been explored.
Herein, we explored the effects of DEX on VSMC phenotype switch and EC dysfunction in Angiotensin II (Ang II)-induced in vitro AAA models. Additionally, DEX impact on the HMGB1/TRL4/NF-κB signaling in AAA was also investigated. The findings might provide a new perspective in AAA therapy.
Materials and methods
A more detailed methods section can be found in the additional file supplementary materials and methods
Cell culture and treatment
Primary human VSMCs (CRL-1999) and aortic ECs (PCS-100-011) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). VSMCs were incubated in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo, Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Hyclone, Salt Lake City, UT, USA) and 1% penicillin-streptomycin (Invitrogen, Carlsbad, CA, USA). ECs were cultured in commercial Endothelial Cell Medium (ScienCell, San Diego, CA, USA). Cells were maintained at 37℃ with 5% CO2 in a humidified incubator and cells at passage 3–5 were used in the study. Human acute monocytic leukemia cell line (THP-1) was obtained from ATCC and incubated in RPMI 1640 (Thermo) containing 10% FBS. THP-1 cells at passage 5–6 were used in subsequent analysis.
VSMCs or ECs were pretreated with 100 ng/mL DEX (Sigma-Aldrich, St. Louis, MO, USA) or dimethyl sulfoxide (DMSO; Sigma-Aldrich) for 24 h [Citation18,Citation19], followed by adding Ang II (1 μmol/L) and culturing for another 24 h [Citation20].
Bromodeoxyuridine (BrdU) incorporation assay
VSMCs with indicated treatment were inoculated in 24-well plates and treated with 200 μL BrdU solution (5 μmol/L; Beyotime, Shanghai, China) for 4 h, followed by incubation with anti-BrdU antibody (ab6326, Abcam, Cambridge, MA, USA) overnight at 4℃. Then, cells were incubated with FITC-conjugated secondary antibody (Abcam) at 37℃ for 1 h away from light and counterstained with DAPI (Sigma-Aldrich). Eventually, cells were observed under a microscope (Nikon, Tokyo, Japan) and the percentage of BrdU positive (BrdU+) cells was measured by ImageJ software (National Institutes of Health, NIH, Bethesda, MD, USA).
Wound healing assay
VSMCs were seeded into 6-well plates (2.5 × 105 cells/well) and grown to 80% confluence. Afterward, a sterile pipette tip was utilized to scratch the cells, and the images were captured with a light microscope (Nikon). After incubation for 24 h at 37℃, images of the wound were captured again, and wound healing percentage was calculated using ImageJ software (NIH).
Transwell assay
VSMC migration was examined using 24-well Transwell chambers (8 μm; Corning Inc., Corning, NY, USA). VSMCs were starved in serum-free medium for 12 h. Then cells (1 × 105/ml) were suspended in 200 μL serum-free medium and placed into the upper chamber. Additionally, 700 μL of culture medium containing 10% FBS was added to the lower chamber. After 24 h of incubation at 37℃ with 5% CO2, a cotton swab was utilized to gently wiped off the non-migratory cells on the upper chamber, while the migratory cells were fixed with 4% formaldehyde, stained with 1% crystal violet solution (Solarbio, Beijing, China) and photographed with a phase-contrast microscope (Nikon).
Western blotting
Proteins were extracted from VSMCs and ECs using RIPA buffer (Thermo) and quantified with a BCA assay kit (Bio-Rad, Hercules, CA, USA). Protein samples (20 μg) were separated on 10% SDS-PAGE and blotted on polyvinylidene difluoride membranes (Thermo). Afterward, the membranes were blocked with 5% skim milk powder and cultured at 4℃ overnight with primary antibodies (Abcam) followed by incubation with the secondary antibody (Abcam) for 1 h at room temperature. An enhanced chemiluminescence kit (Solarbio) was utilized for protein band visualization and Image-Pro Plus 6.0 software (Media Cybernetics, Shanghai, China) was utilized for quantification.
Gelatin zymography assay
The activities of MMP-2 and MMP-9 were detected using gelatin zymography assay. VSMCs were seeded in six-well plates followed by indicated treatment and incubation in serum-free DMEM for 24 h. Cell culture medium was collected, and protein samples were separated by 10% SDS-PAGE containing 0.1% gelatin. Gels were washed with 2.5% Triton X-100 and incubated in reaction buffer at 37°C for 24 h. Then, gels were stained with Coomassie Brilliant Blue R-250 (Merck; Darmstadt, Germany) in 10% acetic acid and 30% methanol, visualized with ChemiDoc MP imaging system (Bio-Rad) and analyzed with ImageJ software (NIH).
Immunofluorescence (IF) staining
ECs were fixed with 4% paraformaldehyde and permeabilized in 0.1% Triton X-100. After being washed with phosphate buffer saline (PBS) and blocked with 5% bovine serum albumin, ECs were incubated at 4℃ overnight with anti-intercellular adhesion molecule 1 (ICAM-1) antibody (Abcam) and then incubated with the corresponding secondary antibody (Abcam) at 37℃ for 1 h. Afterward, cells were rinsed with PBS three times and counterstained with DAPI. Stained cells were mounted with anti-fade reagent, covered with a coverslip and photographed with a fluorescence microscope (Nikon). Image-Pro Plus 6.0 software (Media Cybernetics) was used for data analysis.
Adhesion assay
ECs were inoculated into 12-well plates with indicated treatment. Subsequently, THP-1 cells were co-incubated with indicated ECs for 30 min at 37℃. Cells were then washed with PBS and bounded THP-1 cells were photographed and counted using a light microscope (Nikon).
Flow cytometry
EC apoptosis was determined by flow cytometry using Annexin V-FITC/PI Apoptosis Detection Kit (Becton Dickinson Biosciences, San Jose, CA, USA). Cells were subjected to centrifugation at 250 × g for 5 min and the supernatant was discarded. Cells were washed twice with pre-cooled PBS and resuspended in 100 μL 1×binding buffer. Then, 5 μL Annexin V-FITC and PI were added and incubated for 15 min at room temperature in the dark. A total of 400 μL 1×binding buffer was added, and cell apoptosis was measured in 1 h using flow cytometry (Becton Dickinson Biosciences).
Ang II-induced AAA mouse model and treatment
Fifty ApoE−/− male mice (8–10 weeks, 20–22 g) with a C57BL/6J background were purchased from Vital River (Beijing, China). All mice were housed in a pathogen-free animal care facility on a 12-h light/dark cycle at a temperature of 25 ± 1°C with free access to food and water. All procedures were conducted following the guidelines of Guide for the Care and Use of Laboratory Animals and approved by the Ethics Committee of The First Affiliated Hospital of Nanchang University (Nanchang, China).
Mice were anesthetized by intraperitoneal injection with 0.3% pentobarbital sodium (50 mg/kg). Mice were randomly divided into four groups: control + DMSO, control + DEX, Ang II + DMSO, Ang II + DEX, Ang II + TA606 (n = 10 mice/group). Mice were subcutaneously implanted with Ang II solution (1000 ng/kg/min) to induce AAA or normal saline as a control for 28 days using osmotic minipumps (Alzet Model 2004; DURECT, Cupertino, CA, USA) [Citation21]. On day 14 after Ang II induction, mice were administrated with DEX (50 µg/kg/day) [Citation22,Citation23], TA-606 (an angiotensin II type I receptor blocker, ARB; 10 mg/kg/day) [Citation24] or DMSO for 2 weeks. Two weeks after drug treatment, mice were euthanized by cervical dislocation under anesthesia and murine abdominal aortic tissue was collected for subsequent analysis.
Hematoxylin-Eosin (HE) staining
Murine abdominal aortic tissue was fixed in 4% paraformaldehyde, embedded in paraffin and sectioned (5-μm-thick). Then, tissue sections were stained with hematoxylin and eosin (Solarbio, Beijing, China) following the instructions of the manufacturer. Afterward, the stained sections were dehydrated, cleaned with ethanol and xylene, sealed with neutral gum (Solarbio) and imaged under an optical microscope (Leica DM750, Nussloch, Germany).
Enzyme-linked immunosorbent assay (ELISA)
Concentrations of cytokines in VSMCs or murine abdominal aortic tissue were evaluated using ELISA kits (Abcam) according to the manufacturer’s protocols.
Statistical analysis
Data are presented as the mean ± standard deviation. SPSS 21.0 software (SPSS, Chicago, IL, USA) was used for statistical analysis. Differences between two groups were analyzed with Student’s t-test, while those among more groups were evaluated using analysis of variance (ANOVA) followed by Turkey post hoc analysis. P˂0.05 was considered as statistically significant.
Results
DEX regulates phenotype switch of VSMCs
First, we examined DEX effect on the phenotypes of VSMCs. BrdU incorporation assay was utilized to assess cell proliferative ability. As displayed by the results, DEX treatment markedly abated Ang II-induced proliferation of VSMCs (). Moreover, wound healing and Transwell assays demonstrated that compared with the control group, Ang II promoted the migratory capability of VSMCs; however, this effect was partially reversed by DEX (). Phenotype switch of VSMCs is characterized by a reduction in contractile markers and an increase in synthetic markers. Notably, protein levels of contractile markers (SM-22α and α-SMA) in Ang II+ DMSO group were decreased, whereas the marker levels were elevated in Ang II+ DEX group (). In contrast, Ang II significantly induced the increase in protein levels of synthetic markers (vimentin and osteopontin) in VSMCs, while DEX treatment abated the effect induced by Ang II (). Additionally, DEX treatment significantly attenuated Ang II-induced upregulation of cytokines (TNF-α, IL-6) in VSMCs (). These results indicated that DEX restrains VSMC phenotype switch induced by Ang II.
Figure 1. DEX inhibits VSMC phenotype switch. (a-b) BrdU incorporation assay for assessing VSMC proliferation in each group. (c-f) Wound healing and Transwell assays for evaluating VSMC migratory ability in each group. (g-h) Western blotting for examining protein levels of contractile markers (α-SMA and SM-22α) and synthetic markers (vimentin, osteopontin). (i-j) ELISA for measuring production of cytokines (TNF-α, IL-6) in VSMCs. Each experiment was performed in triplicate.Each experiment was performed in triplicate. **p˂0.01 vs. control+ DMSO group; #p˂0.05 vs. Ang II+ DMSO group; &p˂0.05 vs. control+ DEX group.
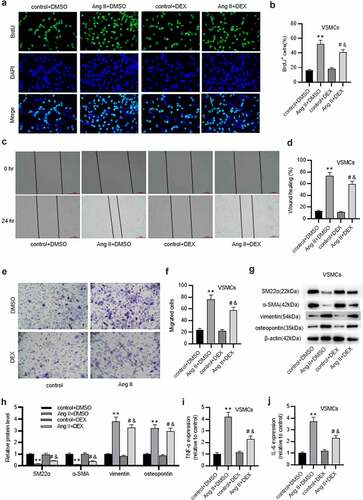
DEX protects against ECM degradation in VSMCs
VSMCs in a synthetic phenotype secrete MMPs that degrade the components of ECM [Citation9]. Results from western blotting presented that in comparison to those in control+ DMSO group, protein levels of collagens were decreased and those of MMPs (MMP-2, MMP-9) were enhanced in Ang II+ DMSO group ( (a-f), confirming that Ang II stimulated VSMC phenotype switch. Notably, the effects induced by Ang II were markedly counteracted by DEX ( (a-f)). Similar results were observed in gelatin zymography assay that showed that DEX markedly decreased Ang II-induced increase in MMP-2 and MMP-9 activities ( (g-j2)). Thus, these data revealed that DEX can protect against Ang II-evoked ECM degradation in VSMCs.
Figure 2. DEX protects against ECM degradation in VSMCs. A-C. Western blotting of collagen protein levels in VSMCs of each group. D-F. Western blotting of MMP-2 and MMP-9 protein levels in VSMCs of each group. G-I. Gelatin zymography assay for detecting MMP-2 and MMP-9 activities in VSMCs. Each experiment was performed in triplicate. **p˂0.01 vs. control+ DMSO group; #p˂0.05 vs. Ang II+ DMSO group; &p˂0.05 vs. control+ DEX group.
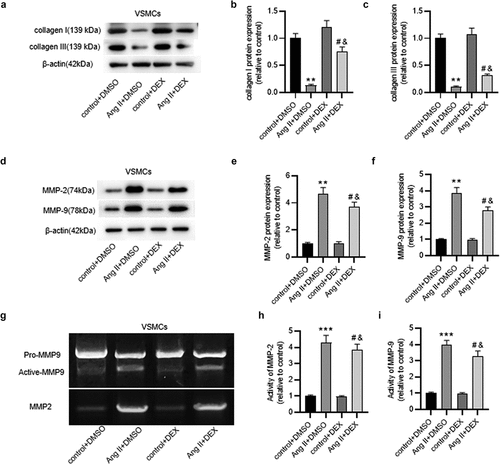
DEX attenuates inflammatory response in ECs
To detect the inflammation in ECs, IF staining was implemented to analyze expression of ICAM-1, a critical indicator of endothelial inflammation and injury. Notably, high staining intensity was shown in Ang II+ DMSO group, while mild staining intensity was observed in Ang II + DEX group (), suggesting the suppressive effect of DEX on Ang II-induced ICAM-1 expression. The results were confirmed by quantification of ICAM-1 protein expression in ECs (). Furthermore, we evaluated DEX effect on monocyte-endothelial cell adhesion. As depicted in ), DEX significantly alleviated Ang II-mediated adhesion of THP-1 monocytes to ECs. Collectively, DEX can attenuate the inflammatory response in ECs induced by Ang II.
Figure 3. DEX attenuates inflammation in Ang II-treated ECs. (a) if staining for detecting ICAM-1 expression in ECs. (b) Quantification of ICAM-1 expression in each group. (c) Adhesion of THP-1 monocytes to ECs observed under a light microscope. (d) Quantification of THP-1 cell adhesion in each group. Each experiment was performed in triplicate. **p˂0.01 vs. control+ DMSO group; #p˂0.05 vs. Ang II+ DMSO group; &p˂0.05 vs. control+ DEX group.
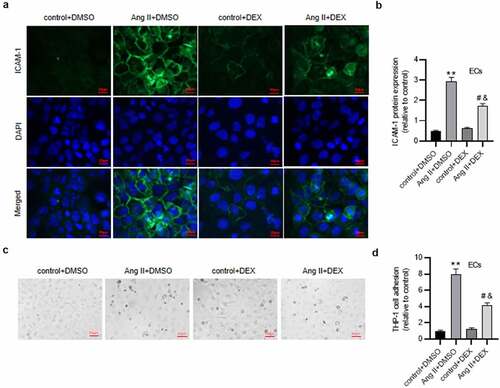
DEX alleviates Ang II-mediated EC apoptosis
Subsequently, to further examine the influence of DEX on EC function, cell apoptosis was measured. As revealed by flow cytometry, the rate of apoptotic ECs was markedly higher in Ang II+ DMSO group than that in the control group (). Contrarily, DEX treatment mitigated Ang II-induced high percentage of apoptotic ECs (). These were consistent with the results of western blotting. Ang II elevated levels of pro-apoptotic proteins (Bax, C-caspase3 and C-caspase9) and decreased anti-apoptotic protein (Bcl-2) level in ECs, however, the effect was attenuated by DEX (). Hence, DEX attenuates Ang II-evoked apoptosis of ECs.
Figure 4. DEX alleviates the apoptosis of ECs. (a-b) Flow cytometry analysis for detecting EC apoptosis in each group. Quadrants 2 and 3 (Q2, Q3) were considered as apoptotic cells. (c) Western blotting for measuring levels of apoptosis-associated proteins in ECs. Each experiment was performed in triplicate. *p˂0.05, **p˂0.01.
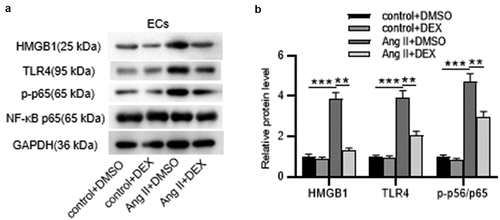
DEX suppresses the expression of HMGB1, TLR4 and p-p65 in ECs
To reveal the potential mechanism of DEX underlying AAA progression, we tested whether DEX affects the expression of HMGB1, TLR4 as well as phosphorylation of NF-κB in Ang II-stimulated ECs. As displayed by western blotting, protein levels of HMGB1, TLR4 and p-NF-κB p65 were markedly elevated by Ang II; however, the elevated protein levels were reduced by DEX treatment (), indicating that DEX suppresses the expression of HMGB1 and TLR4 and the phosphorylation of NF-κB in Ang II-treated ECs.
Figure 5. DEX suppresses the expression of HMGB1, TLR4 and p-p65 in ECs. (a-b) Western blotting for analyzing protein levels of HMGB1, TLR4, p-p65 and NF-κB p65 in ECs of each group. Each experiment was performed in triplicate. *p˂0.05, **p˂0.01.
DEX affects the inflammation and apoptosis of ECs via HMGB1/TLR4/NF-κB signaling pathway
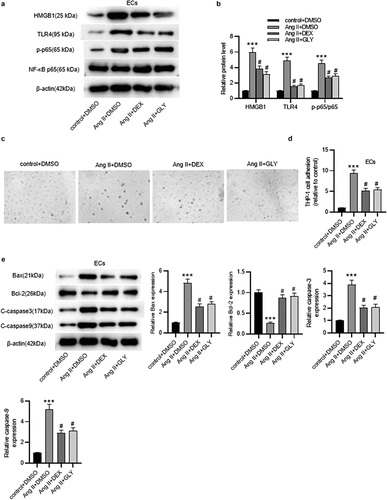
Subsequently, GLY (glycyrrhizin), a direct HMGB1 inhibitor which can inhibit the chemoattractant and mitogenic activities of HMGB1, was used to inhibit HMGB1 signaling. A shown in ), treatment of DEX and GLY significantly downregulated Ang II-induced upregulation of HMGB1 and TLR4 as well as phosphorylation of NF-κB, indicating that DEX could suppress the activation of HMGB1/TLR4/NF-κB signaling pathway. The inflammatory response in ECs was tested. Notably, DEX and GLY both attenuated Ang II-mediated adhesion of THP-1 monocytes to ECs (). Likewise, the results of western blotting displayed that DEX and GLY markedly reduced the protein levels of pro-apoptotic proteins (Bax, C-caspase3 and C-caspase9) and enhanced Bcl-2 protein level in ECs (). Collectively, these results indicate that DEX alleviates Ang II-induced inflammation and apoptosis of ECs by inhibiting HMGB1/TLR4/NF-κB signaling pathway.
Figure 6. DEX affects the inflammation and apoptosis of ECs via HMGB1/TLR4/NF-κB signaling pathway. (a-b) Western blotting for assessing HMGB1/TLR4/NF-κB signaling pathway-related proteins in ECs. (c) Adhesion of THP-1 monocytes to ECs observed under a light microscope. (d) Quantification of THP-1 cell adhesion in each group. (e) Western blotting measuring levels of apoptosis-associated proteins in ECs. Each experiment was performed in triplicate. ***p˂0.001 vs. control+ DMSO group; #p˂0.05 vs. Ang II+ DMSO group.
DEX attenuates Ang II-induced AAA and inflammation in mice
To further verify the effect of DEX on the development of AAA, in vivo experiments were conducted. HE staining was performed to examine the histopathological changes of murine aortic samples. As a result, in comparison to the control group, Ang II infusion markedly increased the abdominal aortic diameter and media/lumen ratio, while DEX administration significantly attenuated the increased aortic diameter and media/lumen ratio ( (a-cb)). Additionally, it has been suggested that angiotensin II type-1 (AT1) receptor blockers (ARBs) can attenuate the development of AAA in animal models [Citation25,Citation26]. Based on these, we used TA-606, an AT1 receptor antagonist, as a positive control in our study. The results revealed that DEX had a similar effect as TA-606 on Ang II-induced AAA. Moreover, we detected the effect of DEX on inflammatory response in Ang II-infused mice. Cytokine production in murine aortic tissue was examined by ELISA, which showed that Ang II significantly enhanced the concentrations of proinflammatory cytokines (TNF-α, MCP-1 and IL-6), whereas administration of DEX or TA-606 alleviated the effect caused by Ang II ( (c-e7d-f)). This indicated that DEX could attenuate Ang II-induced inflammatory response in mice.
Figure 7. DEX attenuates Ang II-induced AAA and inflammation in mice. (a) Representative images of HE staining for histopathological observation of murine abdominal aortic tissue. (b-c) Abdominal aortic diameter and media/lumen ratio of each group. (d-f) ELISA for determining the concentrations of proinflammatory cytokines in murine abdominal aortic samples. n = 10 mice/group. Each experiment was performed in triplicate. **p˂0.01 vs. control+ DMSO group; #p˂0.05, &p˂0.05vs. Ang II+ DMSO group.
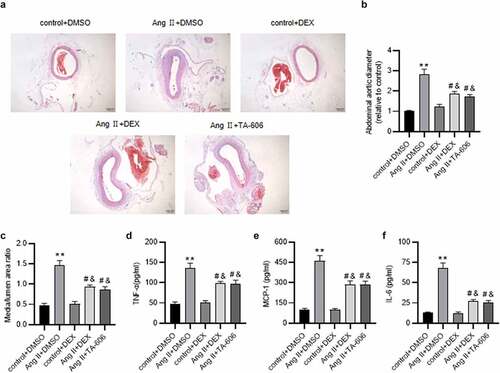
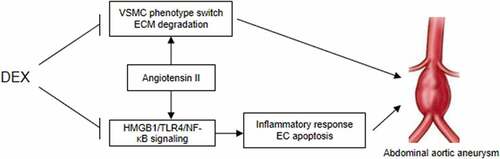
Figure 8. A schematic view of the in vitro model investigated in this study. VSMCs and ECs were treated with Ang II to mimic the pathological condition of AAA in vitro. DEX can suppress Ang II-evoked VSMC phenotype switch and ECM degradation, and it inactivates the HMGB1/TLR4/NF-κB signaling pathway to attenuate inflammatory response and the apoptosis of ECs, thereby ameliorating the condition of AAA.
Discussion
AAA is a degenerative process of abdominal aortas that seriously threatens the life of the elderly population [Citation27]. Nevertheless, there is currently no available effective treatment for AAA since the existing two therapies are limited to aneurysms that larger than 5.5 cm in diameter, a state with increased risk of aneurysm rupture [Citation28]. DEX is an α2-adrenergic receptor agonist that has been indicated to exert anti-inflammatory and neuroprotective effects [Citation29]. Previous evidence showed that DEX can suppress the progression of AAA in a rat model by targeting the miR-21/PDCD4 axis [Citation17]. However, the signaling pathways that mediated by DEX in AAA progression have not been studied. Ang II, the key effector of the renin-angiotensin system (RAS), has been demonstrated to act as a powerful proinflammatory mediator via stimulation of AT1 receptor [Citation30]. In the course of inflammation, excessive local Ang II concentration enhances vascular permeability by stimulating the production of prostaglandins and the vascular endothelial cell growth factor (VEGF), consequently initiating the inflammatory response and activating the immune system [Citation31]. Herein, we established in vitro AAA models by Ang II induction in VSMCs and ECs. We detected DEX role in VSMC phenotype switch, ECM degradation and EC dysfunction. A schematic view of the in vitro model invetiagted in our study was shown in .
VSMC phenotype switch exerts an indispensable effect on the initiation of AAA pathogenesis [Citation32]. VSMCs are responsible for maintaining blood vessel elasticity [Citation33]. VSMCs switching from a contractile phenotype to a synthetic type can release extracellular vehicles that facilitate local inflammatory response [Citation34]. Moreover, the phenotype switch of VSMCs is featured with the decreased levels of contractile markers and the elevated levels of synthetic markers as well as MMPs that can degrade ECM components like elastin and collagen [Citation35]. Dedifferentiated VSMCs possess increased proliferative and migratory abilities, which contributes to the deterioration of AAA [Citation10]. Presently, it was found that DEX significantly suppressed the proliferation and migration of Ang II-treated VSMCs. Moreover, DEX was shown to enhance levels of contractile VSMC markers and collagens, but reduced levels of MMPs in Ang II-stimulated VSMCs. These results revealed that DEX could restrain Ang II-evoked VSMC phenotype switch and ECM component degradation, indicating its protective role in AAA. These were consistent with previous reports that DEX suppressed the expression of MMP-3/9, thereby inhibiting the degradation of ECM [Citation36]. On the other hand, it was reported that DEX attenuated the expression of MMP-9 and collogen I in TGF-β-treated airway smooth muscle cells, which was partially conflicted with our results, highlighting the need of further research on the effect of ECM degradation.
Increasing evidence has elucidated that ECs play a pivotal role in the formation of AAA [Citation37,Citation38]. ECs are critical in maintaining vascular homeostasis; ECs cam release diverse substances that affect the functions of VSMCs, leukocytes and other cells in the aortic wall [Citation39]. Many cardiovascular risk factors such as proinflammatory cytokines can induce EC dysfunction that subsequently further promotes vascular inflammation and aggravates the pathological conditions [Citation40]. Here, we explored the effect of DEX on EC dysfunction induced by Ang II. Many studies have illustrated that DEX exerts a suppressive effect on inflammation [Citation41,Citation42]. Consistently, the results in the present study depicted that DEX markedly reduced levels of cell adhesion molecules (ICAM-1) and suppressed the adhesion of THP-1 monocytes to ECs, confirming the anti-inflammatory role of DEX in Ang II-treated ECs. Moreover, DEX significantly restrained Ang II-evoked EC apoptosis. Collectively, DEX mitigates Ang II-induced EC dysfunction.
To understand the potential regulatory mechanism of DEX underlying the pathogenesis of AAA, we examined DEX impact on the HMGB1/TLR4/NF-κB signaling pathway in Ang II-treated ECs. Emerging evidence has illustrated the prominent role of the HMGB1/TLR4/NF-κB in multiple disorders, such as contrast-induced nephropathy, liver injury, and acute pancreatitis [Citation43–45]. HMGB1 is a highly conserved nuclear, nonhistone DNA‑binding protein that acts as a proinflammatory factor in the pathogenesis of multiple disorders by promoting proinflammatory cytokine secretion, inflammatory cell infiltration and vascular permeability [Citation16]. HMGB1 exerts its regulatory functions by interacting with pivotal transmembrane receptors including TLR4 [Citation15,Citation46]. Furthermore, NF-κB, an important inflammatory regulator, is implicated in various pathological processes [Citation47]. NF-κB signaling can be triggered by TLR4 activation. HMGB1 binds with TLR4 and activates downstream NF-κB, consequently promoting the inflammatory response that is the primary inducement of AAA [Citation4,Citation48]. Importantly, DEX was shown to exert a protective effect in ischemia/reperfusion injury by inhibiting HMGB1/TLR4/NF-κB signaling [Citation49,Citation50]. Here, we found that DEX treatment led to the downregulation of HMGB1, TLR4 and p-NF-κB p65 proteins. Additionally, the HMGB1 inhibitor GLY treatment downregulated HMGB1 and TLR4 as well as inhibited phosphorylation of NF-κB p65. These results indicated that DEX suppressed Ang II-induced activation of the HMGB1/TLR4/NF-κB signaling in ECs, which was in accord with previous studies.
Moreover, to elucidate the effect of DEX on the development of AAA, we conducted in vivo experiments using Ang II-infused mice. It was previous reported that DEX could attenuate the formation of AAA in surgery-induced AAA rats [Citation17]. In the present study, we confirmed that DEX administration could alleviate Ang II-induced histopathological changes and inflammatory response in murine abdominal aortas, which was in accord with the results of the previous report.
In conclusion, we examined the functions of DEX and its potential mechanism underlying AAA development in vitro and in vivo. The results revealed that DEX inhibits Ang II-induced AAA and inflammation in mice and suppresses phenotype switch and ECM degradation of VSMCs. It also attenuates Ang II-induced EC dysfunction by inactivating HMGB1/TLR4/NF-κB signaling pathway. Our findings might develop a new perspective for ameliorating AAA. More signaling pathways that might be mediated by DEX in AAA need to be further investigated in the future.
Supplemental Material
Download MS Word (31.5 KB)Supplemental Material
Download MS Word (31.3 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384101.2022.2124489
Additional information
Funding
Related Research Data
References
- Golledge J. Abdominal aortic aneurysm: update on pathogenesis and medical treatments. Nature Reviews Cardiology. 2019;16(4):225–242.
- Chang Z, Zhao G, Zhao Y, et al. Baf60a deficiency in vascular smooth muscle cells prevents abdominal aortic aneurysm by reducing inflammation and extracellular matrix degradation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2020;40(10):2494–2507. DOI:10.1161/ATVBAHA.120.314955.
- Nordon IM, Hinchliffe RJ, Loftus IM, et al. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nature Reviews Cardiology. 2011;8(2):92–102.
- Bernal S, Lopez-Sanz L, Jimenez-Castilla L, et al. Protective effect of suppressor of cytokine signalling 1-based therapy in experimental abdominal aortic aneurysm. British Journal of Pharmacology. 2021;178(3):564–581. DOI:10.1111/bph.15330.
- Yang H, Zhou T, Sorenson CM, et al. Myeloid-derived TSP1 (Thrombospondin-1) contributes to abdominal aortic aneurysm through suppressing tissue inhibitor of metalloproteinases-1. Arteriosclerosis, Thrombosis, and Vascular Biology. 2020;40(12):e350–e66.
- Chiang MT, Chen IM, Hsu FF, et al. Gal-1 (Galectin-1) upregulation contributes to abdominal aortic aneurysm progression by enhancing vascular inflammation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2021;41(1):331–345. DOI:10.1161/ATVBAHA.120.315398.
- Kokot M, Biolik G, Ziaja D, et al. Endothelium injury and inflammatory state during abdominal aortic aneurysm surgery: scrutinizing the very early and minute injurious effects using endothelial markers – a pilot study. Archives of Medical Science : AMS. 2013;3(3):479–486. DOI:10.5114/aoms.2013.34412.
- Peng H, Zhang K, Liu Z, et al. VPO1 modulates vascular smooth muscle cell phenotypic switch by activating extracellular signal-regulated kinase 1/2 (ERK 1/2) in abdominal aortic aneurysms. Journal of the American Heart Association. 2018;7(17):e010069. DOI:10.1161/JAHA.118.010069.
- Zhang Z, Zou G, Chen X, et al. Knockdown of lncRNA PVT1 inhibits vascular smooth muscle cell apoptosis and extracellular matrix disruption in a murine abdominal aortic aneurysm model. Molecules and Cells. 2019;42(3):218–227. DOI:10.14348/molcells.2018.0162.
- Li P, Zhu N, Yi B, et al. MicroRNA-663 regulates human vascular smooth muscle cell phenotypic switch and vascular neointimal formation. Circulation Research. 2013;113(10):1117–1127. DOI:10.1161/CIRCRESAHA.113.301306.
- Okabe T, Takeda S, Akada S, et al. Postoperative intensive care unit drug fever caused by dexmedetomidine. Anesthesia and Analgesia. 2009;108(5):1589–1591.
- Wen W, Gong X, Cheung H, et al. Dexmedetomidine alleviates microglia-induced spinal inflammation and hyperalgesia in neonatal rats by systemic lipopolysaccharide exposure. Frontiers in Cellular Neuroscience. 2021;15:725267.
- Wang X, Liu Q. Dexmedetomidine relieved neuropathic pain and inflammation response induced by CCI through HMGB1/TLR4/NF-κB signal pathway. Biological & Pharmaceutical Bulletin. 2021. DOI:10.1248/bpb.b21-00329
- Yang YF, Peng K, Liu H, et al. Dexmedetomidine preconditioning for myocardial protection in Ischaemia-reperfusion injury in rats by downregulation of the high mobility group box 1-toll-like receptor 4-nuclear factor κB signalling pathway. Clinical and Experimental Pharmacology & Physiology. 2017;44(3):353–361.
- Lai CH, Wang KC, Lee FT, et al. Toll-like receptor 4 is essential in the development of abdominal aortic aneurysm. PloS One. 2016;11(1):e0146565. DOI:10.1371/journal.pone.0146565.
- Renfeng Q, Shuxiao C, Peixian G, et al. ADAM10 attenuates the development of abdominal aortic aneurysms in a mouse model. Molecular Medicine Reports. 2021;24(5). DOI:10.3892/mmr.2021.12414.
- Yu Q, Li Q, Yang X, et al. Dexmedetomidine suppresses the development of abdominal aortic aneurysm by downregulating the mircoRNA‑21/PDCD 4 axis. International Journal of Molecular Medicine. 2021;47(5). DOI:10.3892/ijmm.2021.4923.
- Zhou XY, Liu J, Xu ZP, et al. Dexmedetomidine inhibits the lipopolysaccharide-stimulated inflammatory response in microglia through the pathway involving TLR4 and NF-κB. The Kaohsiung Journal of Medical Sciences. 2019;35(12):750–756.
- Liu F, Zhu S, Ni L, et al. Dexmedetomidine alleviates insulin resistance in hepatocytes by reducing endoplasmic reticulum stress. Endocrine. 2020;67(1):87–94.
- Chen G, Xu Y, Yao Y, et al. IKKε knockout alleviates angiotensin II-induced apoptosis and excessive autophagy in vascular smooth muscle cells by regulating the ERK1/2 pathway. Experimental and Therapeutic Medicine. 2021;22(4):1051. DOI:10.3892/etm.2021.10485.
- Wu G, Chen T, Shahsafaei A, et al. Complement regulator CD59 protects against angiotensin II-induced abdominal aortic aneurysms in mice. Circulation. 2010;121(11):1338–1346. DOI:10.1161/CIRCULATIONAHA.108.844589.
- Tao L, Guo X, Xu M, et al. Dexmedetomidine ameliorates high-fat diet-induced nonalcoholic fatty liver disease by targeting SCD1 in obesity mice. Pharmacology Research & Perspectives. 2021;9(1):e00700. DOI:10.1002/prp2.700.
- Shi J, Yu T, Song K, et al. Dexmedetomidine ameliorates endotoxin-induced acute lung injury in vivo and in vitro by preserving mitochondrial dynamic equilibrium through the HIF-1a/ho-1 signaling pathway. Redox Biology. 2021;41:101954.
- Fukuda D, Enomoto S, Nagai R, et al. Inhibition of renin-angiotensin system attenuates periadventitial inflammation and reduces atherosclerotic lesion formation. Biomedicine & Pharmacotherapy = Biomedecine & Pharmacotherapie. 2009;63(10):754–761.
- Bruemmer D, Daugherty A, Lu H, et al. Relevance of angiotensin II-induced aortic pathologies in mice to human aortic aneurysms. Annals of the New York Academy of Sciences. 2011;1245:7–10.
- Lu H, Rateri DL, Bruemmer D, et al. Involvement of the renin-angiotensin system in abdominal and thoracic aortic aneurysms. Clinical Science (London, England : 1979). 2012;123(9):531–543.
- Lai CH, Shi GY, Lee FT, et al. Recombinant human thrombomodulin suppresses experimental abdominal aortic aneurysms induced by calcium chloride in mice. Annals of Surgery. 2013;258(6):1103–1110. DOI:10.1097/SLA.0b013e31827df7cb.
- Wang KC, Li YH, Shi GY, et al. Membrane-bound thrombomodulin regulates macrophage inflammation in abdominal aortic aneurysm. Arteriosclerosis, Thrombosis, and Vascular Biology. 2015;35(11):2412–2422. DOI:10.1161/ATVBAHA.115.305529.
- Guo B, Chen C, Yang L, et al. Effects of dexmedetomidine on postoperative cognitive function of sleep deprivation rats based on changes in inflammatory response. Bioengineered. 2021;12(1):7920–7928.
- Marchesi C, Paradis P, Schiffrin EL. Role of the renin-angiotensin system in vascular inflammation. Trends in Pharmacological Sciences. 2008;29(7):367–374.
- Chang Y, Wei W. Angiotensin II in inflammation, immunity and rheumatoid arthritis. Clinical and Experimental Immunology. 2015;179(2):137–145.
- Qin X, He L, Tian M, et al. Smooth muscle-specific Gsα deletion exaggerates angiotensin II-induced abdominal aortic aneurysm formation in mice in vivo. Journal of Molecular and Cellular Cardiology. 2019;132:49–59.
- Moxon JV, Parr A, Emeto TI, et al. Diagnosis and monitoring of abdominal aortic aneurysm: current status and future prospects. Current Problems in Cardiology. 2010;35(10):512–548.
- Schurgers LJ, Akbulut AC, Kaczor DM, et al. Initiation and propagation of vascular calcification is regulated by a concert of platelet- and smooth muscle cell-derived extracellular vesicles. Frontiers in Cardiovascular Medicine. 2018;5:36.
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiological Reviews. 2004;84(3):767–801.
- Qi S, Li C, Kong X, et al. Dexmedetomidine suppresses oxidative stress and inflammation of nucleus pulposus cells by activating the PI3K/Akt signaling pathway. Die Pharmazie. 2020;75(10):505–509.
- Franck G, Dai J, Fifre A, et al. Reestablishment of the endothelial lining by endothelial cell therapy stabilizes experimental abdominal aortic aneurysms. Circulation. 2013;127(18):1877–1887. DOI:10.1161/CIRCULATIONAHA.113.001677.
- Sun J, Deng H, Zhou Z, et al. Endothelium as a potential target for treatment of abdominal aortic aneurysm. Oxidative Medicine and Cellular Longevity. 2018;2018:6306542.
- Godo S, Shimokawa H. Endothelial Functions. Arteriosclerosis, thrombosis, and vascular biology. Arteriosclerosis, Thrombosis, and Vascular Biology. 2017;37(9):e108–e14.
- Zhao G, Chang Z, Zhao Y, et al. KLF11 protects against abdominal aortic aneurysm through inhibition of endothelial cell dysfunction. JCI Insight. 2021;6(5). DOI:10.1172/jci.insight.141673.
- Bao Y, Zhu Y, He G, et al. Dexmedetomidine attenuates neuroinflammation in LPS-stimulated BV2 microglia cells through upregulation of miR-340. Drug Design, Development and Therapy. 2019;13:3465–3475.
- He Y, Yang Z, Li J, et al. Dexmedetomidine reduces the inflammation and apoptosis of doxorubicin-induced myocardial cells. Experimental and Molecular Pathology. 2020;113:104371.
- Zhou S, Lu S, Guo S, et al. Protective effect of ginsenoside Rb1 nanoparticles against contrast-induced nephropathy by inhibiting high mobility group box 1 Gene/toll-like receptor 4/NF-κB signaling pathway. Journal of Biomedical Nanotechnology. 2021;17(10):2085–2098.
- Lu JM, Jin GN, Lu YN, et al. Resveratrol modulates Toxoplasma gondii infection induced liver injury by intervening in the HMGB1/TLR4/NF-κB signaling pathway. European Journal of Pharmacology. 2021;910:174497.
- Abdelmageed ME, Nader MA, Zaghloul MS. Targeting HMGB1/TLR4/NF-κB signaling pathway by protocatechuic acid protects against l-arginine induced acute pancreatitis and multiple organs injury in rats. European Journal of Pharmacology. 2021;906:174279.
- Ortiz-Espinosa S, Morales X, Senent Y, et al. Complement C5a induces the formation of neutrophil extracellular traps by myeloid-derived suppressor cells to promote metastasis. Cancer Letters. 2022;529:70–84.
- Xu T, Wang S, Li X, et al. Lithium chloride represses abdominal aortic aneurysm via regulating GSK3β/SIRT1/NF-κB signaling pathway. Free Radical Biology & Medicine. 2021;166:1–10.
- Gendy AM, Elnagar MR, Allam MM, et al. Berberine-loaded nanostructured lipid carriers mitigate warm hepatic ischemia/reperfusion-induced lesion through modulation of HMGB1/TLR4/NF-κB signaling and autophagy. Biomedicine & Pharmacotherapy = Biomedecine & Pharmacotherapie. 2022;145:112122.
- Zhai Y, Zhu Y, Liu J, et al. Dexmedetomidine post-conditioning alleviates cerebral Ischemia-reperfusion injury in rats by inhibiting high mobility group protein B1 Group (Hmgb1)/toll-like receptor 4 (Tlr4)/nuclear factor kappa B (NF-κB) signaling pathway. Medical Science Monitor : International Medical Journal of Experimental and Clinical Research. 2020;26:e918617.
- Liu J, Zhang S, Fan X, et al. Dexmedetomidine preconditioning ameliorates inflammation and blood-spinal cord barrier damage after spinal cord Ischemia-reperfusion injury by down-regulation high mobility group box 1-toll-like receptor 4-nuclear factor κB signaling pathway. Spine. 2019;44(2):E74–e81.