
ABSTRACT
Disruption of the complex network that regulates redox homeostasis often underlies resistant phenotypes, which hinder effective and long-lasting cancer eradication. In addition, the RNA methylome-dependent control of gene expression also critically affects traits of cellular resistance to anti-cancer agents. However, few investigations aimed at establishing whether the epitranscriptome-directed adaptations underlying acquired and/or innate resistance traits in cancer could be implemented through the involvement of redox-dependent or -responsive signaling pathways. This is unexpected mainly because: i) the effectiveness of many anti-cancer approaches relies on their capacity to promote oxidative stress (OS); ii) altered redox milieu and reprogramming of mitochondrial function have been acknowledged as critical mediators of the RNA methylome-mediated response to OS. Here we summarize the current state of understanding on this topic, as well as we offer new perspectives that might lead to original approaches and strategies to delay or prevent the problem of refractory cancer and tumor recurrence.
Introduction
Cancer is undoubtedly a primary cause of death in the world, with approx. 10 million people deceasing from cancer every year, and 19 million new cases in 2020 [Citation1]. Despite the efforts made by governments, law makers, and public health authorities, cancer prevention is often achievable only in part, thus leaving in the hands of physicians and oncologists, most of the responsibility for the patient’s treatment and recovery. In this context, the extraordinary efforts of generations of cancer researchers led to significant improvements in handling neoplasms and malignancies, with remarkable advances in surgical management, radio-, chemo- and immune-therapy, which are widely credited as the few effective methods that are able to delay or counteract the spreading of tumors and cancers [Citation2–4]. Regrettably, resistance to therapy, both innate and acquired, still represents a critical complication for successful cancer patients. This phenomenon is multifactorial, but altered redox homeostatic capacity, genetic mutations, and the disruption of epigenetic regulatory mechanisms seem to be three constants in this multitude of factors [Citation5–7]. Only in the last few decades, with the advent of advanced techniques for accurately studying a number of biologically relevant modifications occurring in RNA molecules, the role of epitranscriptomic diversity has gained a primary position on cancer researchers’ radar, especially in terms of methyl-RNA-mediated response to therapy through redox-dependent pathways. This review article aims at summarizing the current state of understanding on this topic, while attempting to provide new perspectives that might help future research to offer answers to still open questions. From a search methodology standpoint, Scopus and Web of Science were considered as the major databases for literature screening, with no publication period limits. Regarding the most specific part of the review, the process for identifying and critically appraising relevant research was conducted after using the following keywords “RNA methylation”, “methylome”, “oxidative stress”, “cancer”, “resistance”, and combinations of them.
RNA methylation and other post-transcriptional modifications
Beyond the well-known epigenetics mechanisms that control gene expression without changes to the DNA nucleotide sequence [Citation8], over the past decades many studies have discovered that more than a hundred different functional post-transcriptional modifications (PTMs) are able to alter the chemical structure of RNA in vivo [Citation9]. PTMs in RNA are considered a powerful means by which cells can modify the functions and fate of RNA in vivo, and such covalent chemical modifications represent critical regulatory events that are involved in both physiological and pathological conditions [Citation10]. In particular, RNA methylation is able to significantly affect both the function and fate of virtually all the classes of RNA. For example, PTMs can alter the stability of transcripts, which is emerging as a key mechanism through which cancer cell biology can adapt to external stimuli, thus relevantly modifying the responsiveness to therapies [Citation11].
The class of RNA that experiences the largest set of PTMs is arguably tRNA, in which about one nucleoside out of ten, on average, is chemically modified [Citation12]. A typical modification, which is present on 67% of the 34 tRNAs, is the methylation at N(1) of adenosine 58. This type of chemical modification is phylogenetically conserved, occurring in bacterial, archaeal, and eukaryotic tRNAs [Citation13]. N1-methylation and de-methylation of A58 is managed by tRNA methyltransferase 6 non-catalytic subunit (TRMT6) and tRNA methyltransferase 61A (TRMT61A) methylases and AlkB homolog 1, histone H2A dioxygenase (ALKBH1) demethylase, and this PTM seems to serve as a crucial determinant of tRNA stability, being its absence a cause of premature tRNA degradation [Citation13]. Another very common modification that is often encountered in tRNAs is the isomerization of uridine to pseudouridine (ψ), which is frequently observed even in the tRNA anticodon region (also known as, nodoc), along with another non-ACUG nucleoside inosine (I), which is often detected in the wobble residue of the nodoc. Other common PTMs of tRNAs are methylations, acetylations, carbonylations and other chemical modifications that form: 2′-O-methyladenosine (Am), 2′-O-methylcytidine (Cm), 1-methylguanosine (m1G), 2-methylguanosine (m2G), 4-acetylcytidine (ac4C), dihydrouridine (D), 2′-O-methylguanosine (Gm), N2,N2-dimethylguanosine (m22G), 3-methylcytidine (m3C), 5-methylcytidine (m5C), 5-methoxycarbonylmethyluridine (mcm5U), 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U), 5-carbamoylmethyluridine (ncm5U), 5-carbamoylmethyl-2′-O-methyluridine (ncm5Um), 1-methylinosine (m1I), N6-isopentenyl adenosine (i6A), wybutosine (yW), N6-threonylcarbamoyladenosine (t6A), 2′-O-methyluridine (Um), 7-methylguanosine (m7G), ribothymidine (rT), and 2′-O-ribosyladenosine (phosphate) (Ar(p)) [Citation12]. Of course, nature rarely wastes, and most tRNA modifications have been demonstrated to play some role in cell growth and survival, especially when the modified residues are missing in or in close proximity to the nodoc loop [Citation14–17].
rRNAs are also heavily modified during their synthesis, both by small nucleolar RNA (snoRNA)-dependent and snoRNA-independent enzymes [Citation18]. It is interesting to note that rRNA modifications cluster in and around functionally critical ribosomal sites, such as the decoding (small ribosomal subunit) and peptidyl-transferase (large ribosomal subunit) centers [Citation19,Citation20], where their modifications seem to be crucial to ensure accurate and efficient protein synthesis, thus allowing a fine-tuned management of gene expression, also in response to external stimuli [Citation18]. It is also usually recognized that rRNA modifications serve as major determinants of secondary and tertiary rRNA structures, by mediating folding and increasing base stacking [Citation18,Citation20]. The most common rRNA modifications are 2”O-methylations on the ribose moiety (with no preference for a specific nucleotide) and the U-ψ isomerization, however, other modifications, such as m1acp3ψ, ac4C, m1A, m5C, m7G, ac4C, m26A, and m3U were identified, for a total of almost 100 modifications found on human rRNA [Citation21–24]. Studies on yeast mutants for enzymes that are responsible for such rRNA modifications revealed that 2”-O-methylations and pseudouridylations are fundamental, but while other modifications seem less important when taken singularly, the cumulative removal of multiple modifications leads to a strong growth impairment, thus indicating that all modifications are important for ribosomal function, but some redundancy exists [Citation25–29].
Messenger RNAs also undergo several enzymatical PTMs during its maturation process from pre-mRNA to mature transcript [Citation30]. More in detail, a common mRNA modification that is found in all mature mRNAs is the so-called 5’ capping, which consists in a very peculiar m7G methylated guanosine bound to the first nucleotide of the RNA through a reverse 5′ to 5′ triphosphate linkage. The classical cap structure also requires the first two nucleotides in the transcript to be 2’O-methylated [Citation31]. Other common nucleotide modifications in mRNAs include added nitro groups and enzymatically-modified ribose moiety [Citation13], but the most common modifications in mRNAs are undoubtedly methylations and U-ψ isomerization [Citation32–34]. The latter has been proven to respond to heat shock and nutrient deprivation, thus suggesting that its occurrence is not random but may have a specific biological function, especially upon environmental changes [Citation13]. However, while pseudouridine and other PTMs may be biologically relevant in specific circumstances, methylation represents the most prevalent internal chemical modification occurring in eukaryotic mRNA [Citation35]. Although this was revealed almost half a century ago, thanks to the pioneering work of Desrosiers and colleagues [Citation36], we had to wait much longer to understand to a greater extent the biological meaning of such modifications. Today, we know that mRNA methylations are common, reversible, and allow for fine-tuning control of gene expression [Citation37]. Different types of mRNA methylation can be found in eukaryotes, both on the nitrogenous base moiety and on the ribose moiety [Citation38]. Such modifications include N6- and N1-methyladenosine (m6A and m1A, respectively), 5-methylcytidine (m5C), and ribose 2′-O-methylation (2′OMe, also known as Nm). Such methylations are reversible and regulated by the so-called “writers”, that include the METTL3-METTL14 methyltransferase complex [Citation39], as well as by the “erasers”, that include the fat mass and obesity-associated protein (FTO) and the alpha-ketoglutarate-dependent dioxygenase AlkB Homolog 5 (ALKBH5) [Citation39,Citation40]. Importantly, the m6A RNA modification is enriched at the 3’UTR of thousands of transcripts and is thought to downregulate translation efficiency via the “reader” protein YTH N6-methyladenosine RNA Binding Protein 2 (YTHDF2) [Citation41], which recruits the CCR4-NOT de-adenylase complex to the methyl-modified mRNAs, thus ultimately reducing their half-life [Citation42]. Intriguingly, the location of m6A seems to be essential for the overall effect of methylation, since it has been suggested that if N6-methylated adenosines are abundant in the 5’UTR, the opposite effect is observed, with a cap-independent induction of translation mediated by eIF3 [Citation43].
N1-methyladenosines (m1As) are quite rare and not as finely characterized as m6A, partially due to the technical difficulties arising from their spontaneous isomerization to m6A [Citation44]. Nevertheless, since this modification bears a positive electrostatic charge, it has been proposed as a factor that influences the secondary structure in the 5’UTR, where it more likely tends to localize, thus altering translation [Citation45].
The biological role of m5C RNA methylation is still highly debated. Some of the methyltransferases involved in their generation were identified as the DNA(cytosine-5)-methyltransferase 2 (DNMT2) and the NOP2/Sun RNA methyltransferase 2 (NSUN2) [Citation46]. Intriguingly, an impaired regulation of m5C RNA levels has an impact on the structural stability of the 60S ribosomal subunits [Citation47]. In addition, experimental evidence indicated that loss of m5C2278 modification led to the recruitment to polysomes of mRNAs specifically linked to the oxidative stress-induced response [Citation48], thus pointing to a possible role of m5C modification in the response to stressors.
The situation is even less defined for 2′OMe modifications, which have the very peculiar feature to localize on the ribose moiety of nucleotides rather than on the nucleobase. So far, little is known about the biological relevance of such modification, that was identified at least in abundant RNA species [Citation49]. Ribosomal RNA 2′OMe was shown to be a key determinant of ribosomal heterogeneity [Citation21], a concept that claims that not all ribosomes are equivalent in cells, with some ribosomes being regulated in terms of translation capacity and efficiencies (ribosomal plasticity) [Citation50]. In particular, 2′OMe patterns in rRNA modulate their ability to interact with internal ribosome entry sites (IRES), as demonstrated in conditions in which U-ψ isomerization was impaired in rRNAs [Citation51]. As a confirmation of the importance of 2′OMe-based PTM in eukaryotic cells, the inhibition of the molecular machinery responsible for such modification resulted in strongly impaired translation [Citation26].
As mentioned above, m6A is the most conserved and prevalent internal chemical modification in eukaryotic RNA molecules [Citation52], and next-generation sequencing (NGS) demonstrated that each eukaryotic mRNA has 3–5 m6A nucleoside modifications [Citation53,Citation54], thus implying that the role played by RNA methylome might be functionally relevant to cell biology. Today, the metabolism of m6A RNA is believed to be involved in developmental processes of vertebrates, especially in the hematopoietic milieu [Citation55,Citation56]. In addition, the deletion of the m6A “reader” protein YTHDF2 was found to promote not only the expansion of human hematopoietic stem cells (HSCs), but also their regenerative capacity upon stress conditions [Citation57]. In addition, the repression of METTL3 in mouse fetal liver was reported to promote the generation of double-stranded RNAs (dsRNAs), whose downstream signaling via melanoma differentiation-associated protein 5- retinoic acid-inducible gene I (MDA5-RIG-I), protein kinase RNA-activated-eukaryotic initiation factor 2α (PKR-eIF2α), and 2”−5”-oligoadenylate synthetase (OAS)-RNase L eventually resulted in hematopoietic development failure [Citation58]. Interestingly, higher m6A levels were observed in the central nervous system (CNS), compared to other systems and organs. Moreover, m6A levels increase as the embryonic brain develops, and this highly suggests that RNA methylome may play an important role during CNS development [Citation52]. Importantly, upon hypoxic stress the downregulation of the m6A “eraser” ALKBH5 enhanced differentiation in postnatal mouse cerebellum via imbalanced RNA m6A methylation in several messenger RNAs involved in cell fate determination [Citation59]. In addition, the loss of the fat mass- and obesity-associated (FTO) “eraser” protein resulted in a reduction of neuronal differentiation in mouse neural stem cells via disruption of the brain-derived neurotrophic factor (BDNF)-dependent signaling, leading to weakened cognitive function [Citation60]. Other researchers confirmed that the correct development of neuronal components is crucially regulated by RNA methylome [Citation61], and others observed that neuron regeneration is critically governed by m6A RNA epitranscriptome [Citation62], thus supporting the notion of an essential participation of m6A RNAs in the regulation of critical neuronal physiological and stress-induced processes.
Another class of RNA that is importantly involved in regulating gene expression is that of miRNAs. Although miRNAs are already considered as crucial epigenetic regulators, it has been discovered that miRNA methylation controls their maturation. It has been demonstrated that miRNA processing is repressed by an atypical dimethylation of the 5’-P group, which is able to block the RNase III endonuclease DICER-mediated miRNA processing in vitro and in vivo, by a mechanism that is dependent on the Bicoid interacting 3 domain (BCDIN3D)-containing RNA methyltransferase [Citation63]. Other miRNA modifications were reported to include m5C, which is also demonstrated to be biologically active [Citation64]. Indeed, m5C is an RNA chemical modification that impairs the hybridization process in the miRNA-mRNA duplex, thus preventing miRNA/RISC-mediated silencing [Citation64]. m6A is also represented in miRNAs and this chemical modification plays an important role for their processing [Citation65,Citation66]. Heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1) was reported to recognize pri-miRNA m6A methylation, and to mediate its interaction with DiGeorge syndrome critical region 8 protein (DGCR8) to promote miRNA processing, which conversely was impaired when m6A was lost in pri-miRNAs [Citation65,Citation67]. Furthermore, an opposite methylation-dependent mechanism that represses DICER-mediated pre-miRNA cleavage into mature miRNAs was also identified. In fact, BCDIN3D, which O-methylates the terminal 5′-mono-phosphate on pre-miRNAs, negatively regulates its binding with DICER, thus blocking its maturation and affecting gene expression, at least in specific miRNAs [Citation63].
Other classes of RNAs have also been found to be methylated, such as circRNAs (m6A) [Citation68,Citation69], piRNA (2’OMe) [Citation70], snRNAs (m6Am) [Citation71], and this highlights the fact that this conserved and maybe underestimated mechanism may be of utmost importance in controlling gene expression within cells via multiple pathways.
A comprehensive examination of the whole landscape depicting the various methyl-based RNA PTMs is beyond the scope of this article, and readers interested in this general aspect of methylepitranscriptomics should consider consulting broader review articles previously published [Citation72–83]. However, in we reported a schematic representation of the most common and best studied methyl-based modifications of the various classes of RNA, along with their main human methylases (“writers”), demethylases (“erasers”) and “readers”, which produce the cellular response when a methylated ribonucleotide is detected.
Figure 1. Schematic representation of the most common and best studied methyl-based modifications of the various classes of RNA, along with their main human methylases (“writers”), demethylases (“erasers”) and “readers”, which drive cellular responses when methylated ribonucleotides are detected. ALKBHs, AlkB Homolog 1, histone H2A dioxygenase; ALYREF, Aly/REF export factor; CMTR1–2, Cap methyltransferases 1 and 2; DNMT, DNA methyltransferases; FBL, fibrillarin; FTO, Fat mass and obesity-associated protein; FTSJ1–2, FtsJ RNA 2’-O-methyltransferases 1 and 2; HENMT1, HEN methyltransferase 1; IGF2BPs, insulin-like growth factor 2 mRNA binding proteins; METTLs, methyltransferase complex subunits; MRM1–3, mitochondrial rRNA methyltransferases 1–3; NML, nucleomethylin; NSUN, NOP2/Sun RNA methyltransferases; PCIF1, phosphorylated CTD interacting factor 1; RNMT, RNA guanine-7 methyltransferase; TRMT, tRNA methyltransferase; WTAP, Wilms tumor 1-associating protein; YBX1, Y-Box Binding Protein 1; YTHDCs, YTH Domain Containing proteins; YTHDFs, YTH N6-methyladenosine RNA binding proteins; ZCCHC4, zinc finger CCHC-type containing 4.
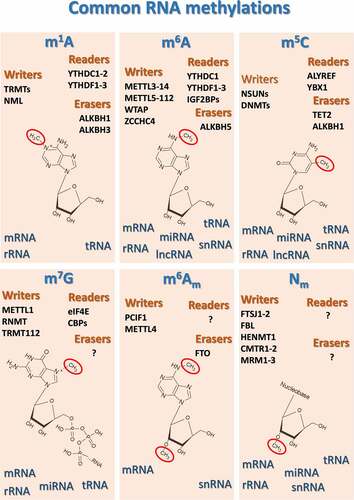
In summary, although how the methylome of tRNAs and other RNAs modulates protein and gene expression represents an emerging research field, it is reasonable to consider the hypothesis that the methyl-based epitranscriptomic landscape might play a significant role in foreseeing or driving different cell responses to external triggers and stimuli.
RNA methylation, redox homeostasis, and oxidative stress
Perturbation of redox balance and alteration of the activity of redox-dependent signaling pathways are among the major triggers of critical cell-fate decisions upon stress [Citation84]. The so-called redox homeostasis depends on the accurate balance between the generation and elimination of redox-active radical and nonradical chemical species in a biological system [Citation85]. Among the most biologically relevant chemical species, there are nonradical reactive oxygen species (ROS), such as hydrogen peroxide (H2O2) and peroxynitrite (ONOO−), whereas radical ROS include the superoxide radical anion (O2•−), the hydroxyl radical (OH•), the nitric oxide (NO•), and a number of alkoxyl/alkyl peroxyl (RO•/ROO•) molecules [Citation85].
Redox homeostasis permits a precise control of local and systemic concentrations of ROS, such as H2O2, which, when present in strictly regulated amounts, serve as critical players that govern metabolic regulation and stress responses, thus permitting adaptation to environmental stimuli and stressors [Citation86]. Conversely, uncontrolled buildup of ROS is today known to lead to oxidative chemical modification of all major macromolecules inside and outside cells and organelles, including lipids, proteins and nucleic acids. If prolonged, such a condition may lead to severe oxidative stress (OS), which is mechanistically linked to serious cell dysfunction and irreversible loss of homeostasis [Citation87]. On this basis, living matter has evolved a complex network that is responsible for scavenging ROS in excess and for accurately controlling the redox homeostatic balance. The clearance of ROS involves coupled and uncoupled enzymatic systems that include superoxide dismutases (SODs), glutathione peroxidases (GPXs), catalase (CAT), thioredoxins (TRXs) and peroxiredoxins (PRDXs), together with non-enzymatic antioxidant species that include reduced glutathione (GSH), several water- and lipid-soluble vitamins and oligoelements [Citation72,Citation88].
Ribosomal RNAs and transfer RNAs
Collectively, the literature suggests that RNA is the most abundant cellular nucleic acid, with 10–20 times higher levels of ROS-mediated damage, as compared to DNA [Citation73–75]. RNA is more susceptible to oxidative damage than DNA mostly due to its single-stranded structure, and due to the absence of protective proteins [Citation76]. Oxidatively modified RNA may lead to significant changes in the most critical biological processes within the cell. For example, oxidative modification of rRNA has been linked to errors during codon-anticodon pairing [Citation77] and altered protein synthesis [Citation78]. Impaired protein biosynthesis could also derive from the presence of oxidatively inactivated tRNAs [Citation79].
The presence of methylated residues in ribonucleic acids has an impact on the structure of the molecule, and some evidence indicates that RNA methylation modifies the susceptibility of RNAs to ROS. Very interestingly, Estevez and collaborators recently reported that structure-stabilizing methylation in E. coli rRNAs protected bacterial 23S ribosomal ribonucleic acids from ROS-induced damage [Citation80]. In this context, 7-methylguanine (m7G), methylguanine (m2G), and guanosine ribose methylation are believed to be particularly important [Citation80].
Remarkably, not only the presence of methylated nucleotides is able to affect RNA susceptibility to ROS, but also RNA methylation might serve as a means through which cells can respond to stimuli. Based on this assumption, and as mentioned above, changes in the epitranscriptomic pattern have been in the radar of researchers for decades. As early as the 1960s, the levels of methylated nucleotides in tRNA, which is the type of RNA on which early research was focused because of the higher abundancy and relative stability [Citation81], were significantly altered by environmental stresses [Citation82]. However, only in the last twenty years a great wave of interest has attracted biomedical researchers, with many groups actively investigating and fully recognizing the importance of the role of post-transcriptional internal methylation of RNAs as a central element in the dynamic regulatory network that is responsible for controlling gene expression through which cells adapt to external stimuli.
As reviewed by Wilkinson et al., the stress response in cells often involves an orchestrated pattern of RNA modifications that are mobilized to modulate a plethora of transduction pathways aimed at counteracting heat shock, metabolic imbalance, loss of proteostasis, and DNA damage [Citation83]. Regarding this latter aspect, RNA modifications were found to participate to the maintenance of genome integrity and repair of nucleic acid lesions by modulating the formation of DNA-RNA hybrids (R-loops) at damage sites on DNA [Citation89].
One of the most important mechanisms through which environmental agents interact with living matter is oxidative stress (OS) [Citation90], and the integration of redox signaling processes often serve as transduction pathways through which cells modify gene expression and phenotype to respond to internal and external stimuli [Citation91].
Unfortunately, some studies have provided potentially interesting but limited information on how RNA methylation-related metabolism is able to affect the cellular and organismal responses to OS, due to some weaknesses in the rationale or methodology (such as, the measurement of total m6A rather than the evaluation of the contribution of distinct RNA classes), however today much more is known as to how internal RNA methylations has a pivotal function in the cellular response to OS, as compared to just a couple of decades ago.
It is worthy of note that the catalysis of RNA methyltransferases does not rely on redox reactions, therefore the discovery of an iron/sulfur-dependent methyltransferase that methylates 23S rRNA in E. coli raised extreme interest twenty years ago. Such findings suggested that RNA methylation events could be affected by the cellular redox status and, more importantly, that the redox-responsive site of some RNA methyltransferases may undergo oxidative-dependent inactivation [Citation92]. It was even hypothesized that methylation of 23S rRNA in E. coli could be regulated by cellular oxidative stress [Citation93]. In 2015, Kyuma and collaborators reported that defective methylation of adenosine and cytidines at several positions in 16S rRNA was able to induce increased sensitivity to oxidative stress in S. aureus, thus leading to its attenuated virulence in a silkworm infection model [Citation94,Citation95]. This straightly led to the hypothesis that the regulatory activity of the methyltransferases the are required to such rRNA modifications may become important upon OS.
Other researchers outstandingly confirmed this idea in eukaryotes. Using bisulfite-based sequencing methods, Schosserer and collaborators observed that gene silencing of the “writer” NSUN5 rendered C. elegans worms resistant to the ROS-generating compound paraquat (PQ) [Citation48]. Similar effects were documented in a H2O2-treated strain of S. cerevisiae with the NSUN5 homologue knocked out [Citation48]. Such effects were linked to the lack of methylated specific cytosines within worm and yeast rRNAs, and this could be due to changes in the core ribosomal function that may allow the cells to modulate ribosome biogenesis faster, permitting a more efficient protein synthesis in case of stress [Citation48].
Other important targets of methylation are tRNAs, and some of these chemical modifications have been critically associated with processes that govern OS-related changes in gene expression. In the early 2000s, Begley and colleagues, who have been among the most active researchers in this field, used high-throughput screening of gene deletion libraries and found that tRNA methyltransferase 9 (Trm9), an enzyme that specifically methylates tRNA, could be critical to prevent DNA damage and to increase resistance of S. cerevisiae against exposure to reactive oxygen species (ROS)-generating agents [Citation96,Citation97]. A few years later, Begley’s team demonstrated that some of the effects elicited by Trm9 were mediated by ribonucleotide reductases 1 and 3, which are key elements that respond to DNA damage via enhancing the production of deoxyribonucleotides needed for DNA synthesis [Citation98]. More recently, a brilliant application of bioanalytical and bioinformatic tools by Chan et al. proposed a model which explained how the exposure of yeast to hydrogen peroxide was able to induce selective translation of mRNAs coding for proteins needed for the oxidative stress response, such as the ribosomal 60S subunit protein RPL22A, via increased levels of m5C at the anti-codon 5’ end of tRNALeu [Citation99].
The idea that tRNA methylation was linked to cellular OS was independently confirmed by Russian researchers, who in 2009 demonstrated that the generation of m6A in tRNAVal conferred a significant growth advantage upon oxidative stress to E. coli [Citation100]. In 2011, Osterman and coworkers showed that E. coli was able to survive extreme OS only when a specific residue of 23S rRNA was methylated (i.e. m2G1835) [Citation101], and this further research corroborated that resistance of prokaryotes to OS was mediated by RNA methylation. Later research on the opportunistic pathogen P. aeruginosa showed that an high resistance to OS could be obtained by abolishing the expression of the trmJ homolog tRNA (cytidine(32)/uridine(32)/adenosine(32)-2′-O)-methyltransferase, thus reducing the formation of Nm in tRNAs [Citation102].
These findings led to the solid idea that the complex system of chemical tRNA modifications, and specifically methylation processes, may represent a powerful means through which cellular responses to the environment could be regulated post-transcriptionally [Citation103].
In 2014, such idea was corroborated by the intriguing observations of Barroso and colleagues, who showed that hypomethylation of tRNA[Ser]Sec, a particular type of transfer RNA that allows the biosynthesis of key antioxidant selenium-containing proteins, was able to induce oxidative stress in human umbilical vein endothelial cells (HUVECs) [Citation104].
This idea was somewhat supported by the findings reported by Schaefer and colleagues, who demonstrated that Dnmt2 null mutant fruit flies showed reduced viability upon H2O2- and paraquat-induced OS [Citation105]. As mentioned in the sections above, DNMT2 is an evolutionary conserved multisubstrate enzyme that is critically involved in the regulation of genomic stability via methylation of nucleic acids [Citation106]. Originally considered as a pure DNA methyltransferase, DNMT2 was demonstrated to be a tRNA methyltransferase in 2006, when DNMT2 was mechanistically linked to methylation of tRNAAsp [Citation107]. In their experiments, Schaefer and coworkers provided an exquisite proof that DNMT2-mediated methylation of tRNAs was able to protect tRNAGlyGCC and tRNAAspGTC from stress-induced ribonuclease cleavage, which represents a conserved response to several stress stimuli in eukaryotes [Citation105]. Taken together, these results point out to a specific role for the DNMT2-dependent regulation of tRNA structure and fate in the response of organisms to pro-oxidant conditions.
Conversely, a few years later, Mytych and coworkers found that human HeLa cells undergo oxidative stress after exposure to nanodiamond particles, possibly via upregulation of DNMT2. The same authors found also that vascular smooth muscle cells responded to a pro-oxidant insult by arresting the cell cycle, and this was associated with the upregulation of DNMT2 as a part of cellular stress response [Citation108]. However, only a speculative stabilizing role of DNMT2 on RNA was hypothesized by the authors [Citation108], without providing clear evidence of the actual mechanism underlying such response.
As already mentioned, NSUN2 is another important regulator of tRNA methylation [Citation109]. Interestingly, increased apoptotic death was observed by Blanco et al. in Nsun2−/− mice and human skin cells in response to sodium arsenite- or UV-induced OS, and this was linked to the loss of NSun2-mediated RNA methylation and to the subsequent accumulation of 5′ tRNA-derived small RNA fragments (tRFs) [Citation110], which are known to inhibit protein synthesis [Citation110,Citation111]. Therefore, the Nsun2-dependent methylation of RNA should be considered as a key step in the OS-induced dynamic post-transcriptional regulation of gene expression.
In 2014, high levels of methylated tRNAAspGUC were associated with high expression of OS-responsive proteins, such as peroxyredoxins (PRXs) [Citation112]. More in detail, methylated C38 in tRNAAspGUC was linked to selective biosynthesis of proteins specifically involved in the response to OS, and whose genes are highly enriched with GAC triplets [Citation113]. The same year, Dewe and coworkers showed that human embryonic kidney cells in which the mammalian ortholog of yeast tRNA(guanine 26,N2,N2)-dimethyltransferase Trm1p (TRMT1) was knocked out or nonfunctional, exhibited hypersensitivity to several pro-oxidant compounds, such as tert-butyl-hydroperoxide (t-bu-OOH) and hydrogen peroxide, and this was associated with defective formation of dimethylguanosine (m2,2G) in cytoplasmic and mitochondrial tRNAs [Citation114]. More recently, S. cerevisiae strains that lacked specific tRNA methyltransferases (namely, Trms 3, 7, 13, and 44) exhibited higher sensitivity to H2O2 and rotenone, as compared to the wild type strain, and in some cases this was linked to increased ROS production and redox imbalance [Citation115]. By using bioinformatics, the authors concluded that UUC-enriched gene transcripts, which encode proteins that play a functional role in recovering from OS, could be regulated by Trm at translational level [Citation115]. Such results further confirmed that tRNA methylation may serve as a critical determinant for the response of eukaryotes to redox imbalance-promoting treatments. Recently, Trixl and colleagues inactivated the methyltransferase Nsun3 in mouse embryonic stem cells (ESCs) via a CRISPR/Cas9 technology, and found that H2O2 induced a weaker mitochondrial ROS production in Nsun3-mutant, with respect to that observed in wild type cells [Citation116]. In the same year, Cosentino and coworkers showed in rat pancreatic islet beta cells that defective methylation of tRNA guanine 9 (m1G9) was able to induce a controlled tRNAGln fragmentation, thus causing oxidative stress via production of both OH• and peroxynitrite [Citation117]. These results confirmed that tRNA-targeting methylation reactions can initiate the execution of cellular redox signaling programs.
Among the pathways involved in cell adaptation to OS is the control of cell cycle progression. In particular, the cell cycle is often altered upon OS, and part of the rationale for this is that the molecular oxidative damage needs to be repaired before proceeding through the various phases [Citation39]. Interestingly, Gkatza and colleagues have recently demonstrated that NSUN2 was essential for human dermal fibroblasts to adapt their cell cycle phases to an arsenite-induced pro-oxidant stimulus, and this was associated with an OS-induced decrease of methylated C34 in tRNALeuCAA [Citation40]. Arsenite was used as a OS-promoting treatment also in other studies. Rashad et al. have recently reported that the m1A “eraser” ALKBH1 participated to the cellular response of rat neuroblasts to sodium meta-arsenite in terms of cytosolic and mitochondrial tRNA methylation state and cleavage, even though the authors themselves claimed that further efforts were needed to better characterize the relationship between specific tRNA cleavage and cellular fate in terms of stress and death [Citation118].
Messenger RNAs
As extensively discussed in the previous sections, ribosomal and transfer RNAs are not the only ribonucleic acids that are targeted by methylation reactions. Several methylated sites were also found in messenger RNAs. However, mRNA internal methylations are more difficult to identify and characterize due to their relatively low abundance [Citation83]. Moreover, how specifically mRNA methylation may have an impact on transcript fate upon cellular stress is still under intense debate. In 2015, Zhou and coworkers published a pioneering work in which heat shock was reported to induce in mouse embryonic fibroblasts (MEFs) a 90-fold increase in the mRNA levels of the Hsp70-related gene hspa1a, and this was correlated to a strong increase in m6A levels within the 5′UTR of the corresponding transcript [Citation119]. In the more specific context of oxidative stress, in 2012, Zhang and colleagues revealed that the response of HeLa cells to hydrogen peroxide relied on the stabilizing action of methylation on the 3′-UTR of p16 mRNA [Citation120]. Four years later, the same research group demonstrated that oxidative stress and high glucose induced accelerated senescence in HUVECs by enhancing the translation of the adaptor protein Shc via NSUN2-dependent methylation of Shc mRNA at multiple sites, both in coding and non-coding regions [Citation121]. Shc mRNA is known to produce three protein isoforms that are crucially involved in redox signaling and mitochondrial ROS production [Citation122], and this strengthens the importance of RNA methylation as a key regulator operating in the complex network that governs how cells respond to OS.
A few years later, it was established that OS could influence mammalian microRNA methylome, as well. In particular, Yuan and coworkers found that OS was able to increase the methylation levels of microRNA 125b (miR-125b), reducing its capacity to recruit (RNA-induced silencing complex) RISC, thus increasing the expression of miR-125b-regulated target transcripts [Citation123]. The transcription factor nuclear factor erythroid-derived 2-like 2 (Nrf2) is certainly one of the most important miR-125b-regulated determinant in cell response to OS [Citation124]. This further supports the idea that post-transcriptional RNA methylation is able to affect cell phenotype in terms of redox homeostatic capacity. To the best of our knowledge, no other studies have attempted to clarify the role of methylation in mammalian microRNAs as epitranscriptomic factors serving as regulators to drive a response to OS. Hence, this definitely represents an under-investigated field that might hide secrets and potential targets that for future benefits.
More recently, Zhao and colleagues have observed increased levels of OS and higher levels of m6A nrf2 mRNA in Sprague-Dawley rats that were exposed to the ROS-generating compound di-(2-ethylhexyl) phthalate [Citation125], thus corroborating the notion that the post-transcriptional control of NRF2 may link mRNA-based epitranscriptomic regulation of gene expression to the cellular effects elicited by pro-oxidant. The involvement of the Kelch like ECH associated protein 1 (KEAP1)-NRF2 axis in the mRNA methylation-dependent cytoprotective response to OS has been recently observed by Arumugam and coauthors, who found that the ROS-generating compound fumonisin B1 (FB1) induced in immortalized human liver cells an aberrant m6A modification in total mRNAs via differential regulation of the major methyltransferases and demethylases that participate to m6A RNA metabolism (namely, METTL3/14 and ALKBH5) [Citation126]. Such effect was shown to be causatively linked to the FB1-induced increase in the levels of m6A mRNA and to the subsequent reduction in the protein expression of the repressor Keap1 [Citation126]. Keap1 downregulation is known to facilitate NRF2 translocation to the nucleus, where in the form of a heterodimer with the small Maf protein it binds to the antioxidant responsive elements (AREs) of several antioxidant genes, thus promoting their transcriptional activation [Citation127]. On this basis, the identification of the Keap1-Nrf2 axis as a pathway affected by OS through selective changes in the levels of specific methylated mRNAs sheds new light on the molecular details underlying the epitranscriptome-dependent regulation of the redox homeostatic capacity in mammals. Other experimental evidence supports such hypothesis. Indeed, NSUN2 and METTL3/METT14 were found to cooperatively and synergistically methylate p21 mRNA at m5C and m6A, thus enhancing p21 expression at the level of translation in a model of H2O2-induced cellular senescence [129,p.2]. P21 is a cyclin dependent kinase (CDK) inhibitor that exerts its protective effect against OS through multiple pathways, including the upregulation of the Nrf2 downstream signaling pathway [Citation128,Citation129].
Interesting results from a recent work by Zhao and coauthors showed that the underexpression of METTL3 and the subsequent reduction of m6A RNA levels could participate to the neurotoxic effect of Aβ oligomers in rodent and human brains, thus linking the regulation of methyl-epitranscriptome to the OS-based pathogenetic mechanisms of Alzheimer disease (AD) [Citation130].
Hypoxia/reoxygenation (H/R) is a well-known phenomenon that causes overproduction of ROS and significant OS [Citation131]. In this context, Song and coworkers have recently reported that rodent cardiomyocytes and hearts respond to H/R with repressed autophagic flux and increased apoptosis, as well as with a parallel upregulation of METTL3 and increased levels of METTL3-dependent methylation (m6A) in mRNAs [Citation132]. These authors demonstrated that the OS-induced overexpression of METTL3 led to the hyper-methylation and destabilization of the transcriptional factor EB (TFEB) mRNA, a master regulator of autophagy, thus preventing its accumulation and translation in cardiomyocytes [Citation132]. A few months later, Wang and colleagues shed further light on the response of the mammals’ cardiovascular system to the H/R-dependent OS. In fact, these authors observed an increased apoptotic death both in human cardiomyocytes exposed to H/R and in a rat model of myocardial ischemia/reperfusion (I/R) [Citation133]. Interestingly, such effect was strictly dependent on the promotion of endoplasmic reticulum (ER) stress via a m6A-based upregulation of the activating transcription factor 4 (ATF4) mRNA [Citation133].
Interestingly, in 2021, Pang et al. reported that increased levels of METTL14 served as a protecting factor against OS-induced reduction in m6A RNA after I/R insult, both in vivo and in vitro [Citation134]. This was accompanied by the activation of the Wnt/β-catenin axis via increased levels of m6A in Wnt1 mRNA [Citation134]. Considered the acknowledged role of the Wnt/β-catenin axis [Citation135], Pang and colleagues’ results seem to suggest that the OS-triggered modulation of the m6A epitranscriptome occurs via regulation of one of the most important and conserved signal transduction pathways in tissue homeostasis. This idea was further confirmed by Guo and coworkers, who recently found that the inhibition of RNA demethylase activity of ALKBH5 was sufficient to improve the function of H/R-treated human trophoblast cells by activating the Wnt/β-catenin pathway [Citation136]. These authors also found that the repression of the activity of ALKBH5 was essential to alleviate OS-dependent apoptosis in H/R-treated human trophoblast cells, and this effect was closely correlated to increased levels of m6A in peroxisome proliferator-activated receptor gamma (PPARγ) mRNA, as well as this effect was linked to augmented levels of the corresponding protein [Citation136], thus strengthening the idea that in mammals there exists a sophisticated ensemble of epitranscriptome-mediated post-transcriptional regulatory mechanisms that is able to modulate cellular resistance to OS-promoting insults.
As discussed above, regarding the exact molecular mechanisms by which the rewiring of the mRNA methylation program can be functional to the response to OS, no conclusive idea has been developed so far. However, in 2018, Anders and colleagues revealed a novel pathway through which OS may lead to changes in the methyl-mRNA landscape. In fact, arsenite (AS)-treated human embryonic kidney cells exhibited increased levels of N6-methyladenosine in the 5′ UTRs and in the 5′ proximity of coding sequences of messenger RNAs, and this was mechanistically linked to the segregation of transcripts in stress granules (SGs) [Citation137]. SGs are cytoplasmic RNA-protein complexes in which mRNAs are thought to be sequestered in a translationally stalled initiation form [Citation138]. On this basis, recent discoveries depict a totally new function of mRNA N6-methyladenosines in the intracellular processes underlying the modulation of cell response to stress in mammals. In this context, the major m6A-binding proteins, YTHDF1–3, were recently indicated as possible critical players in the process of SG formation. In particular, double knockdown-based experiments on mammalian bone cells showed that YTHDF1 and YTHDF3 are essentially required to form SGs [Citation139]. Another interesting observation was recently reported by Du and colleagues, who found that I/R-injury in liver and hepatocytes proceeds via OS and Drp1-dependent mitochondrial fragmentation due to a reduction of the enzymatic activity of N6-methyladenosine demethylase FTO [Citation140].
lncRNAs
Another potentially interesting finding comes from a recent work by Qu et al., who reported that m6A-modified long noncoding RNAs (lncRNAs) may also have a role in mediating the effects of cadmium-induced oxidative damages [Citation141]. Few other researchers have confirmed that methyl-based modification of lncRNAs may be involved in the spectrum of molecular changes activated by OS in living cells. Su and colleagues observed that exposure to the ROS-generating PQ induced a hypermethylation of total RNA in mouse neuroblasts, and this was associated with impaired expression of m6A methyltransferases and demethylases, along with an altered pattern of m6A-modified lncRNAs [Citation142]. In particular, the authors identified two specific lncRNAs that were differentially expressed in PQ-treated cells, compared to controls: the cell division cycle 5-like (lncRNA CDC5L) and the signal transducer and activator of transcription 3 (lncRNA STAT3), and this seems to suggest that the regulation of the autophagic process could be closely involved in the downstream signaling activated by OS via lncRNA methylation [Citation142]. Unfortunately, some of these reports tend to rely on relatively simple statistical analyses, but do not always provide a clear explanation as to how the findings observed might have biological relevance. More specifically, in some cases, a limited molecular explanation was provided as to how the participation of methyl-lncRNAs could determine the cytotoxic effects of OS or could prevent the OS-dependent biological effects, therefore researchers with documented and strong expertise in this field should study more in depth this intriguing phenomenon.
Oxidative stress and anticancer therapy
As briefly discussed above, at strictly controlled concentrations, ROS serve as functional signaling entities in cells, regulating crucial physiological processes such as proliferation, differentiation, survival, and adaptive response to external stimuli [Citation86].
However, an unbalanced production of ROS may lead to an important loss of redox homeostasis, metabolic dysregulation, molecular damage, genetic instability, and cell dysfunction, thus contributing to the onset of several disorders and diseases, including tumor and cancer [Citation86,Citation143].
ROS and chronic OS not only can promote carcinogenesis, but also mediate tumor survival and progression [Citation144,Citation145]. In fact, an increasing number of reports have established that high levels of ROS are often required by cancer cells to escape cell death, to promote mass growth, to adapt metabolic needs, and to modify the surrounding environment to facilitate tumor expansion and migration.
For example, both superoxide anion and H2O2 appear to be linked to tumor and cancer cell phenotype and behavior. Specifically, in the 1990s, the metastatic capacity of rodent fibrosarcoma cells was reduced by increasing the mitochondrial production of O2.- [Citation146]. In addition, a strong O2.--dependent mitogenic activity was found in fibroblasts expressing a constitutively active isoform of the oncogene p21 [Citation147]. In more recent years, Lee et al. showed that the ROS-induced inhibition of tyrosine phosphatases was able to sustain the activation of antiapoptotic pathways in pancreatic cancer cells [Citation148]. In addition, enhanced ROS levels were linked to drastically diminished apoptotic death in bladder cancer cells [Citation149]. Likewise, the selective upregulation of mitochondrial ROS was demonstrated to stimulate cell proliferation and cell survival in carcinoma cells, along with epithelial – mesenchymal transition (EMT) through the activation of mitogen-activated protein kinase (MAPK) and Ras-ERK axis [Citation150]. In 2016, Cao and coworkers reported that the overproduction of ROS and the consequent activation of ERK and p38 MAPK signaling pathways were essential for hyperglycemic stress to induce migration and invasion of pancreatic cancer cells [Citation151]. Similarly, proliferation of colon cancer cells was found to be promoted by EGF-induced stimulation of ROS via activation of NADPH oxidase and heme oxygenase-1 (HO-1) [Citation152]. In addition, inducible nitric oxide synthase (iNOS)-dependent NO• production boosted the growth of oral squamous carcinoma cells, also through the stimulation of angiogenesis [Citation153]. In 2017, Aydin and colleagues reported that ROS promoted the metastatic ability of murine melanoma cells by repressing the NK cell- and lymphocyte-mediated anti-cancer activity [Citation154]. Furthermore, the ROS-induced hyper-proliferation of human melanoma cells was found to be facilitated by the generation of high local concentrations of extracellular ROS via modulation of signaling pathways involving HIF-1α and Akt/GSK3β/p27Kip1 [Citation155].
On this basis, biomedical research was solicited to find a way to scavenge ROS as a tool to prevent tumor formation or, at least, to delay the progression of neoplasms. As early as the 1950s, dietary intake of the antioxidant vitamin E was found to be associated with protection from the tumorigenic effect of 3’-methyl-4-dimethylamino azobenzene [Citation156]. Over the following years, several studies and randomized trials have confirmed that in some cases antioxidant chemicals could serve as co-therapeutics in anticancer treatments [Citation157–159].
However, modern literature seems to support the notion that a strong and persistent increase in ROS levels leads to cell death in many cancer cell types. This explains why some of the most effective cancer therapeutics that are used today by oncologists undoubtedly rely on the ability to upregulate ROS concentrations within cancer cells [Citation160,Citation161]. As a corollary to this concept, it should be noted that both increased antioxidant protection and enhanced redox buffering capacity have been frequently linked to a more aggressive and therapy-resistant behavior in cancers [Citation162]. For example, as discussed previously, the NRF2-KEAP1 system plays a critical role in preserving redox homeostasis, and the activation of such a molecular switch has a primary function to adapt to cellular OS [Citation163]. Accordingly, a prolonged activation of NRF2 was hypothesized to promote the survival of precancerous cells harboring oncogenic mutations [Citation164], and analogous findings help to understand why NRF2 is highly expressed in a variety of cancers [Citation165]. The improvement of the redox homeostatic capacity is certainly one of the most important adaptive responses through which tumor progression/dissemination can be achieved, and one of the most central mechanisms by which therapy resistance traits can be developed in malignancies [Citation161,Citation166,Citation167]. Other means include: i) reduced drug intake and/or enhanced drug inactivation; ii) adaptation of metabolic pathways; iii) increased DNA repair; iv) suppression of proapoptotic signaling and/or activation of prosurvival signaling; v) enhanced autophagy; vi) rewiring of mitochondrial dynamics [Citation161,Citation166–170].
Mitochondrial ROS production and m6A-modified RNAs
Mitochondria represent one of the major sources of ROS generation within cells [Citation171]. Therefore, some researchers have investigated whether and how RNA methylation could modulate the activity of ROS-producing mitochondrial pathways. In 2019, Zhuang and colleagues reported that the FTO demethylase was able to reduce m6A levels in the PGC-1α transcript, thus increasing the expression of this master regulator of mitochondrial biogenesis and boosting both mitochondrial activity and ROS production in a model of clear cell renal cell carcinoma [Citation172]. However, FTO was found to “erase” m6Am, not m6A, in cells [Citation173,Citation174], thus suggesting that Zhuang and coworkers’ findings might be read in terms of reversible dimethyladenosine modification to PGC-1α mRNA.
In 2019, some researchers found that dopaminergic PC12 cells overexpressing FTO were characterized by lower activities of superoxide dismutase and mitochondrial complex II, thus strengthening the idea that the dynamics of dimethyladenosines in RNA could regulate ROS-generating processes, especially in mitochondria, interfering with the activation of the apoptotic pathway [Citation175]. A year later, Liu and coworkers demonstrated that the activity of p38 mitogen-activated protein (MAP) kinase (MAPK), which undergoes phosphorylation and subsequent activation upon mitochondrial ROS generation, was regulated by the expression of adenylate kinase (AK4), and the expression of AK4 was higher in tamoxifen-resistant MCF-7 cells with increased levels of METTL3-induced m6A in the 5′ and 3′ UTR of adenylate kinase (AK4) mRNA [Citation176]. In 2021, melatonin was found to attenuate ROS generation in a reproductive model of cromium-induced mitochondrial redox imbalance, and the protective role of melatonin were exerted via restoration of METTL3-mediated m6A RNA modification, as well as through the activation of mitochondrial fusion and inhibition of mitophagic pathway [Citation177]. Other researchers provided strong evidence that the regulation of the RNA methylome is able modulate mitochondrial ROS production. In fact, in 2022, Sun and collaborators found in a liver fibrosis model an impaired translation of the mRNA of peroxiredoxin 3 (PRDX3), which serves as a master regulator of mitochondrial oxidative stress, and this was mechanistically linked to a diminished interaction between m6A and the m6A “reader” YTHDF3, thus leading to increased mitochondrial ROS and hepatic stellate cell (HSC) activation [Citation178]. A very interesting paper from Xu and colleagues reported that METTL3 was able to enhance the methylation of cytochrome c oxidase subunit (NDUFA4) mRNA, and this was sufficient to stimulate the expression of NDUFA4 in gastric cancer cells, with consequent inhibition of mitochondrial ROS accumulation [Citation179].
These results strongly point out to RNA methylation as a powerful regulator of mitochondrial redox homeostasis and activity, and explain the raising interest of researchers toward the methylepitranscriptome as a biological means through which important mitochondrial redox alterations can occur in cells leading to significant changes in cellular phenotype. This aspect certainly deserves further investigation, as mitochondria serve as a critical crossroads in cells where ROS homeostasis meets metabolic and energy needs, thus representing a potential hub that may integrate redox sensing with adaptive changes in gene expression via RNA post-transcriptional dynamic and reversible chemical modifications, both in physiology and disease. In this context, Zhang et al. found that oxidized low-density lipoprotein (oxLDL) activate OS- and flogosis-related responses in monocytes via a METTL3-YTHDF2 cooperative interaction that causes the degradation of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) mRNA, increased ROS leakage and, ultimately, reduced ATP production [Citation180]. Considered that PGC-1α critically regulates mitochondrial biogenesis [Citation181], Zhang and coworkers’ findings seem to confirm that the OS-activated pathways in mammalian cells could be intimately linked to dynamic changes in methylepitranscriptome, altered mitochondrion-dependent energy metabolism, ROS sensing and production.
RNA methylation and cancer cell response to clinically relevant OS-generating treatments: factual evidence and missing pieces of the puzzle
As discussed above, on one hand, the fine-tuned control of ROS production and metabolism, along with the redox homeostatic buffering capacity, is a key factor that contributes to define the susceptibility of neoplasms and malignancies to many anticancer therapeutics. On the other hand, adaptive processes and cellular response to OS-generating stimuli are governed by a multilevel molecular network that reacts to exogenous stimuli, via regulatory events that include changes in the RNA methylation pattern.
As discussed by others [Citation182], it might be conceived that specific methylated RNA residues or even the overall RNA methylation diversity could predict or influence the effect of redox-based anticancer treatments on tumor and cancer cells.
5-fluorouracil
In this context, one of the first evidence was reported by Cori and coauthors in the late 70s, when the resistance of hepatoma cells to 5-fluorouracil (5-FU), a pyrimidine analog that is widely used to treat a variety of solid cancers, was reversed by adding inosine to the incubation mixture, and this was surprisingly associated with re-shaping of the methylation pattern of ribosomal RNA [Citation183]. It is important to note that the antiblastic mechanism of action of 5-FU, which is a drug often used to treat cancers that are unresponsive to targeted therapy, is at least in part mediated by its ROS-generating capacity [Citation184]. This should have probably pushed redox researchers to investigate this topic further. However, after such a pioneering report, this field remained relatively dormant for many decades in the shadow cast by the limited knowledge and technical difficulties surrounding the epigenetics field [Citation185,Citation186]. Only in April 2008, Marie Gustavsson and Hans Ronne proposed a model to explain how 5-FU-induced effects in eukaryotes may be influenced also by biological processes that involved RNA methylation. The authors hypothesized that cellular responsiveness to 5-FU could depend on the reduced activity of several tRNA modifying enzymes, among which the tRNA 5-methyluridine methyltransferase would promote RNA destabilization, especially at higher temperature [Citation187]. Interestingly, this would explain why hyperthermia is able to enhance the effect of 5-FU in some anticancer therapies [Citation188]. A few years later, Okamoto et al. found that the sensitivity of human cancer cells to 5-FU could be decreased by co-overexpressing both tRNA methyltransferases NSUN2 and METTL1 in HeLa cells; accordingly, the cytotoxic effect of 5-FU was increased through a double knockdown of both NSUN2 and METTL1, and such potentiated action of 5-FU was associated with a rapid degradation of tRNAValAAC [Citation189]. This finding is important because it is known that cleavage of tRNAs is increasingly considered as an effective mechanism through which protein synthesis can be regulated during specific stress conditions [Citation190–192]. A few years ago, the role of the m6A “writer” METTL3 in chemoresistance toward 5-FU was also investigated. In their work, Taketo and coauthors showed that the viability of 5-FU-treated pancreatic cancer cells was dramatically reduced when METTL3 was stably knocked down [Citation193]. The extreme importance of mutant versions of the tumor suppressor protein p53 is fully acknowledged by modern molecular oncologists [Citation194]. In this context, Uddin and coworkers recently found that human colon cancer cells with specific methylated adenosines in p53 pre-mRNA generated a R273H missense p53 mutant, thus conferring drug resistance toward 5-FU [Citation195]. p53, which is also known as the “guardian of genome”, serves as an important regulator for dual cellular responses to OS, activating the major antioxidant defense system to remove pro-oxidants and oxidative damage, thus ensuring cell survival in mild OS, while exhibiting pro-oxidant effects that further increase the levels of stresses, leading to apoptosis, in distress conditions [Citation196]. Therefore, the participation of p53 to the epitranscriptome-dependent regulatory network that determines the response to chemotherapeutics should motivate redox biologists to investigate further and with great effort this fascinating research field. In this regard, Ke and coworkers found that the p53 mRNA sequence presents 24 DRACH sequence motifs [Citation197], which are the most common subset of methylation sites [Citation35], thus strongly suggesting that methylation processes may play a strategic role in regulating p53 expression.
More recently, Ma et al. reported that higher expression levels of the lncRNA ladybird homeobox 2 antisense RNA 1 (LBX2-AS1) were correlated with lower responsiveness to 5-FU, and this was mechanistically caused by the METTL3-dependent m6A RNA methylation [Citation198]. Pan and colleagues’ results confirmed that METTL3 plays a key role in resistance to 5-FU, more importantly in exosomal-mediated transfer of cancer resistance traits. In fact, the authors showed that the upregulation of METTL3‑dependent m6A methylation in colorectal cancer (CRC) enhanced the processing and expression of exosomal miR‑181d‑5p, which in turn inhibited the sensitivity of CRC cells to 5‑FU by targeting neurocalcin δ [Citation199]. Unfortunately, most of these studies were carried out excluding any parameters clearly linked to redox homeostasis (e.g. DNA oxidative damage, lipid peroxidation, ROS levels etc.), therefore only suggestions can be provided as to whether the redox-responsive signaling pathways can be modulated or OS-related phenomena can be involved in the epitranscriptome-driven adaptive response triggered in cancer cells by the exposure to chemotherapeutics. In this context, LBX2-AS1 has been found to co-express with other genes frequently expressed upon OS-generating conditions, such as flogosis [Citation200]. In addition, neurocalcin δ has been recently reported to downregulate inflammation via inhibition of the IKK/IκBα/NF-κB signaling pathway [Citation201]. Moreover, it was recently reported that miR-181d-5p overexpression significantly repressed apoptosis in a model of ischemia/reperfusion oxidative injury [Citation202]. On this basis, flogosis-related biological signaling might represent a putative target to be investigated further by molecular oncologists that are interested in clarifying the role of redox biology in modifying the response of cancer to chemotherapy in general, and to 5-FU in particular. This aspect gains particular importance for colorectal cancer patients, in which 5-FU is known to elicit a significant pro-inflammatory effect [Citation203].
Doxorubicin
RNA methylation seems to be associated with resistance to other commonly used anticancer chemotherapeutics. He and coworkers reported that resistance to doxorubicin (DOXO) in human breast cancer was mechanistically due to the activation of the GlcNAc N-deacetylase/N-sulfotransferase-1 (NDST1)-dependent downstream signaling pathway, and this was found to be caused by the hypermethylation of the 5′-UTR of miR-149 and its consequent downregulation [Citation204]. DOXO, whose pharmacological action is also elicited by promoting a condition of strongly increased oxidative load [Citation205], is a powerful antitumor drug that is often administered to breast cancer patients. Accordingly, DOXO-treated triple-negative breast cancer cells undergo massive H2O2 overproduction and caspase 3/8-dependent apoptosis [Citation206]. In this context, it is worth to note that interleukin 6 (IL-6) is one of most interesting ROS- and flogosis-promoting targets of miR-149 [Citation207]. Therefore, the few indications found in literature might suggest that a link between cancer cell resistance to ROS-promoting chemotherapeutics and the hypermethylation of miR-149 may exist. Unfortunately, no direct evidence for such a cause-effect relationship was provided so far.
Uddin and coworkers recently reported that colon cancer cells expressing the missense mutant p53 due to RNA m6A methylation at codon 273 exhibited resistance toward DOXO [Citation195]. Interestingly, when the authors silenced METTL3, drug-resistant cells were re-sensitized to DOXO, thus suggesting that METTL3 was essential for colon cancer cells to develop drug resistance through transited adenosines in the p53 pre-mRNA [Citation195]. p53 activation is often observed as a response to ROS over-production and DNA damage [Citation208], however Uddin and coworkers’ study did not investigate whether ROS or OS may have a role in the cytoprotective effect observed in mutant p53-haboring cancer cells, leaving this question open to further research. In 2021, Pan et al. showed in breast cancer cells that resistance to DOXO was associated with an increased expression of METTL3, that in turn upregulated the expression of miR-221-3p [Citation209]. In their work, METTL3 over-expression was intriguingly linked to enhanced maturation of pri-miR-221-3p via increased levels of m6A methylation in its RNA [Citation209]. Unluckily, no OS-related parameters or redox-specific molecular pathways were investigated in this study, however, the authors demonstrated that one major target of miR-221-3p was the homeodomain interacting protein kinase-2, which has been recently acknowledged as a critical stress-responsive kinase whose activity plays a key role in promoting cell survival upon OS [Citation210].
Platinum-containing agents
Platinum-containing drugs (e.g. cisplatin, carboplatin, oxaliplatin, etc.) are also ROS-generating drugs that are widely employed in cancer patients with several types of solid tumors [Citation211,Citation212]. In this context, some Japanese researchers observed a strongly reduced viability in pancreatic cancer cells treated with cisplatin after METTL3 knockdown, and similar results were obtained treating cells with gemcitabine [Citation193]. A microarray-based gene ontology (GO)-based analysis revealed that shMETTL3 cancer cells had several genes associated with the MAPK cascade significantly down-regulated [Citation193]. Importantly, MAPK-dependent signaling is involved in vital biological processes, such as proliferation and stress response, along with DNA repair and apoptosis [Citation213,Citation214], thus highlighting the important fil rouge among RNA methylation, gene expression and response to stressors. In 2018, Li and colleagues studied biopsies from patients with esophageal squamous cell carcinoma (ESCC), and identified an overexpressed NSUN2-methylated lncRNA (NMR) that was able to promote migration and invasion of ESCC cells in vitro, and to prevent cisplatin-induced apoptosis [Citation215]. Surprisingly, such a protective effect of NMR was presumably related to its direct binding to a component of the nucleosome remodeling factor (NURF) chromatin-remodeling complex [Citation215]. A high resistance to cisplatin was recently revealed in non-small-cell lung cancer cells with METTL3-dependent m6A over-methylation within the pre-mRNA of Yes-associated protein (YAP) [Citation216], whose aberrant activation is able to drive a broad transcriptional program intimately connected to cell cycle progression and DNA repair [Citation217]. Also in this case, no information was unfortunately provided about the possible participation of redox-dependent pathways to the METTL3-YAP axis. This identifies an important knowledge gap, also taking into account the recognized strict co-operation between YAP and NRF2, probably the most important master regulator of cell response to OS, in contributing to chemoresistance in cancer cells [Citation218]. In 2019, a brilliant work from Zhang and coauthors demonstrated that METTL3-induced m6A over-methylation in the mRNA of Chromobox 8 (CBX8) was able to enhance the stability of CBX8 mRNA, thus leading to CBX8 overexpression and development of chemoresistance in colon cancers [Citation219]. Again, no details were provided as to whether redox-dependent factors or redox-dependent signaling pathways participated in such an effect. Interestingly, some China-based researchers recently showed that the expression of CBX8 can be regulated by redox stress [Citation220], and this identified another potential signaling pathway through which cancer chemoresistance traits could be developed by rewiring the control program that defines the state of the RNA methylome. In this regard, Wei and others reported that seminoma cells could be rendered resistant to cisplatin by stabilizing the mRNA of the transcription factor AP-2 gamma (TFAP2C) via METTL3-mediated upregulation of m6A methylation levels [Citation221]. Even though no specific OS-related endpoints were included by the authors in their study, such findings are appealing also in the context of cellular responsivity to OS-promoting conditions. In fact, in 2013, Kulak et al. showed that TFAP2C was able to transcriptionally regulate the expression of glutathione peroxidase (GPX1) through the AP-2 regulatory sequence in the promoter region of the gpx1 gene [Citation222]. This might represent a possible link through which METTL3 overexpression could increase the redox homeostatic capacity of cancer cells, thus promoting the development of cisplatin-resistant phenotypes. In 2021, Song and colleagues confirmed that methyl-RNA “writers” had an extremely relevant role in cancer drug resistance. In their paper, the authors demonstrated that inhibition of METTL3 dramatically reduced the sensitivity of human lung adenocarcinoma cells to cisplatin, whereas its overexpression sensitized cells to the drug [Citation223]. Very interestingly, mettl3 was proved to be a direct target gene for miR-4443, which resulted to be enriched in resistance-transferring exosomes from cisplatin-resistant non-small cell lung carcinoma (NSCLCs) cells [Citation223]. Fascinatingly, Song and colleagues showed also that the miR-4443/METTL3 axis could drive resistance in NSCLC via increasing the level of the ferroptosis suppressor protein 1 (FSP1) [Citation223], and this crucially links the METTL3-dependent regulation of the methylation state of RNA to the iron-dependent and lipid peroxidation-induced regulated death observed in cells exposed to certain chemotherapeutics, such as some platinum-containing drugs [Citation224]. Others have confirmed the participation of m6A “writer” METTL3 in the regulation of ferroptosis. In fact, Sun et al. recently reported that METTL3-dependent methylation of the ferroptosis defense protein cystine/glutamate antiporter mRNA serves to generate binding sites for the ferroptosis suppressor NF-κB activating protein [Citation225]. In this regard, it is worthy to note that recent reviews have extensively highlighted how the regulation of ferroptosis could represent a powerful therapeutic tool through which cancer resistance to conventional therapies and immunotherapy may be effectively reversed [Citation226,Citation227].
5-azacytidine
Some types of hematologic malignancies, such as acute myeloid leukemia, are treated with 5-azacytidine (5-AZA), whose antimetabolic activity leads to an effective chemotherapeutic action [Citation228]. In 2018, Cheng and coauthors found that RNA 5-methylcytosine (m5C) and its modifying enzymes are strongly overexpressed in 5-AZA-resistant leukemia cells, both in vitro and ex vivo [Citation229], however neither direct nor indirect participation of OS or ROS-involving pathways were investigated by the authors. This left open the intriguing hypothesis that the resistance of leukemia cells toward 5-AZA could be based on an epitranscriptomic-induced alteration of redox-dependent signaling and metabolism. Recent literature seems to corroborate such a hypothesis, since RNA m5C methyltransferases include NSUN2 and DNMT2, both of which were demonstrated to affect cell resistance to OS-promoting conditions. In fact, NSUN2 serves as a key promoter of the OS-induced downregulation of protein synthesis through the formation of tRNA-derived noncoding fragments (tRFs) [Citation230], and DNMT2-depleted fibroblasts were found to be more sensitive to oxidative stress-generating conditions [Citation231].
PARP inhibitors
As discussed above, the cytotoxic action of many anticancer drugs is known to be mediated by increased OS and DNA damage. This is also the case of poly (ADP-ribose) polymerase inhibitors (PARPi), whose antitumor effect in ovarian cancer cells has been shown to depend on NADPH oxidase-dependent ROS over-production [Citation232]. In 2020, Fukumoto and coworkers reported that the response of epithelial ovarian cancer cells to PARPi was reduced when cells had m6A modifications in the frizzled class receptor 10 (FZD10) mRNAs [Citation233]. This resulted in the upregulation of the Wnt/β-catenin pathway, which is known to belong to the adaptive response pathway through which cancer cells adapt to OS-promoting treatments [Citation234,Citation235]. Again, the study did not include endpoints related to OS or ROS-dependent signaling, thus not answering to the important question as to whether methyl-based epitranscriptome could have a role in protecting malignant cells from the redox perturbation induced by the clinically important class of PARP inhibitors, which are often used to treat BRCA mutant malignancies [Citation236].
Tyrosine kinase inhibitors
Oxidative stress also underlies some of the antitumor effects of other compounds, such as tyrosine kinase inhibitors (TKIs). In 2006, Balko and colleagues carried out an Affymetrix GeneChip array-based gene expression study and revealed that lung adenocarcinoma cells resistant to the representative epidermal growth factor receptor (EGFR) TKI erlotinib exhibited increased levels of METTL3 mRNA [Citation237], whose protein product is one of the major m6A “writer” on RNA [Citation126]. Unluckily, no indication of the involvement of redox responsive signaling pathways emerged from the research. Resistance toward other TKIs has also been studied in terms of RNA methylation-related pathways. Sorafenib is known to promote OS and mitochondrial dysfunction in hepatocellular carcinoma cells [Citation238]. In this context, in 2020, Lin and coworkers reported that METTL3 was down‐regulated in sorafenib‐resistant human hepatocellular carcinomas (HCCs), as well as METTL3 downregulation induced sorafenib resistance in cultured HCC cells in vitro [Citation239]. Interestingly, such effect was tightly linked to reduced m6A levels in the 3′‐UTR of the FOXO3 mRNA, whose stability was subsequently impaired [Citation239]. FOXO3 is a Forkhead transcription factor critically required by cells to respond to oxidative stress [Citation240], also via the transcriptional activation of manganese superoxide dismutase (MnSOD, alias SOD2) and catalase (CAT), whose participation in the first line of antioxidant defense ensures adequate ROS scavenging and antioxidant protection within cells [Citation241,Citation242]. Apparently, and on the basis of what has been discussed so far, this may be seen as a counterintuitive finding. However, the authors demonstrated that the downregulated activity of the METTL3/FOXO3 axis induced autophagy in sorafenib‐resistant HepG‐2 cells [Citation239], thereby pointing to the autophagic flux as a novel target to be considered in the context of epitranscriptome-dependent regulation of cellular response to OS-promoting chemotherapeutics. Another clinically relevant OS-promoting tyrosine kinase inhibitor is gefitinib [Citation243]. In 2020, Liu and coworkers confirmed that the METTL3-dependent modulation of the autophagic process was critically involved in the cellular and molecular mechanisms responsible for resistance to gefitinib in lung cancer [Citation244], although also in this case, no endpoint specifically linked to redox-dependent pathways was included in the study, thus leaving unexplored the potential participation of ROS-related signaling pathways to the RNA methylome-dependent re-programming of the autophagic process in cancer resistance. Extending the knowledge on this topic, Chen and colleagues found that resistant non-small cell lung cancer cells could be re-sensitized to gefitinib-induced apoptosis by upregulating the main m6A “eraser” FTO, and the levels of m6A in myc mRNA were considered as crucial factors in such epitranscriptome-dependent response to this particular TKI [Citation245]. Taking into account the acknowledged function of MYC in the modulation of the epigenetic redox changes that are critical for the development of therapy resistance [Citation246], this finding should be reconsidered to design more in-depth studies and ascertain whether some cancers could respond to gefitinib or other TKIs by re-programming specific redox-related pathways through a complex interplay between methyl-epitranscriptome and MYC-dependent signaling.
As discussed in the sections above, several research reports have revealed that lncRNAs are emerging as key mediators of the response that chemotherapies trigger in various cancer types [Citation247,Citation248]. In 2021, Chen et al. found in hepatocellular carcinoma cells that the upregulation of lncRNA NIFK-AS1 led to an increased resistance to sorafenib, along with enhanced growth and invasion potential, and this effect was mechanistically linked to the METTL3-induced m6A hyper-methylation and stabilization of lncRNA NIFK-AS1 [Citation249]. Also in this case, no authors’ interest was evident in evaluating whether the major redox-responsive pathways could be involved in the effects observed. However, Chen et al. identified AKT serine/threonine kinase 1 (AKT1) as a key target gene whose induction by NIFK-AS1could be crucial upon development of drug resistance [Citation249], and this may importantly link the METTL3/NIFK-AS1/AKT1 pathway to the molecular network responsible for redox homeostasis. In fact, some researchers showed that AKT1 crucially participates in critical prosurvival signaling pathways that ensure the restoration of adequate oxidative protection upon OS stimuli [Citation250,Citation251].
Tamoxifen
Tamoxifen (TMX) is often used to treat estrogen receptor-positive breast cancers, even though the development of drug resistance still remains one of the major challenges for TMX-based therapies [Citation252]. TMX is known to generate in breast cancer cells an overproduction of ROS, whose chemical reactivity has been shown to contribute significantly to the drug-induced cytotoxic effects [Citation253]. In 2020, Liu and colleagues reported that TMX-resistant breast adenocarcinoma cells exhibited an increased METTL3-dependent methylation at several m6A consensus motif sites in the 5′-UTR of adenylate kinase 4 (AK4) mRNA, and this was associated with the over-expression of its protein product [Citation176]. AK4 is extremely important to maintain energy homeostasis in mitochondria [Citation254], as well as it is a possible indirect activator of NRF2 [Citation255]. In their work, Liu and colleagues found increased ROS concentrations in TMX-resistant cells, compared to their parental counterpart [Citation176]. Unfortunately, only basal ROS levels were studied by the authors, while it would be extremely interesting to verify whether the two cell sublines may respond differently in terms of ROS production and/or scavenging after treatment with TMX. This would contribute to establish whether the epitranscriptome-dependent regulation of AK4 metabolic activity in cancer cells could result in different responsiveness profiles to OS-generating chemotherapeutics. In 2021, the same research group provided evidence that breast cancer cells resistant to TMX had their activating transcription factor 3 (ATF3) protein overexpressed, and this was due to the stabilizing effect of lower levels of m6A at residue 131 in the 5′-UTR of ATF3 mRNA [256,p.3]. ATF3 is widely considered as a strong modulator of the inflammatory response and as a powerful protective factor in OS-based conditions, being able to prevent NRF2 degradation [Citation256]. Moreover, ATF3 participates in the unfolded protein response (UPR) [Citation257,Citation258], and it is well known that in the endoplasmic reticulum (ER) stress and oxidative stress reciprocally inter-communicate upon UPR [Citation259,Citation260]. For example, ROS promote chemical modifications of ER stress sensors, which, in turn, activate the NRF2-dependent antioxidative response [Citation261]. Interestingly, Liu and coworkers proposed ATF3 as a downstream effector of AK4 activation [Citation257]3]. On this basis, and despite the absence of a distinctive endpoints related to the redox-dependent landscape in their cancer model, Liu and colleagues have possibly identified a common redox-responsive epitranscriptome-controlled pathway responsible for the resistance of breast cancer to TMX-based antiblastic therapy. Further experimental studies would help researchers to clarify the precise molecular mediators underlying such regulatory pathways.
I summarizes the main literature reports and findings on how changes in the RNA methylation status affect resistance to anticancer drugs potentially via interfering with the activity of pathways and regulators of ROS production and metabolism.
Table 1. RNA methylation and cancer resistance traits potentially related to redox rewiring.
Radiation-based therapy
Along with chemotherapy, radiation-based therapy also helps clinicians to deal with the cure of several neoplasms, especially in the case of localized tumors, with no migrating cells spread to secondary sites [Citation262]. Moreover, the combined use of radiotherapy and chemotherapy is still considered an effective approach to treat many solid tumors [Citation263]. It is worthy of note that important cytotoxic effects of radiotherapy are mediated by the overgeneration of ROS, with the consequent activation of intracellular OS being a powerful proapoptotic challenge for tumor cells [Citation264,Citation265]. Therefore, and on the basis of what we discussed so far, it would be reasonable to suspect that the altered methylation patterns in RNAs could respond differently to radiation-based cancer therapies, as well as it could be possible to hypothesize that cancer cells may respond adaptively to radiotherapies by regulating the status of RNA methylome. In this regard, the role of N6-methyladenosine metabolism on tumor and cancer response to radiation-based stress was recently investigated by Kowalski-Chauvel and coworkers, who found that stem cells from human glioblastoma biopsies could be sensitized to radiotherapy by downregulating ALKBH5, one of the main m6A “erasers” that remove N6-methylation from adenosines in RNA [Citation266]. Interestingly, such effect was associated with an orchestrated downregulation of genes specifically involved in DNA homologous recombination repair. Accordingly, in glioblastoma cells with alkbh5 silenced, the irradiation (IR)-dependent increase in DNA damage persisted even 24 hours post-IR, thus suggesting that radiotherapy efficacy could be improved by repressing ALKBH5 expression [Citation266].
Future perspectives
As discussed above, the activity of several research teams has been increasing tremendously over the last two decades to clarify whether and how the RNA methylation diversity may serve as a key driver for the development of resistant traits in tumor and cancer cells.
Cancer stem cells (CSCs)
Particular attention should be paid to the role of RNA methylation in reshaping the tumor microenvironment, particularly in terms of resident and circulating CSCs. CSCs are a subpopulation of quiescent self-renewing tumorigenic cells that occupy tumor niches in most neoplasms and hematopoietic malignancies [Citation267]. CSC subsets within neoplasms and cancers undoubtedly represent a serious challenge for oncology specialists, since a major driver of resistance to therapy resides in the genetic and nongenetic heterogeneity that characterizes CSCs. Such heterogeneity leads to phenotypic differences that often reduce the susceptibility to several anticancer therapies [Citation268,Citation269]. More importantly, chemotherapeutic drugs often enrich cancers for CSC population via selection of clones that are resistant to treatment [Citation270]. Some interesting evidence has been provided regarding the emerging role of deregulated RNA methylation processes in CSCs, with important consequences on cancer biology, such as self-renewal and survival. In particular, m6A methylation signature has been identified as a clinical biomarker for CSC generation and cancer progression [Citation271], with m6A promoting the translation of diverse specific oncogene products, such as the epidermal growth factor receptor (EGFR) and the transcriptional co-activator with PDZ-binding motif (TAZ) [Citation272]. In 2020, Gao and coauthors showed that m6A levels in RNA were higher in bladder cancer stem cells (BCSCs), compared to bladder cancer cells with no stemlike phenotype [Citation273]. In addition, the authors demonstrated that METTL3 was essentially required for self-renewal of BCSCs, via a pathway that upregulates m6A levels, along with the expression of AF4/FMR2 family member 4 (AFF4), thus increasing the transcription of SOX2 and MYC [Citation273]. RNA adenosine demethylases also seem to affect stemness-related molecular factors in cancer. In fact, in 2020, some Indian researchers found in cisplatin-resistant squamous carcinoma cells an ALKBH5-dependent decrease in m6A levels within the nascent transcripts of FOXM1 and NANOG, and such regulation was linked to increased resistance to cisplatin [Citation274]. FOXM1 and NANOG are two major stemness markers, as well as these two proteins are the main regulators of aggressiveness, invasive properties, and chemoresistance in several cancers [Citation275]. [Citation276–278]Very recently, Yang et al. found that a stem cell-like phenotype can be promoted in liver cancer by upregulating the levels of m6A modifications in the alpha-1,6-mannosylglycoprotein 6-beta-N-acetylglucosaminyltransferase mRNA, thus enhancing transcript stability [Citation279]. In the same year, Wang and colleagues explored the role of m1A methylation in tRNA in determining profound changes in cancer stem cell biology. More in detail, the authors found a TRMT6/TRMT61A-mediated m1A hypermethylation in tRNA of liver CSCs, and this was mechanistically linked to overexpression of PPARδ and increased cholesterol biosynthesis, along with a dramatic activation of Hedgehog (HH) signaling [Citation280]. HH targets numerous genes whose differential activation is important for stem cell maintenance, proliferation, and successful embryonic development, however, altered HH signaling has been associated with the development and progression of different cancer types [Citation281]. Finally, Liu and coworkers have recently examined whether the methyl-RNA landscape could play a role in CSCs derived from lung cancer, finding that high levels of the p53-dependent up-regulation of m6A demethylase ALKBH5 in CSCs support the overexpression of critical stem hallmarks, such as NANOG and OCT4 [Citation282]. Unexpectedly, this field of research is still underinvestigated. However, given the potential importance of this research topic in terms of translation to clinic, much greater effort should be devoted to clarify in depth whether and how the reprogramming of RNA methylation could affect CSCs biology in terms of aggressive phenotype and responsiveness to treatment.
Immunotherapy
Another promising field is represented by the application of techniques that modulate RNA methylation states with the aim of increasing the efficacy of cancer immunotherapy, which today significantly prolongs the survival of many patients affected by otherwise fatal cancers. The hypothesis to use the physiological capacity of the immune system as a tool to exploit to treat neoplasms appeared in researchers’ minds in the nineteenth century, as reviewed by Oiseth and Aziz [Citation283], but only in the last decades specific knockout mouse models and rapidly advancing molecular and biochemical techniques have led to the identification of tumor-specific immune responses to use against tumors [Citation4,Citation284,Citation285]. In this context, Li and colleagues brilliantly demonstrated that deletion of the m6A demethylase ALKBH5 was sufficient to sensitize tumors to cancer immunotherapy, eventually prolonging mouse survival [Citation286], thus paving the way to potentially ground-breaking innovations for cancer medicine and healthcare providers. The capacity of RNA methylation to influence cancer response to immunotherapy was recently confirmed by Zhang and coworkers, who found that m6A levels in RNA could regulate the expression of metadherin [Citation287], a well-established oncogene whose upregulation promotes EMT in various cancers [Citation288,Citation289]. Very recently, Yin and colleagues confirmed the idea that key enzymes for m6A RNA metabolism could represent a potential therapeutic target for tumor immunotherapy, finding that the loss of METTL3 led to increased tumor growth and metastasis via activation of NF-kB and STAT3 [Citation290], with NF-kB and STAT3 being important players in cancer cell-induced immunosuppression [Citation291]. Bioinformatics analyses and experimental data from other researchers supported the notion that METTL3-dependent signaling may serve as an important immune evasion mechanism for tumors [Citation292].
Novel molecular pathways
As mentioned in the previous sections, the reorganization of the autophagic process and metabolic rewiring play important roles in cellular differentiation, resistance to stressors, and adaptation to (micro)environmental stimuli.
Certainly, sirtuins (SIRTs) serve as key redox-active regulators of autophagic flux and metabolic reprogramming, also due to the reliance on NAD+ for their de-ac(et)ylase activity toward multiple protein targets [Citation293]. In fact, today the role of SIRTs in cancer resistance to therapy is acknowledged and intensely studied. In this context, to the best of our knowledge, no investigation has attempted so far to establish whether the methyl epitranscriptomic control of cancer cell response to chemotherapeutics or other anticancer interventions could be implemented by modulating the activity of sirtuins in mammals. This is a serious lack of knowledge, also taking into account that some researchers recently found that knockdown of METTL14 facilitated autophagy and alleviated apoptosis and inflammation in stressed podocytes in vitro, and such effect was mechanistically due to the upregulation of SIRT1 via m6A hypo-methylation of sirt1 mRNA [Citation294]. Interestingly, SIRT1 modifies the activity of ATP-binding cassette (ABC) transporters in cancer cells, thus significantly affecting drug penetration properties [Citation295]. In addition, SIRT1 is able to confer anti-apoptotic advantages in cancer cells via deacetylation of p53/p73, thus repressing p53/p73-dependent programmed cell death pathways [Citation296,Citation297]. Moreover, SIRT1 relocates to the nucleus to promote repair and cell survival upon DNA damage-promoting stress [Citation298]. Finally, SIRT1 is a well-known activator of antioxidant protection in OS-promoting conditions, such as the ischemia/reperfusion [Citation299]. On such a basis, it is evident that further and more targeted studies should be carried out to provide detailed information on how the adaptive responses of resistant cancer cells toward anticancer interventions could be deployed by an interplay between the methyl epitranscriptome and the SIRT-based stress-responsive molecular network.
Another pathway that is largely underinvestigated is that responsible for the clearance of methylglyoxal (MG), which is an endogenous cancer-static and glycating α-oxoaldehyde [Citation300]. More specifically, MG is frequently overproduced by the increased glycolytic flux in tumor and cancer cells (i.e. Warburg effect), and this requires neoplastic and malignant cells to strongly upregulate the activity of the glyoxalase system [Citation301,Citation302]. The process of MG enzymatic removal is tightly interconnected with both redox homeostasis and antioxidant protection, mainly because glyoxalase 1 (GLO1) essentially requires glutathione to convert MG into the less toxic S-D-lactoylglutathione [Citation303]. Accordingly, GLO1 was reported as highly overexpressed in the apoptosis-resistant human leukemia cells [Citation304]. In addition, the knockdown of GLO1 sensitized adenocarcinoma cells toward irradiation-induced apoptosis [Citation305]. Moreover, the inhibition of GLO1 was found to improve the antiproliferative effect of sorafenib in carcinoma [Citation306]. On such a basis, it would not be unreasonable to hypothesize that cancer-resistant phenotypes may also develop via an epitranscriptome-dependent upregulation of glyoxalases. Surprisingly, it would seem that no study has ever attempted to verify this so far. However, Kulkarni and coworkers recently showed that alkbh7 null mice had elevated levels of GLO1 [Citation307], thus providing a proof-of-concept that at least one of the Fe(II)/α-ketoglutarate-dependent dioxygenases that are able to de-methylate RNA can serve as important regulators of the MG-targeting system. Clearly, this should be further investigated with more specifically targeted experiments, especially in tumor and cancer cells. However, a better comprehension of such phenomena could help researchers and oncologists to circumvent or prevent the development of resistant traits in cancer cells upon antiblastic treatment.
Innovative technologies
An in-depth exploration of the role of the epitranscriptome in physiology and disease is hindered by the limited availability of techniques for accurate mapping of specific RNA modifications throughout the transcriptome. Indeed, conventional methods for RNA modifications-targeting analysis require great amounts of material and are relatively labor intensive. This is particularly true for the detection and quantitation of small RNAs, such as microRNAs (miRNAs) and Piwi-interacting RNAs (piRNAs) [Citation308]. On this basis, over the last few years, some truly inspired researchers have presented convenient, sensitive, and reliable methods for quantifying a wide range of RNA modifications. In the standard methylated RNA immunoprecipitation and sequencing (MeRIP-Seq) method, m6A-specific antibodies are used to immunoprecipitate RNA, then post-reverse-transcription cDNA is deep sequenced providing high-resolution reads of m6A-methylated RNA [Citation54]. Some improvements have also been developed to circumvent typical limitations of MeRIP-Seq, such as PA-m6A-seq, miCLIP, and m6A-CLIP [Citation309]. However, some authors have recently proposed a novel high-throughput next-generation sequencing (NGS)-based approach (i.e. RiboMethSeq method) [Citation308], whereas others have presented a technique that combines oxidative deep sequencing and stem-loop RT-qPCR to quantify accurately the levels of 3′-terminal 2′-O-methylated small RNAs in biological samples [Citation310]. Another interesting method was recently developed by US-based researchers, who used a hydrazine-aniline cleavage sequencing (HAC-seq) to detect and profile the RNA m3C methylome at single-nucleotide resolution [Citation311]. In addition, great interest is emerging for developing GC-MS/MS and LC-MS/MS to identify epitranscriptomic biomarker signatures that could help cancer diagnosis, detecting modified nucleosides by monitoring multiple reactions [Citation312]. Very recently, Cui and coworkers proposed a sensitive platform for simple simultaneous detection of both m6A and FTO protein. This method is based on the FTO-dependent conversion of the methyl group of m6A to hydroxymethyl, whose covalent reaction with dithiotreitol allows the interaction with a Cd-based electrode thus generating a detectable photocurrent [Citation313]. Further development of such innovative methods may help to circumvent the great limitations and shortcomings that still impede or delay the progress of biomedical research, thus allowing future investigations to follow complex patterns of RNA methylation, to establish and validate specific epitranscriptomic signatures, and to study how the dynamics of RNA methylation landscape could lead to different responses to stressors, including cancer chemo- and radio-therapy. This would certainly open new interesting perspectives in terms of personalized anticancer medicine.
Bioinformatics
In parallel with the escalating impact of cancer as a major global burden to human health, the interest in classifying high or low responders and in predicting the effect of chemotherapy in the perspective of personalized medicine, has been increasing formidably [Citation314]. However, with the advent of new technologies and multi-omics-based approaches, great amounts of data have started to overwhelm the analysis capacity of biomedical researchers, thus urging innovative techniques and methods for data handling and processing. 21st century technology and artificial intelligence (AI) may serve as a game-changer and powerful tool for this task. In this regard, in 2019, the novel approach of some French researchers allowed to develop the GECKO algorithm, which in this perspective could pave the way to the exploration of large amounts of high-throughput sequencing data to identify new sequences of interest without the intensive use of highly specialized bioinformatics workflows [Citation315]. Three years later, Sui and colleagues created machine learning-based models that helped to identify and validate in the lab some important genes and miRNA related to the IC50 value of cisplatin [Citation316]. Even though no gene or miRNA was clearly linked to methyl RNA metabolism, Sui and coauthors proposed a new workflow for future investigations, thus contributing to bridge the gap that still exists between bioinformatics and translational research. In this perspective, clinicians may benefit from multi-omics-based approaches, and the therapeutic arsenal of multimodal therapies would be greatly increased in view of highly personalized medicine.Translational research
RNA methylation is being explored not only by basic researchers, but also by scientific teams that are more oriented to translational research. In fact, some researchers proposed that much greater efficacy of antisense-based clinical approaches would be obtained by using methylated versions of anti-miRNA oligonucleotides. More specifically, Nahar and coworkers found that 2’-O-methyl RNAs directed against miR-21, an oncogenic miRNA whose increased activity is linked to a more aggressive behavior, was able to reduce migration capacity in metastatic adenocarcinoma cells [Citation317]. Although being a preliminary report, this kind of approach clearly represents a suggestive indication that studies on methyl-RNA-based gene regulation in the field of oncology might be exploited in the future to improve cancer disease control or even to prevent the development of therapy resistance.
A schematic representation of the future directions research may take to reveal if and how the regulation of methylepitranscriptome could affect cancer sensitivity to treatments, especially in terms of redox-related cellular adaptive response, is reported in .
Figure 2. Schematic representation of the possible directions that future research activities and efforts may take to reveal in detail the role of the methylepitranscriptome in affecting or determining cancer cell response to treatments, also in terms of potential participation of redox-sensitive and redox-dependent biomolecular mechanisms.
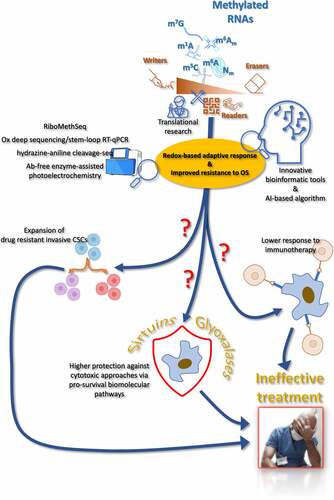
Conclusions
Over the last forty years, biomedical research has been focused on revealing the genetic and epigenetic nature of resistance traits in tumors and cancers. Nevertheless, only in the last two decades the importance of modifications and structural rearranging of RNA has gained the attention of the most important molecular oncologists and biologists worldwide. More specifically, the literature offers substantial evidence that the RNA methylome is a powerful biological tool through which cancer cells achieve a fine-tuned control of gene expression in response to anticancer treatments. Such rewiring of gene expression-related processes are realized by operating and modulating a complex molecular network of enzymes responsible for “writing”, “reading” and “erasing” methylated residues within substantially all classes of RNA, both coding and noncoding. However, it is surprising that a small number of investigations attempted to study in-depth whether the epitranscriptome-directed adaptations underlying the acquired and/or innate resistance traits in cancer could be deployed or arranged through the involvement of redox-dependent or -responsive signaling pathways. Somehow, this is curious since the effectiveness of many, if not most, antiblastic approaches available today relies on the powerful generation of reactive oxygen species within cancer and tumor cells. This renders the existing knowledge gap even more unexpected, since the alteration of the redox milieu, as well as the functional reprogramming of mitochondria, which represents a major critical determinant for cellular oxidative metabolism and a contributor to intracellular ROS formation, have already been documented as key mediators of the RNA methylome-mediated response to OS. In addition, many recent evidence suggested that a number of molecular pathways that are regulated by the RNA methylome are deeply linked to ROS-targeting enzymatic clearance and DNA repair, as well as to the expression and localization of transcription factors whose downstream signaling crucially governs the activity of key antioxidant protection systems.
On this basis, from a critical review of the existing literature corpus, we believe that a greater effort should be spent to explore and verify whether the activation of redox-active pathways may underlie the methyl epitranscriptome-controlled development of resistance to therapy in neoplasms and malignancies. Also taking into account the new high-throughput techniques and AI-based methods available today and possibly achievable in the next future, we would suggest that all researchers interested in this topic and most active and dedicated redox biologists should consider to dedicate more effort to explore this field more in depth, essentially because this topic may still reserve the possibility of breakthrough discoveries. Such striking innovations may help future biomedical researchers to understand in detail how cancer cell response unfolds upon treatment, as well as clinicians and oncologists may benefit from these advances to develop new therapeutic approaches and strategies, with the specific aim of delaying or preventing the severe problem of refractory cancer and tumor recurrence.
Disclosure statement
The authors declare that there is no relevant financial or non-financial conflict of interest to report.
Additional information
Funding
References
- Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: gLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249.
- Bernier J, Hall EJ, Giaccia A. Radiation oncology: a century of achievements. Nat Rev Cancer. 2004;4:737–747.
- Kachalaki S, Ebrahimi M, Mohamed Khosroshahi L, et al. Cancer chemoresistance; biochemical and molecular aspects: a brief overview. Eur J Pharm Sci off J Eur Fed Pharm Sci. 2016;89:20–30.
- Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20:651–668.
- Guo L, Lee Y-T, Zhou Y, et al. Targeting epigenetic regulatory machinery to overcome cancer therapy resistance. Semin Cancer Biol. 2022;83:487–502.
- Mani DR, Krug K, Zhang B, et al. Cancer proteogenomics: current impact and future prospects. Nat Rev Cancer. 2022;22:298–313.
- Liu Y, Li Q, Zhou L, et al. Cancer drug resistance: redox resetting renders a way. Oncotarget. 2016;7:42740–42761.
- Zhang L, Lu Q, Chang C. Epigenetics in Health and Disease. In: Chang C Lu Q editors. Epigenetics Allergy Autoimmune [Internet]. Singapore: Springer Singapore; 2020pp. 3–55. cited 2022 Jul 11. Available from. https://link.springer.co
- Machnicka MA, Milanowska K, Osman Oglou O, et al. MODOMICS: a database of RNA modification pathways—2013 update. Nucleic Acids Res. 2012;41:D262–267.
- Barbieri I, Kouzarides T. Role of RNA modifications in cancer. Nat Rev Cancer. 2020;20:303–322.
- Song H, Liu D, Dong S, et al. Epitranscriptomics and epiproteomics in cancer drug resistance: therapeutic implications. Signal Transduct Target Ther. 2020;5:193.
- Phizicky EM, Hopper AK. tRNA biology charges to the front. Genes Dev. 2010;24:1832–1860.
- Nachtergaele S, He C. The emerging biology of RNA post-transcriptional modifications. RNA Biol. 2017;14:156–163.
- Lecointe F, Simos G, Sauer A, et al. Characterization of Yeast Protein Deg1 as Pseudouridine Synthase (Pus3) Catalyzing the Formation of Ψ38 and Ψ39 in tRNA Anticodon Loop. J Biol Chem. 1998;273:1316–1323.
- Bjork GR. A primordial tRNA modification required for the evolution of life? EMBO J. 2001;20:231–239.
- Gerber AP, Keller W. An Adenosine Deaminase that Generates Inosine at the Wobble Position of tRnas. Science. 1999;286:1146–1149.
- Dihanich ME, Najarian D, Clark R, et al. Isolation and characterization of MOD5, a gene required for isopentenylation of cytoplasmic and mitochondrial tRnas of Saccharomyces cerevisiae. Mol Cell Biol. 1987;7:177–184.
- Sloan KE, Warda AS, Sharma S, et al. Tuning the ribosome: the influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017;14:1138–1152.
- Ben-Shem A, Garreau de Loubresse N, Melnikov S, et al. The Structure of the Eukaryotic Ribosome at 3.0 Å Resolution. Science. 2011;334:1524–1529.
- Decatur WA, Fournier MJ. rRNA modifications and ribosome function. Trends Biochem Sci. 2002;27:344–351.
- Krogh N, Jansson MD, Häfner SJ, et al. Profiling of 2′- O -Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic Acids Res. 2016;44:7884–7895.
- Birkedal U, Christensen-Dalsgaard M, Krogh N, et al. Profiling of Ribose Methylations in RNA by High-Throughput Sequencing. Angew Chem Int Ed. 2015;54: 451–455.
- Maden BEH, Corbett ME, Heeney PA, et al. Classical and novel approaches to the detection and localization of the numerous modified nucleotides in eukaryotic ribosomal RNA. Biochimie. 1995;77:22–29.
- Bakin A, Ofengand J. Four newly located pseudouridylate residues in Escherichia coli 23S ribosomal RNA are all at the peptidyltransferase center: analysis by the application of a new sequencing technique. Biochemistry. 1993;32:9754–9762.
- King TH, Liu B, McCully RR, et al. Ribosome Structure and Activity are Altered in Cells Lacking snoRnps that Form Pseudouridines in the Peptidyl Transferase Center. Mol Cell. 2003;11:425–435.
- Liang X, Liu Q, Fournier MJ. Loss of rRNA modifications in the decoding center of the ribosome impairs translation and strongly delays pre-rRNA processing. RNA. 2009;15:1716–1728.
- Baxter-Roshek JL, Petrov AN, Dinman JD. Optimization of Ribosome Structure and Function by rRNA Base Modification. In: Preiss T, editor. PLoS ONE. Vol. 2. 2007. p. e174.
- Liang X, Liu Q, Fournier MJ. rRNA Modifications in an Intersubunit Bridge of the Ribosome Strongly Affect Both Ribosome Biogenesis and Activity. Mol Cell. 2007;28:965–977.
- Baudin-Baillieu A, Fabret C, Liang X, et al. Nucleotide modifications in three functionally important regions of the Saccharomyces cerevisiae ribosome affect translation accuracy. Nucleic Acids Res. 2009;37:7665–7677.
- Andreassi C, Crerar H, Riccio A. Post-transcriptional Processing of mRNA in Neurons: the Vestiges of the RNA World Drive Transcriptome Diversity. Front Mol Neurosci. 2018;11:304.
- Furuichi Y. Discovery of m7G-cap in eukaryotic mRnas. Proc Jpn Acad Ser B. 2015;91:394–409.
- Carlile TM, Rojas-Duran MF, Zinshteyn B, et al. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014;515:143–146.
- Lovejoy AF, Riordan DP, Brown PO. Transcriptome-Wide Mapping of Pseudouridines: pseudouridine Synthases Modify Specific mRnas in S. cerevisiae. In: Preiss T, editor. PLoS ONE. Vol. 9. 2014. p. e110799.
- Li X, Zhu P, Ma S, et al. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat Chem Biol. 2015;11:592–597.
- He L, Li H, Wu A, et al. Functions of N6-methyladenosine and its role in cancer. Mol Cancer. 2019;18:176.
- Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71:3971–3975.
- Jia G, Fu Y, He C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. 2013;29:108–115.
- Roundtree IA, Evans ME, Pan T, et al. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017;169:1187–1200.
- Liu J, Yue Y, Han D, et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–95.
- Jia G, Fu Y, Zhao X, et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–887.
- Wang X, Lu Z, Gomez A, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120.
- Du H, Zhao Y, He J, et al. YTHDF2 destabilizes m6a-containing RNA through direct recruitment of the CCR4–NOT deadenylase complex. Nat Commun. 2016;7:12626.
- Meyer KD, Patil DP, Zhou J, et al. 5′ UTR m6a Promotes Cap-Independent Translation. Cell. 2015;163:999–1010.
- Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, et al. The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016;530:441–446.
- Zhou H, Kimsey IJ, Nikolova EN, et al. M1a and m1g disrupt A-RNA structure through the intrinsic instability of Hoogsteen base pairs. Nat Struct Mol Biol. 2016;23:803–810.
- Xue C, Zhao Y, Li L. Advances in RNA cytosine-5 methylation: detection, regulatory mechanisms, biological functions and links to cancer. Biomark Res. 2020;8:43.
- Sharma S, Yang J, Watzinger P, et al. Yeast Nop2 and Rcm1 methylate C2870 and C2278 of the 25S rRNA, respectively. Nucleic Acids Res. 2013;41:9062–9076.
- Schosserer M, Minois N, Angerer TB, et al. Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat Commun. 2015;6:6158.
- Aschenbrenner J, Marx A. Direct and site-specific quantification of RNA 2′-O-methylation by PCR with an engineered DNA polymerase. Nucleic Acids Res. 2016;44:3495–3502.
- Erales J, Marchand V, Panthu B, et al. Evidence for rRNA 2′-O-methylation plasticity: control of intrinsic translational capabilities of human ribosomes. Proc Natl Acad Sci. 2017;114:12934–12939.
- Basu A, Das P, Chaudhuri S, et al. Requirement of rRNA Methylation for 80S Ribosome Assembly on a Cohort of Cellular Internal Ribosome Entry Sites. Mol Cell Biol. 2011;31:4482–4499.
- Jiang X, Liu B, Nie Z, et al. The role of m6a modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6:74.
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6a RNA methylomes revealed by m6a-seq. Nature. 2012;485:201–206.
- Meyer KD, Saletore Y, Zumbo P, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149:1635–1646.
- Zhang C, Chen Y, Sun B, et al. M6a modulates haematopoietic stem and progenitor cell specification. Nature. 2017;549:273–276.
- Lv J, Zhang Y, Gao S, et al. Endothelial-specific m6a modulates mouse hematopoietic stem and progenitor cell development via Notch signaling. Cell Res. 2018;28:249–252.
- Wang H, Zuo H, Liu J, et al. Loss of YTHDF2-mediated m6a-dependent mRNA clearance facilitates hematopoietic stem cell regeneration. Cell Res. 2018;28:1035–1038.
- Gao Y, Vasic R, Song Y, et al. M6a Modification Prevents Formation of Endogenous Double-Stranded RNAs and Deleterious Innate Immune Responses during Hematopoietic Development. Immunity. 2020;52:1007–1021.e8.
- Ma C, Chang M, Lv H, et al. RNA m6a methylation participates in regulation of postnatal development of the mouse cerebellum. Genome Biol. 2018;19:68.
- Li L, Zang L, Zhang F, et al. Fat mass and obesity-associated (FTO) protein regulates adult neurogenesis. Hum Mol Genet. 2017;26:2398–2411.
- Zhuang M, Li X, Zhu J, et al. The m6a reader YTHDF1 regulates axon guidance through translational control of Robo3.1 expression. Nucleic Acids Res. 2019;47:4765–4777.
- Weng Y-L, Wang X, An R, et al. Epitranscriptomic m6a Regulation of Axon Regeneration in the Adult Mammalian Nervous System. Neuron. 2018;97:313–325.e6.
- Xhemalce B, Robson SC, Kouzarides T. Human RNA Methyltransferase BCDIN3D Regulates MicroRNA Processing. Cell. 2012;151:278–288.
- Cheray M, Etcheverry A, Jacques C, et al. Cytosine methylation of mature microRnas inhibits their functions and is associated with poor prognosis in glioblastoma multiforme. Mol Cancer. 2020;19:36.
- Alarcón CR, Lee H, Goodarzi H, et al. N6-methyladenosine marks primary microRnas for processing. Nature. 2015;519:482–485.
- Berulava T, Rahmann S, Rademacher K, et al. N6-Adenosine Methylation in MiRnas. In: Antoniewski C, editor. PLOS ONE. Vol. 10. 2015. p. e0118438.
- Zhou Y, Kong Y, Fan W, et al. Principles of RNA methylation and their implications for biology and medicine. Biomed Pharmacother. 2020;131:110731.
- Wu J, Guo X, Wen Y, et al. N6-Methyladenosine Modification Opens a New Chapter in Circular RNA Biology. Front Cell Dev Biol. 2021;9:709299.
- Zhang C, Cui H, Huang C, et al. Interactions of circRnas with methylation: an important aspect of circRNA biogenesis and function (Review). Mol Med Rep. 2022;25:169.
- Pastore B, Hertz HL, Price IF, et al. Pre-piRNA trimming and 2′-O-methylation protect piRnas from 3′ tailing and degradation in C. elegans. Cell Rep. 2021;36:109640.
- Goh YT, Koh CWQ, Sim DY, et al. METTL4 catalyzes m6am methylation in U2 snRNA to regulate pre-mRNA splicing. Nucleic Acids Res. 2020;48:9250–9261.
- Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol. 1997;82:291–295.
- Shen Z, Wu W, Hazen SL. Activated leukocytes oxidatively damage DNA, RNA, and the nucleotide pool through halide-dependent formation of hydroxyl radical. Biochemistry. 2000;39:5474–5482.
- Hofer T, Badouard C, Bajak E, et al. Hydrogen peroxide causes greater oxidation in cellular RNA than in DNA. Biol Chem. 2005;386:333–337.
- Liu M, Gong X, Alluri RK, et al. Characterization of RNA damage under oxidative stress in Escherichia coli. Biol Chem. 2012;393:123–132.
- Chen X, Yu H, Li Z, et al. Oxidative RNA Damage in the Pathogenesis and Treatment of Type 2 Diabetes. Front Physiol. 2022;13:725919.
- Tanaka M, Chock PB, Stadtman ER. Oxidized messenger RNA induces translation errors. Proc Natl Acad Sci, USA. 2007;104:66–71.
- Willi J, Küpfer P, Evéquoz D, et al. Oxidative stress damages rRNA inside the ribosome and differentially affects the catalytic center. Nucleic Acids Res. 2018;46:1945–1957.
- Leiva LE, Pincheira A, Elgamal S, et al. Modulation of Escherichia coli Translation by the Specific Inactivation of tRnagly Under Oxidative Stress. Front Genet. 2020;11:856.
- Estevez M, Valesyan S, Jora M, et al. Oxidative Damage to RNA is Altered by the Presence of Interacting Proteins or Modified Nucleosides. Front Mol Biosci. 2021;8:697149.
- Baldridge KC, Contreras LM. Functional implications of ribosomal RNA methylation in response to environmental stress. Crit Rev Biochem Mol Biol. 2014;49:69–89.
- Borek E, Srinivasan PR. The Methylation of Nucleic Acids. Annu Rev Biochem. 1966;35:275–298.
- Wilkinson E, Cui Y-H, He Y-Y. Context-Dependent Roles of RNA Modifications in Stress Responses and Diseases. Int J Mol Sci. 2021;22:1949.
- Cao SS, Kaufman RJ. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid Redox Signal. 2014;21:396–413.
- Zhang H, Gong W, Wu S, et al. Hsp70 in Redox Homeostasis. Cells. 2022;11:829.
- Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21:363–383.
- Juan CA, Pérez de la Lastra JM, Plou FJ, et al. The Chemistry of Reactive Oxygen Species (ROS) Revisited: outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int J Mol Sci. 2021;22:4642.
- Zhang L, Wang X, Cueto R, et al. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019;26:101284.
- Lee SY, Kim JJ, Miller KM. Emerging roles of RNA modifications in genome integrity. Brief Funct Genomics. 2021;20:106–112.
- Samet JM, Wages PA. Oxidative stress from environmental exposures. Curr Opin Toxicol. 2018;7:60–66.
- Sies H. Oxidative eustress: on constant alert for redox homeostasis. Redox Biol. 2021;41:101867.
- Agarwalla S, Kealey JT, Santi DV, et al. Characterization of the 23 S Ribosomal RNA m5u1939 Methyltransferase from Escherichia coli. J Biol Chem. 2002;277:8835–8840.
- Agarwalla S, Stroud RM, Gaffney BJ. Redox Reactions of the Iron-Sulfur Cluster in a Ribosomal RNA Methyltransferase, RumA. J Biol Chem. 2004;279:34123–34129.
- Kyuma T, Kimura S, Hanada Y, et al. Ribosomal RNA methyltransferases contribute to Staphylococcus aureus virulence. FEBS J. 2015;282:2570–2584.
- Kyuma T, Kizaki H, Ryuno H, et al. 16S rRNA methyltransferase KsgA contributes to oxidative stress resistance and virulence in Staphylococcus aureus. Biochimie. 2015;119:166–174.
- Begley TJ, Rosenbach AS, Ideker T, et al. Damage recovery pathways in Saccharomyces cerevisiae revealed by genomic phenotyping and interactome mapping. Mol Cancer Res MCR. 2002;1:103–112.
- Begley TJ, Rosenbach AS, Ideker T, et al. Hot Spots for Modulating Toxicity Identified by Genomic Phenotyping and Localization Mapping. Mol Cell. 2004;16:117–125.
- Begley U, Dyavaiah M, Patil A, et al. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol Cell. 2007;28:860–870.
- Chan CTY, Pang YLJ, Deng W, et al. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat Commun. 2012;3:937.
- Golovina AY, Sergiev PV, Golovin AV, et al. The yfiC gene of E. coli encodes an adenine-N6 methyltransferase that specifically modifies A37 of tRNA 1 val (cmo 5 UAC). RNA. 2009;15:1134–1141.
- Osterman IA, Sergiev PV, Tsvetkov PO, et al. Methylated 23S rRNA nucleotide m2g1835 of Escherichia coli ribosome facilitates subunit association. Biochimie. 2011;93:725–729.
- Jaroensuk J, Atichartpongkul S, Chionh YH, et al. Methylation at position 32 of tRNA catalyzed by TrmJ alters oxidative stress response in Pseudomonas aeruginosa. Nucleic Acids Res. 2016;44:10834–10848.
- Dedon PC, Begley TJ. A system of RNA modifications and biased codon use controls cellular stress response at the level of translation. Chem Res Toxicol. 2014;27:330–337.
- Barroso M, Florindo C, Kalwa H, et al. Inhibition of Cellular Methyltransferases Promotes Endothelial Cell Activation by Suppressing Glutathione Peroxidase 1 Protein Expression. J Biol Chem. 2014;289:15350–15362.
- Schaefer M, Pollex T, Hanna K, et al. RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev. 2010;24:1590–1595.
- Schaefer M, Lyko F. Solving the Dnmt2 enigma. Chromosoma. 2010;119:35–40.
- Goll MG, Kirpekar F, Maggert KA, et al. Methylation of tRnaasp by the DNA methyltransferase homolog Dnmt2. Science. 2006;311:395–398.
- Mytych J, Lewinska A, Bielak-Zmijewska A, et al. Nanodiamond-mediated impairment of nucleolar activity is accompanied by oxidative stress and DNMT2 upregulation in human cervical carcinoma cells. Chem Biol Interact. 2014;220:51–63.
- Chellamuthu A, Gray SG. The RNA Methyltransferase NSUN2 and Its Potential Roles in Cancer. Cells. 2020;9:1758.
- Blanco S, Dietmann S, Flores JV, et al. Aberrant methylation of t RNA s links cellular stress to neuro‐developmental disorders. EMBO J. 2014;33:2020–2039.
- Ivanov P, Emara MM, Villen J, et al. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol Cell. 2011;43:613–623.
- Hertz R, Tovy A, Kirschenbaum M, et al. The Entamoeba histolytica Dnmt2 homolog (Ehmeth) confers resistance to nitrosative stress. Eukaryot Cell. 2014;13:494–503.
- Jeltsch A, Ehrenhofer-Murray A, Jurkowski TP, et al. Mechanism and biological role of Dnmt2 in Nucleic Acid Methylation. RNA Biol. 2017;14:1108–1123.
- Dewe JM, Fuller BL, Lentini JM, et al. TRMT1-Catalyzed tRNA Modifications are Required for Redox Homeostasis to Ensure Proper Cellular Proliferation and Oxidative Stress Survival. Mol Cell Biol. 2017;37:e00214–17.
- Endres L, Rose RE, Doyle F, et al. 2’-O-ribose methylation of transfer RNA promotes recovery from oxidative stress in Saccharomyces cerevisiae. In: Preiss T, editor. PLOS ONE. Vol. 15. 2020. p. e0229103.
- Trixl L, Amort T, Wille A, et al. RNA cytosine methyltransferase Nsun3 regulates embryonic stem cell differentiation by promoting mitochondrial activity. Cell Mol Life Sci. 2018;75:1483–1497.
- Cosentino C, Toivonen S, Diaz Villamil E, et al. Pancreatic β-cell tRNA hypomethylation and fragmentation link TRMT10A deficiency with diabetes. Nucleic Acids Res. 2018;46:10302–10318.
- Rashad S, Han X, Sato K, et al. The stress specific impact of ALKBH1 on tRNA cleavage and tiRNA generation. RNA Biol. 2020;17:1092–1103.
- Zhou J, Wan J, Gao X, et al. Dynamic m(6)a mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–594.
- Zhang X, Liu Z, Yi J, et al. The tRNA methyltransferase NSun2 stabilizes p16ink4 mRNA by methylating the 3′-untranslated region of p16. Nat Commun. 2012;3:712.
- Cai X, Hu Y, Tang H, et al. RNA methyltransferase NSUN2 promotes stress-induced HUVEC senescence. Oncotarget. 2016;7:19099–19110.
- Ciciliot S, Fadini G. Modulation of Obesity and Insulin Resistance by the Redox Enzyme and Adaptor Protein p66shc. Int J Mol Sci. 2019;20:985.
- Yuan S, Tang H, Xing J, et al. Methylation by NSun2 represses the levels and function of microRNA 125b. Mol Cell Biol. 2014;34:3630–3641.
- Lu C, Zhou D, Wang Q, et al. Crosstalk of MicroRnas and Oxidative Stress in the Pathogenesis of Cancer. Oxid Med Cell Longev. 2020;2020:1–13.
- Zhao T-X, Wang J-K, Shen L-J, et al. Increased m6a RNA modification is related to the inhibition of the Nrf2-mediated antioxidant response in di-(2-ethylhexyl) phthalate-induced prepubertal testicular injury. Environ Pollut. 2020;259:113911.
- Arumugam T, Ghazi T, Chuturgoon AA. Fumonisin B1 alters global m6a RNA methylation and epigenetically regulates Keap1-Nrf2 signaling in human hepatoma (HepG2) cells. Arch Toxicol. 2021;95:1367–1378.
- Moldogazieva NT, Lutsenko SV, Terentiev AA. Reactive Oxygen and Nitrogen Species–Induced Protein Modifications: implication in Carcinogenesis and Anticancer Therapy. Cancer Res. 2018;78:6040–6047.
- Li Q, Li X, Tang H, et al. NSUN2-Mediated m5c Methylation and METTL3/METTL14-Mediated m6a Methylation Cooperatively Enhance p21 Translation: nSUN2 and METTL3/METTL14 R EGULATE p21 T RANSLATION. J Cell Biochem. 2017;118:2587–2598.
- Chen W, Sun Z, Wang X-J, et al. Direct Interaction between Nrf2 and p21cip1/WAF1 Upregulates the Nrf2-Mediated Antioxidant Response. Mol Cell. 2009;34:663–673.
- Zhao F, Xu Y, Gao S, et al. METTL3-dependent RNA m6a dysregulation contributes to neurodegeneration in Alzheimer’s disease through aberrant cell cycle events. Mol Neurodegener. 2021;16:70.
- Gonchar O, Mankovska I. Hypoxia/Reoxygenation modulates Oxidative Stress Level and Antioxidative Potential in Lung Mitochondria: possible participation of P53 and NF-KB Target Proteins. Arch Pulmonol Respir Care. 2017;3:035–043.
- Song H, Feng X, Zhang H, et al. METTL3 and ALKBH5 oppositely regulate m 6 a modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy. 2019;15:1419–1437.
- Wang J, Zhang J, Ma Y, et al. WTAP promotes myocardial ischemia/reperfusion injury by increasing endoplasmic reticulum stress via regulating m6a modification of ATF4 mRNA. Aging. 2021;13:11135–11149.
- Pang P, Qu Z, Yu S, et al. Mettl14 Attenuates Cardiac Ischemia/Reperfusion Injury by Regulating Wnt1/β-Catenin Signaling Pathway. Front Cell Dev Biol. 2021;9:762853.
- Liu J, Xiao Q, Xiao J, et al. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther. 2022;7:3.
- Guo Y, Song W, Yang Y. Inhibition of ALKBH5 ‐mediated m 6 a modification of PPARG mRNA alleviates H/R‐induced oxidative stress and apoptosis in placenta trophoblast. Environ Toxicol. 2022;37:910–924.
- Anders M, Chelysheva I, Goebel I, et al. Dynamic m 6 a methylation facilitates mRNA triaging to stress granules. Life Sci Alliance. 2018;1:e201800113.
- Protter DSW, Parker R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016;26:668–679.
- Fu Y, Zhuang X. M6a-binding YTHDF proteins promote stress granule formation. Nat Chem Biol. 2020;16:955–963.
- Du YD, Guo WY, Han CH, et al. N6-methyladenosine demethylase FTO impairs hepatic ischemia-reperfusion injury via inhibiting Drp1-mediated mitochondrial fragmentation. Cell Death Dis. 2021;12:442.
- Qu T, Mou Y, Dai J, et al. Changes and relationship of N6-methyladenosine modification and long non-coding RNAs in oxidative damage induced by cadmium in pancreatic β-cells. Toxicol Lett. 2021;343:56–66.
- Su Q, Chen N, Tang J, et al. Paraquat-induced oxidative stress regulates N6-methyladenosine (m6a) modification of long noncoding RNAs in Neuro-2a cells. Ecotoxicol Environ Saf. 2022;237:113503.
- Prasad S, Gupta SC, Tyagi AK. Reactive oxygen species (ROS) and cancer: role of antioxidative nutraceuticals. Cancer Lett. 2017;387:95–105.
- Marengo B, Nitti M, Furfaro AL, et al. Redox Homeostasis and Cellular Antioxidant Systems: crucial Players in Cancer Growth and Therapy. Oxid Med Cell Longev. 2016;2016:6235641.
- Perillo B, Di Donato M, Pezone A, et al. ROS in cancer therapy: the bright side of the moon. Exp Mol Med. 2020;52:192–203.
- Safford SE, Oberley TD, Urano M, et al. Suppression of fibrosarcoma metastasis by elevated expression of manganese superoxide dismutase. Cancer Res. 1994;54:4261–4265.
- Beehler BC, Przybyszewski J, Box HB, et al. Formation of 8-hydroxydeoxyguanosine within DNA of mouse keratinocytes exposed in culture to UVB and H2O2. Carcinogenesis. 1992;13:2003–2007.
- Lee JK, Edderkaoui M, Truong P, et al. NADPH oxidase promotes pancreatic cancer cell survival via inhibiting JAK2 dephosphorylation by tyrosine phosphatases. Gastroenterol. 2007;133:1637–1648.
- Seo JM, Cho KJ, Kim EY, et al. Up-regulation of BLT2 is critical for the survival of bladder cancer cells. Exp Mol Med. 2011;43:129–137.
- Cheng C-W, Kuo C-Y, Fan C-C, et al. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death Dis. 2013;4:e681.
- Cao L, Chen X, Xiao X, et al. Resveratrol inhibits hyperglycemia-driven ROS-induced invasion and migration of pancreatic cancer cells via suppression of the ERK and p38 MAPK signaling pathways. Int J Oncol. 2016;49:735–743.
- Lien G-S, Wu W, Bien M-Y, et al. Epidermal growth factor stimulates nuclear factor-κB activation and heme oxygenase-1 expression via c-Src, NADPH oxidase, PI3K, and Akt in human colon cancer cells. PLoS ONE. 2014;9:e104891.
- Varghese SS, Sunil PM, Madhavan RN. Expression of inducible nitric oxide synthase (iNOS) in oral precancer and oral squamous cell carcinoma: an immunohistochemical study. Cancer Biomark Sect Dis Markers. 2010;8:155–160.
- Aydin E, Johansson J, Nazir FH, et al. Role of NOX2-Derived Reactive Oxygen Species in NK Cell-Mediated Control of Murine Melanoma Metastasis. Cancer Immunol Res. 2017;5:804–811.
- Antony S, Jiang G, Wu Y, et al. NADPH oxidase 5 (NOX5)-induced reactive oxygen signaling modulates normoxic HIF-1α and p27kip1 expression in malignant melanoma and other human tumors. Mol Carcinog. 2017;56:2643–2662.
- Swick RW, Baumann CA, Miller WL, et al. Tocopherol in tumor tissues and effects of tocopherol on the development of liver tumors. Cancer Res. 1951;11:948–953.
- Zhao H, Zhu H, Huang J, et al. The synergy of Vitamin C with decitabine activates TET2 in leukemic cells and significantly improves overall survival in elderly patients with acute myeloid leukemia. Leuk Res. 2018;66:1–7.
- Jaakkola K, Lähteenmäki P, Laakso J, et al. Treatment with antioxidant and other nutrients in combination with chemotherapy and irradiation in patients with small-cell lung cancer. Anticancer Res. 1992;12:599–606.
- Satoh M, Naganuma A, Imura N. Effect of coadministration of selenite on the toxicity and antitumor activity of cis-diamminedichloroplatinum (II) given repeatedly to mice. Cancer Chemother Pharmacol. 1992;30:439–443.
- Azmanova M, Pitto-Barry A. Oxidative Stress in Cancer Therapy: friend or Enemy? Chembiochem Eur J Chem Biol. 2022;23:e202100641.
- Falone S, Santini S, Cordone V, et al. Extremely low-frequency magnetic fields and redox-responsive pathways linked to cancer drug resistance: insights from co-exposure-based in vitro studies. Front Public Health. 2018;6:33.
- Hempel N, Carrico PM, Melendez JA. Manganese superoxide dismutase (Sod2) and redox-control of signaling events that drive metastasis. Anticancer Agents Med Chem. 2011;11:191–201.
- Wu S, Lu H, Bai Y. Nrf2 in cancers: a double-edged sword. Cancer Med. 2019;8:2252–2267.
- Ikehata H, Yamamoto M. Roles of the KEAP1-NRF2 system in mammalian skin exposed to UV radiation. Toxicol Appl Pharmacol. 2018;360:69–77.
- Xian D, Lai R, Song J, et al. Emerging Perspective: role of Increased ROS and Redox Imbalance in Skin Carcinogenesis. Oxid Med Cell Longev. 2019;2019:8127362.
- Falone S, Santini S, Cordone V, et al. Power frequency magnetic field promotes a more malignant phenotype in neuroblastoma cells via redox-related mechanisms. Sci Rep. 2017;7:11470.
- Chern Y-J, Tai IT. Adaptive response of resistant cancer cells to chemotherapy. Cancer Biol Med. 2020;17:842–863.
- Holohan C, Van Schaeybroeck S, Longley DB, et al. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726.
- Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol. 2005;205:275–292.
- Debatin K-M, Krammer PH. Death receptors in chemotherapy and cancer. Oncogene. 2004;23:2950–2966.
- Tirichen H, Yaigoub H, Xu W, et al. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front physiol. 2021;12:627837.
- Zhuang C, Zhuang C, Luo X, et al. N6-methyladenosine demethylase FTO suppresses clear cell renal cell carcinoma through a novel FTO-PGC-1α signalling axis. J Cell Mol Med. 2019;23:2163–2173.
- Mauer J, Sindelar M, Despic V, et al. FTO controls reversible m6am RNA methylation during snRNA biogenesis. Nat Chem Biol. 2019;15:340–347.
- Mauer J, Luo X, Blanjoie A, et al. Reversible methylation of m6am in the 5’ cap controls mRNA stability. Nature. 2017;541:371–375.
- Chen X, Yu C, Guo M, et al. Down-Regulation of m6a mRNA Methylation is Involved in Dopaminergic Neuronal Death. ACS Chem Neurosci. 2019;10:2355–2363.
- Liu X, Gonzalez G, Dai X, et al. Adenylate Kinase 4 Modulates the Resistance of Breast Cancer Cells to Tamoxifen through an m6a-Based Epitranscriptomic Mechanism. Mol Ther. 2020;28:2593–2604.
- Lv Y, Li T, Yang M, et al. Melatonin Attenuates Chromium (VI)-Induced Spermatogonial Stem Cell/Progenitor Mitophagy by Restoration of METTL3-Mediated RNA N6-Methyladenosine Modification. Front Cell Dev Biol. 2021;9:684398.
- Sun R, Tian X, Li Y, et al. The m6a reader YTHDF3-mediated PRDX3 translation alleviates liver fibrosis. Redox Biol. 2022;54:102378.
- Xu W, Lai Y, Pan Y, et al. M6a RNA methylation-mediated NDUFA4 promotes cell proliferation and metabolism in gastric cancer. Cell Death Dis. 2022;13:715.
- Zhang X, Li X, Jia H, et al. The m6a methyltransferase METTL3 modifies PGC-1α mRNA promoting mitochondrial dysfunction and oxLDL-induced inflammation in monocytes. J Biol Chem. 2021;297:101058.
- Goffart S, Wiesner RJ. Regulation and co-ordination of nuclear gene expression during mitochondrial biogenesis. Exp Physiol. 2003;88:33–40.
- Yang B, Chen Q .Cross-Talk between Oxidative Stress and m6a RNA Methylation in Cancer. Oxid Med Cell Longev. 2021:6545728. 2021. doi:10.1155/2021/6545728.
- Cory JG, Breland JC, Carter GL. Effect of 5-fluorouracil on RNA metabolism in Novikoff hepatoma cells. Cancer Res. 1979;39:4905–4913.
- Chun K-S, Joo SH. Modulation of Reactive Oxygen Species to Overcome 5-Fluorouracil Resistance. Biomol Ther. 2022. DOI:10.4062/biomolther.2022.017
- Adhikari S, Bhattacharya A, Adhikary S, et al. The paradigm of drug resistance in cancer: an epigenetic perspective. Biosci Rep. 2022;42:BSR20211812.
- Stein-O’Brien G, Kagohara LT, Li S, et al. Integrated time course omics analysis distinguishes immediate therapeutic response from acquired resistance. Genome Med. 2018;10:37.
- Gustavsson M, Ronne H. Evidence that tRNA modifying enzymes are important in vivo targets for 5-fluorouracil in yeast. RNA N Y N. 2008;14:666–674.
- Kouloulias V, Plataniotis G, Kouvaris J, et al. Chemoradiotherapy combined with intracavitary hyperthermia for anal cancer: feasibility and long-term results from a phase II randomized trial. Am J Clin Oncol. 2005;28:91–99.
- Okamoto M, Fujiwara M, Hori M, et al. tRNA Modifying Enzymes, NSUN2 and METTL1, Determine Sensitivity to 5-Fluorouracil in HeLa Cells. In: Horwitz M, editor. PLoS Genet. Vol. 10. 2014. p. e1004639.
- Saikia M, Krokowski D, Guan B-J, et al. Genome-wide Identification and Quantitative Analysis of Cleaved tRNA Fragments Induced by Cellular Stress. J Biol Chem. 2012;287:42708–42725.
- Elkordy A, Mishima E, Niizuma K, et al. Stress‐induced tRNA cleavage and tiRNA generation in rat neuronal PC12 cells. J Neurochem. 2018;146:560–569.
- Pereira M, Ribeiro DR, Pinheiro MM, et al. M5u54 tRNA Hypomodification by Lack of TRMT2A Drives the Generation of tRNA-Derived Small RNAs. Int J Mol Sci. 2021;22:2941.
- Taketo K, Konno M, Asai A, et al. The epitranscriptome m6a writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J Oncol [Internet]. 2017 [cited 2022 Jun 23]; Available from: http://www.spandidos-publications.com/10.3892/ijo.2017.4219.
- Mantovani F, Collavin L, Del Sal G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019;26:199–212.
- Uddin MB, Roy KR, Hosain SB, et al. An N-methyladenosine at the transited codon 273 of p53 pre-mRNA promotes the expression of R273H mutant protein and drug resistance of cancer cells. Biochem Pharmacol. 2019;160:134–145.
- Liu D, Xu Y. P53, oxidative stress, and aging. Antioxid Redox Signal. 2011;15:1669–1678.
- Ke W, Zhang L, Zhao X, et al. P53 m6a modulation sensitizes hepatocellular carcinoma to apatinib through apoptosis. Apoptosis. 2022;27:426–440.
- Y-N M, Hong Y-G, G-Y Y, et al. LncRNA LBX2-AS1 promotes colorectal cancer progression and 5-fluorouracil resistance. Cancer Cell Int. 2021;21:501.
- Pan S, Deng Y, Fu J, et al. N6‑methyladenosine upregulates miR‑181d‑5p in exosomes derived from cancer‑associated fibroblasts to inhibit 5‑FU sensitivity by targeting NCALD in colorectal cancer. Int J Oncol. 2022;60:14.
- Venugopal V, Sumi S. Molecular Biomarkers and Drug Targets in Brain Arteriovenous and Cavernous Malformations: where are We? Stroke. 2022;53:279–289.
- Zhang Y, Zhang X, Li H, et al. Antidepressant-like effects of helicid on a chronic unpredictable mild stress-induced depression rat model: inhibiting the IKK/IκBα/NF-κB pathway through NCALD to reduce inflammation. Int Immunopharmacol. 2021;93:107165.
- Zhang Y, Li C, Guan C, et al. MiR-181d-5p Targets KLF6 to Improve Ischemia/Reperfusion-Induced AKI Through Effects on Renal Function, Apoptosis, and Inflammation. Front physiol. 2020;11:510.
- Abdellateif MS, Salem SE, Badr DM, et al. The Prognostic Significance of 5-Fluorouracil Induced Inflammation and Immuno-Modulation in Colorectal Cancer Patients. J Inflamm Res. 2020;13:1245–1259.
- D-X H, X-T G, Y-R L, et al. Methylation-regulated miR-149 modulates chemoresistance by targeting GlcNac N -deacetylase/N -sulfotransferase-1 in human breast cancer. FEBS J. 2014;281:4718–4730.
- Pecoraro M, Pala B, Di Marcantonio M, et al. Doxorubicin‑induced oxidative and nitrosative stress: mitochondrial connexin 43 is at the crossroads. Int J Mol Med. 2020;46:1197–1209.
- Pilco-Ferreto N, Calaf GM. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int J Oncol. 2016;49:753–762.
- Li P, Shan J-X, Chen X-H, et al. Epigenetic silencing of microRNA-149 in cancer-associated fibroblasts mediates prostaglandin E2/interleukin-6 signaling in the tumor microenvironment. Cell Res. 2015;25:588–603.
- Shi T, Dansen TB. Reactive Oxygen Species Induced p53 Activation: dNA Damage, Redox Signaling, or Both? Antioxid Redox Signal. 2020;33:839–859.
- Pan X, Hong X, Li S, et al. METTL3 promotes adriamycin resistance in MCF-7 breast cancer cells by accelerating pri-microRNA-221-3p maturation in a m6a-dependent manner. Exp Mol Med. 2021;53:91–102.
- Dang X, Zhang R, Peng Z, et al. HIPK2 overexpression relieves hypoxia/reoxygenation-induced apoptosis and oxidative damage of cardiomyocytes through enhancement of the Nrf2/ARE signaling pathway. Chem Biol Interact. 2020;316:108922.
- Yu W, Chen Y, Dubrulle J, et al. Cisplatin generates oxidative stress which is accompanied by rapid shifts in central carbon metabolism. Sci Rep. 2018;8:4306.
- He P, Ge R, Mao W, et al. Oxidative stress induced by carboplatin promotes apoptosis and inhibits migration of HN‑3 cells. Oncol Lett [Internet]. 2018 [cited 2022 Jun 23]; Available from: http://www.spandidos-publications.com/10.3892/ol.2018.9563.
- Dhillon AS, Hagan S, Rath O, et al. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–3290.
- Corre I, Paris F, Huot J. The p38 pathway, a major pleiotropic cascade that transduces stress and metastatic signals in endothelial cells. Oncotarget. 2017;8:55684–55714.
- Li Y, Li J, Luo M, et al. Novel long noncoding RNA NMR promotes tumor progression via NSUN2 and BPTF in esophageal squamous cell carcinoma. Cancer Lett. 2018;430:57–66.
- Jin D, Guo J, Wu Y, et al. M6a mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J Hematol OncolJ Hematol Oncol. 2019;12:135.
- Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 2016;29:783–803.
- Grattarola M, Cucci MA, Roetto A, et al. Post-translational down-regulation of Nrf2 and YAP proteins, by targeting deubiquitinases, reduces growth and chemoresistance in pancreatic cancer cells. Free Radic Biol Med. 2021;174:202–210.
- Zhang Y, Kang M, Zhang B, et al. M6a modification-mediated CBX8 induction regulates stemness and chemosensitivity of colon cancer via upregulation of LGR5. Mol Cancer. 2019;18:185.
- Teng B-W, Zhang K-D, Yang Y-H, et al. Genome-wide CRISPR-Cas9 screening identifies that hypoxia-inducible factor-1a-induced CBX8 transcription promotes pancreatic cancer progression via IRS1/AKT axis. World J Gastrointest Oncol. 2021;13:1709–1724.
- Wei J, Yin Y, Zhou J, et al. METTL3 potentiates resistance to cisplatin through m 6 a modification of TFAP2C in seminoma. J Cell Mol Med. 2020;24:11366–11380.
- Kulak MV, Cyr AR, Woodfield GW, et al. Transcriptional regulation of the GPX1 gene by TFAP2C and aberrant CpG methylation in human breast cancer. Oncogene. 2013;32:4043–4051.
- Song Z, Jia G, Ma P, et al. Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6a modification-mediated ferroptosis. Life Sci. 2021;276:119399.
- Guo J, Xu B, Han Q, et al. Ferroptosis: a Novel Anti-tumor Action for Cisplatin. Cancer Res Treat. 2018;50:445–460.
- Sun S, Gao T, Pang B, et al. RNA binding protein NKAP protects glioblastoma cells from ferroptosis by promoting SLC7A11 mRNA splicing in an m6a-dependent manner. Cell Death Dis. 2022;13:73.
- Zhang C, Liu X, Jin S, et al. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer. 2022;21:47.
- Nie Q, Hu Y, Yu X, et al. Induction and application of ferroptosis in cancer therapy. Cancer Cell Int. 2022;22:12.
- Christman JK. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–5495.
- Cheng JX, Chen L, Li Y, et al. RNA cytosine methylation and methyltransferases mediate chromatin organization and 5-azacytidine response and resistance in leukaemia. Nat Commun. 2018;9:1163.
- Gkatza NA, Castro C, Harvey RF, et al. Cytosine-5 RNA methylation links protein synthesis to cell metabolism. PLoS Biol. 2019;17:e3000297.
- Lewinska A, Adamczyk-Grochala J, Kwasniewicz E, et al. Reduced levels of methyltransferase DNMT2 sensitize human fibroblasts to oxidative stress and DNA damage that is accompanied by changes in proliferation-related miRNA expression. Redox Biol. 2018;14:20–34.
- Hou D, Liu Z, Xu X, et al. Increased oxidative stress mediates the antitumor effect of PARP inhibition in ovarian cancer. Redox Biol. 2018;17:99–111.
- Fukumoto T, Zhu H, Nacarelli T, et al. N6-Methylation of Adenosine of FZD10 mRNA Contributes to PARP Inhibitor Resistance. Cancer Res. 2019;79:2812–2820.
- Wang Z, Xia J, Li J, et al. Rg1 Protects Hematopoietic Stem Cells from LiCl-Induced Oxidative Stress via Wnt Signaling Pathway. Evid-Based Complement Altern Med ECAM. 2022;2022:2875583.
- Majchrzak-Celińska A, Kleszcz R, Studzińska-Sroka E, et al. Lichen Secondary Metabolites Inhibit the Wnt/β-Catenin Pathway in Glioblastoma Cells and Improve the Anticancer Effects of Temozolomide. Cells. 2022;11:1084.
- Cong K, Cantor SB. Exploiting replication gaps for cancer therapy. Mol Cell. 2022;82:2363–2369.
- Balko JM, Potti A, Saunders C, et al. Gene expression patterns that predict sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer cell lines and human lung tumors. BMC Genomics. 2006;7:289.
- Rodríguez-Hernández MA, de la Cruz-Ojeda P, Gallego P, et al. Dose-dependent regulation of mitochondrial function and cell death pathway by sorafenib in liver cancer cells. Biochem Pharmacol. 2020;176:113902.
- Lin Z, Niu Y, Wan A, et al. RNA m 6 a methylation regulates sorafenib resistance in liver cancer through FOXO 3‐mediated autophagy. EMBO J. 2020;39 [[cited 2022 Jul 1]]. InternetAvailable from: https://onlinelibrary.wiley.com/doi/10.15252/embj.2019103181
- Essers MAG, de Vries-Smits LMM, Barker N, et al. Functional Interaction Between ß-Catenin and FOXO in Oxidative Stress Signaling. Science. 2005;308:1181–1184.
- Burgering BMT, Medema RH. Decisions on life and death: fOXO Forkhead transcription factors are in command when PKB/Akt is off duty. J Leukocyte Biol. 2003;73:689–701.
- Di Emidio G, Falone S, Vitti M, et al. SIRT1 signalling protects mouse oocytes against oxidative stress and is deregulated during aging. Hum Reprod Oxf Eng. 2014;29:2006–2017.
- Okon IS, Coughlan KA, Zhang M, et al. Gefitinib-mediated reactive oxygen specie (ROS) instigates mitochondrial dysfunction and drug resistance in lung cancer cells. J Biol Chem. 2015;290:9101–9110.
- Liu S, Li Q, Li G, et al. The mechanism of m6a methyltransferase METTL3-mediated autophagy in reversing gefitinib resistance in NSCLC cells by β-elemene. Cell Death Dis. 2020;11:969.
- Chen H, Jia B, Zhang Q, et al. Meclofenamic Acid Restores Gefinitib Sensitivity by Downregulating Breast Cancer Resistance Protein and Multidrug Resistance Protein 7 via FTO/m6A-Demethylation/c-Myc in Non-Small Cell Lung Cancer. Front Oncol. 2022;12:870636.
- Marengo B, Garbarino O, Speciale A, et al. MYC Expression and Metabolic Redox Changes in Cancer Cells: a Synergy Able to Induce Chemoresistance. Oxid Med Cell Longev. 2019;2019:1–9.
- Wang KC, Chang HY. Molecular Mechanisms of Long Noncoding RNAs. Mol Cell. 2011;43:904–914.
- Yan X, Hu Z, Feng Y, et al. Comprehensive Genomic Characterization of Long Non-coding RNAs across Human Cancers. Cancer Cell. 2015;28:529–540.
- Chen Y, Xiang D, Zhao X, et al. Upregulation of lncRNA NIFK-AS1 in hepatocellular carcinoma by m6a methylation promotes disease progression and sorafenib resistance. Hum Cell. 2021;34:1800–1811.
- Singh AK, Kashyap MP, Tripathi VK, et al. Neuroprotection Through Rapamycin-Induced Activation of Autophagy and PI3K/Akt1/mTOR/CREB Signaling Against Amyloid-β-Induced Oxidative Stress, Synaptic/Neurotransmission Dysfunction, and Neurodegeneration in Adult Rats. Mol Neurobiol. 2017;54:5815–5828.
- Chong ZZ, Shang YC, Hou J, et al. Wnt1 neuroprotection translates into improved neurological function during oxidant stress and cerebral ischemia through AKT1 and mitochondrial apoptotic pathways. Oxid Med Cell Longev. 2010;3:153–165.
- Quirke VM. Tamoxifen from Failed Contraceptive Pill to Best-Selling Breast Cancer Medicine: a Case-Study in Pharmaceutical Innovation. Front Pharmacol. 2017;8:620.
- Bekele RT, Venkatraman G, Liu R-Z, et al. Oxidative stress contributes to the tamoxifen-induced killing of breast cancer cells: implications for tamoxifen therapy and resistance. Sci Rep. 2016;6:21164.
- Panayiotou C, Solaroli N, Karlsson A. The many isoforms of human adenylate kinases. Int J Biochem Cell Biol. 2014;49:75–83.
- Jan Y-H, Lai T-C, Yang C-J, et al. Adenylate kinase 4 modulates oxidative stress and stabilizes HIF-1α to drive lung adenocarcinoma metastasis. J Hematol OncolJ Hematol Oncol. 2019;12:12.
- Shan Y, Akram A, Amatullah H, et al. ATF3 protects pulmonary resident cells from acute and ventilator-induced lung injury by preventing Nrf2 degradation. Antioxid Redox Signal. 2015;22:651–668.
- Liu X, Yuan J, Zhang X, et al. ATF3 Modulates the Resistance of Breast Cancer Cells to Tamoxifen through an N 6 -Methyladenosine-Based Epitranscriptomic Mechanism. Chem Res Toxicol. 2021;34:1814–1821.
- Edagawa M, Kawauchi J, Hirata M, et al. Role of Activating Transcription Factor 3 (ATF3) in Endoplasmic Reticulum (ER) Stress-induced Sensitization of p53-deficient Human Colon Cancer Cells to Tumor Necrosis Factor (TNF)-related Apoptosis-inducing Ligand (TRAIL)-mediated Apoptosis through Up-regulation of Death Receptor 5 (DR5) by Zerumbone and Celecoxib. J Biol Chem. 2014;289:21544–21561.
- Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293.
- Higa A, Chevet E. Redox signaling loops in the unfolded protein response. Cell Signal. 2012;24:1548–1555.
- Hourihan JM, Moronetti Mazzeo LE, Fernández-Cárdenas LP, et al. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/nrf2 Antioxidant Response. Mol Cell. 2016;63:553–566.
- Gordon RR, Nelson PS. Cellular senescence and cancer chemotherapy resistance. Drug Resist Updat. 2012;15:123–131.
- Brunner TB. The rationale of combined radiotherapy and chemotherapy - Joint action of Castor and Pollux. Best Pract Res Clin Gastroenterol. 2016;30:515–528.
- Navarro J, Obrador E, Pellicer JA, et al. Blood glutathione as an index of radiation-induced oxidative stress in mice and humans. Free Radic Biol Med. 1997;22:1203–1209.
- Jia S, Dong S, Liu H, et al. Dopamine-derived nanoparticles for the protection of irradiation-induced intestinal injury by maintaining intestinal homeostasis. Biomater Sci. 2022;10:3309–3322.
- Kowalski-Chauvel A, Lacore MG, Arnauduc F, et al. The m6a RNA Demethylase ALKBH5 Promotes Radioresistance and Invasion Capability of Glioma Stem Cells. Cancers (Basel). 2020;13:40.
- Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124–1134.
- Najafi M, Farhood B, Mortezaee K. Cancer stem cells (CSCs) in cancer progression and therapy. J Cell Physiol. 2019;234:8381–8395.
- Shibue T, Weinberg RE. Cscs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611–629.
- Talukdar S, Emdad L, Das SK, et al. Evolving Strategies for Therapeutically Targeting Cancer Stem Cells. Adv Cancer Res [Internet] Elsevier ; 2016 [cited 2022 Jul 6]. p. 159–191. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0065230X16300343.
- Jiang Q, Crews LA, Holm F, et al. RNA editing-dependent epitranscriptome diversity in cancer stem cells. Nat Rev Cancer. 2017;17:381–392.
- Lin S, Choe J, Du P, et al. The m(6)a Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol Cell. 2016;62:335–345.
- Gao Q, Zheng J, Ni Z, et al. The m6a Methylation-Regulated AFF4 Promotes Self-Renewal of Bladder Cancer Stem Cells. Stem Cells Int. 2020;2020:8849218.
- Shriwas O, Priyadarshini M, Samal SK, et al. DDX3 modulates cisplatin resistance in OSCC through ALKBH5-mediated m6a-demethylation of FOXM1 and NANOG. Apoptosis Int J Program Cell Death. 2020;25:233–246.
- Mohan A, R RR, Mohan G, et al. Reporters of Cancer Stem Cells as a Tool for Drug Discovery. Front Oncol. 2021;11:669250.
- Tsao A-N, Chuang Y-S, Lin Y-C, et al. Dinaciclib inhibits the stemness of two subtypes of human breast cancer cells by targeting the FoxM1 and Hedgehog signaling pathway. Oncol Rep. 2022;47:105.
- Li L, Liu Y, Xiao L-M, et al. Induction of cancer cell stemness in glioma through glycolysis and the long noncoding RNA HULC-activated FOXM1/AGR2/HIF-1α axis. Lab Investig J Tech Methods Pathol. 2022;102:691–701.
- Song I-S, Jeong YJ, Jeong SH, et al. FOXM1-Induced PRX3 Regulates Stemness and Survival of Colon Cancer Cells via Maintenance of Mitochondrial Function. Gastroenterology. 2015;149:1006–1016.e9.
- Yang Y, Wu J, Liu F, et al. IGF2BP1 Promotes the Liver Cancer Stem Cell Phenotype by Regulating MGAT5 mRNA Stability by m6a RNA Methylation. Stem Cells Dev. 2021. scd.2021.0153. 10.1089/scd.2021.0153
- Wang Y, Wang J, Li X, et al. N1-methyladenosine methylation in tRNA drives liver tumourigenesis by regulating cholesterol metabolism. Nat Commun. 2021;12:6314.
- Sari IN, Phi LTH, Jun N, et al. Hedgehog Signaling in Cancer: a Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells. 2018;7:E208.
- Liu X, Wang Z, Yang Q, et al. RNA Demethylase ALKBH5 Prevents Lung Cancer Progression by Regulating EMT and Stemness via Regulating p53. Front Oncol. 2022;12:858694.
- Oiseth SJ, Aziz MS. Cancer immunotherapy: a brief review of the history, possibilities, and challenges ahead. J Cancer Metastasis Treat. 2017;3:250.
- Chow MT, Möller A, Smyth MJ. Inflammation and immune surveillance in cancer. Semin Cancer Biol. 2012;22:23–32.
- Halliday GM, Patel A, Hunt MJ, et al. Spontaneous regression of human melanoma/nonmelanoma skin cancer: association with infiltrating CD4+ T cells. World J Surg. 1995;19:352–358.
- Li N, Kang Y, Wang L, et al. ALKBH5 regulates anti–pd-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci. 2020;117:20159–20170.
- Zhang F, Huang H, Qin Y, et al. MTDH associates with m6a RNA methylation and predicts cancer response for immune checkpoint treatment. iScience. 2021;24:103102.
- Emdad L, Das SK, Hu B, et al. AEG-1/MTDH/LYRIC: a Promiscuous Protein Partner Critical in Cancer, Obesity, and CNS Diseases. Adv Cancer Res. 2016;131:97–132.
- Yu C, Liu Y, Tan H, et al. Metadherin regulates metastasis of squamous cell carcinoma of the head and neck via AKT signalling pathway-mediated epithelial–mesenchymal transition. Cancer Lett. 2014;343:258–267.
- Yin H, Zhang X, Yang P, et al. RNA m6a methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat Commun. 2021;12:1394.
- Hayakawa T, Sugiyama J, Yaguchi T, et al. Enhanced anti-tumor effects of the PD-1/PD-L1 blockade by combining a highly absorptive form of NF-kB/STAT3 inhibitor curcumin. J Immunother Cancer. 2014;2:P210. 2051-1426-2-S3-P210.
- Liu Z, Wang T, She Y, et al. N6-methyladenosine-modified circIGF2BP3 inhibits CD8+ T-cell responses to facilitate tumor immune evasion by promoting the deubiquitination of PD-L1 in non-small cell lung cancer. Mol Cancer. 2021;20:105.
- Aventaggiato M, Vernucci E, Barreca F, et al. Sirtuins’ control of autophagy and mitophagy in cancer. Pharmacol Ther. 2021;221:107748.
- Lu Z, Liu H, Song N, et al. METTL14 aggravates podocyte injury and glomerulopathy progression through N6-methyladenosine-dependent downregulating of Sirt1. Cell Death Dis. 2021;12:881.
- Wang Z, Chen W. Emerging Roles of SIRT1 in Cancer Drug Resistance. Genes Cancer. 2013;4:82–90.
- Luo J, Nikolaev AY, Imai S, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148.
- Dai JM, Wang ZY, Sun DC, et al. SIRT1 interacts with p73 and suppresses p73-dependent transcriptional activity. J Cell Physiol. 2007;210:161–166.
- O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4:e1000155.
- Zhang J, Ren D, Fedorova J, et al. SIRT1/SIRT3 Modulates Redox Homeostasis during Ischemia/Reperfusion in the Aging Heart. Antioxid Basel Switz. 2020;9:E858.
- Falone S, Santini S, di Loreto S, et al. Improved mitochondrial and methylglyoxal-related metabolisms support hyperproliferation induced by 50 Hz magnetic field in neuroblastoma cells. J Cell Physiol. 2016;231:2014–2025.
- Thornalley PJ. Protecting the genome: defence against nucleotide glycation and emerging role of glyoxalase I overexpression in multidrug resistance in cancer chemotherapy. Biochem Soc Trans. 2003;31:1372–1377.
- Rabbani N, Xue M, Weickert MO, et al. Multiple roles of glyoxalase 1-mediated suppression of methylglyoxal glycation in cancer biology—involvement in tumour suppression, tumour growth, multidrug resistance and target for chemotherapy. Semin Cancer Biol. 2018;49:83–93.
- Honek JF. Glyoxalase biochemistry. Biomol Concepts. 2015;6:401–414.
- Sakamoto H, Mashima T, Kizaki A, et al. Glyoxalase I is involved in resistance of human leukemia cells to antitumor agent-induced apoptosis. Blood. 2000;95:3214–3218.
- Antognelli C, Palumbo I, Aristei C, et al. Glyoxalase I inhibition induces apoptosis in irradiated MCF-7 cells via a novel mechanism involving Hsp27, p53 and NF-κB. Br J Cancer. 2014;111:395–406.
- Michel M, Hollenbach M, Pohl S, et al. Inhibition of Glyoxalase-I Leads to Reduced Proliferation, Migration and Colony Formation, and Enhanced Susceptibility to Sorafenib in Hepatocellular Carcinoma. Front Oncol. 2019;9:785.
- Kulkarni CA, Nadtochiy SM, Kennedy L, et al. ALKBH7 mediates necrosis via rewiring of glyoxal metabolism. Elife. 2020;9:e58573.
- Marchand V, Pichot F, Thüring K, et al. Next‐generation Sequencing‐Based RiboMethseq Protocol for Analysis of tRNA 2′‐o‐methylation. Biomolecules. 2017;7:13.
- Li X, Xiong X, Yi C. Epitranscriptome sequencing technologies: decoding RNA modifications. Nat Methods. 2016;14:23–31.
- Kong Y, Hu H, Shan Y, et al. Accurate quantification of 3′-terminal 2′-O-methylated small RNAs by utilizing oxidative deep sequencing and stem-loop RT-qPCR. Front Med. 2022;16:240–250.
- Cui J, Liu Q, Sendinc E, et al. Nucleotide resolution profiling of m3c RNA modification by HAC-seq. Nucleic Acids Res. 2021;49: e27–e27.
- Amalric A, Bastide A, Attina A, et al. Quantifying RNA modifications by mass spectrometry: a novel source of biomarkers in oncology. Crit Rev Clin Lab Sci. 2022;59:1–18.
- Cui X, Zhou Y, Zheng Y, et al. Investigation of the enhanced photoactivity of CdS/Bi2MoO6/MoSe2 and its application in antibody-free enzyme-assisted photoelectrochemical strategy for detection of N6-methyladenosine and FTO protein. Mater Today Nano. 2022;20:100269.
- Kourou K, Exarchos TP, Exarchos KP, et al. Machine learning applications in cancer prognosis and prediction. Comput Struct Biotechnol J. 2015;13:8–17.
- Thomas A, Barriere S, Broseus L, et al. GECKO is a genetic algorithm to classify and explore high throughput sequencing data. Commun Biol. 2019;2:222.
- Sui Q, Chen Z, Hu Z, et al. Cisplatin resistance-related multi-omics differences and the establishment of machine learning models. J Transl Med. 2022;20:171.
- Nahar S, Kotikam V, Kumar VA, et al. Inhibition of miR-21 by 3′/5′-Serinyl-Capped 2′- O -Methyl RNA Interspersed with 2′- O -(2-Amino-3-Methoxypropyl) Uridine Units. Nucleic Acid Ther. 2016;26:327–334.