
ABSTRACT
Peroxisome proliferator-activated receptor gamma (PPARγ) is a nuclear receptor and master transcription factor of adipogenesis-related genes, and has been reported as an antitumor target for chondrosarcomas. Herein, we show that the nonsteroidal anti-inflammatory drug, zaltoprofen, induces the expression of PPARγ at the mRNA and protein levels, following the induction of PPARγ-activating factors, such as Krox20, C/EBPβ, and C/EBPα, in human extraskeletal chondrosarcoma H-EMC-SS cells. Upregulation of the cell cycle checkpoint proteins, p21, p27, and p53, was observed upon treatment of H-EMC-SS cells with zaltoprofen, which probably resulted in the inhibition of proliferation of these cells observed in vitro. Zaltoprofen treatment inhibited tumor growth, induced tumor cell apoptosis, and was well tolerated in a mouse model of extraskeletal myxoid chondrosarcoma. Our results provide mechanistic insights into the therapeutic effect of zaltoprofen that should promote further studies on the rational use of this drug for the effective treatment of sarcomas.
Introduction
Extraskeletal myxoid chondrosarcoma (EMC) is a soft tissue sarcoma that arises at various sites including the extremities [Citation1]. Because radiation and chemotherapy are ineffective, wide resection is the only radical treatment available for EMC [Citation1]. Although considered a slow-growing neoplasm, the recurrence and metastatic rates of EMC after radical resection (with values up to 50% for each) are higher than those for soft-tissue sarcomas [Citation1,Citation2].
We recently reported that peroxisome proliferator-activated receptor gamma (PPARγ), a ligand-activated nuclear receptor that controls the differentiation of adipocytes in normal tissues, could be a therapeutic target for inhibiting cell invasion and matrix metalloproteinase-2 (MMP2) expression in chondrosarcoma cells [Citation3]. In addition, we demonstrated that zaltoprofen, a nonsteroidal anti-inflammatory drug (NSAID), could effectively induce and activate PPARγ, and elicit antitumor effects in chondrosarcoma cells [Citation3].
To better understand the therapeutic potential of PPARγ-targeted treatment using zaltoprofen, in the present study, we investigated the mechanism of zaltoprofen-mediated induction of PPARγ and its anti-proliferative effect in vitro and in vivo on human EMC cells (H-EMC-SS).
Materials and methods
Reagents
Zaltoprofen was purchased from Santa Cruz Biotechnology (Dallas, TX, USA) and was dissolved in 10% dimethyl sulfoxide (DMSO) (FUJIFILM Wako Pure Chemical, Osaka, Japan) for in vitro experiments or in 0.2% carboxymethyl cellulose (CMC) sodium (FUJIFILM Wako Pure Chemical) for in vivo experiments.
Cell culture
The human EMC cell line, H-EMC-SS (RIKEN BioResource Research Center, Tsukuba, Japan), was cultured in Dulbecco’s modified Eagle’s medium/Ham’s F-12 (FUJIFILM Wako Pure Chemical) supplemented with fetal bovine serum (10%) (FUJIFILM Wako Pure Chemical), penicillin (100 U/mL) (FUJIFILM Wako Pure Chemical), and streptomycin (100 μg/mL) (FUJIFILM Wako Pure Chemical) at 37°C.
RNA interference
PPARγ knockdown was conducted using human PPARγ short hairpin RNA (shRNA) (SureSilencing shRNA plasmid, Qiagen, Valencia, CA, USA). The Attractene transfection reagent (Qiagen) was used to transfect the PPARγ shRNA (shPPARγ) plasmid or nontargeting shRNA (shNT) control plasmid into H-EMC-SS cells. Neomycin (600 µg/mL, G418 solution, Roche Diagnostics K.K., Tokyo, Japan) was used for selecting shRNA-transfected cells, and the knockdown efficiency was confirmed by the quantitative reverse transcription polymerase chain reaction (RT-qPCR). Thus, PPARγ knockdown (H-EMC-SSshPPARγ) and nontarget control (H-EMC-SSshNT) cell lines were established. The shRNA insert sequences were as follows: PPARγ shRNA, 5′-CCACGAGATCATTACACAAT-3′; nontargeting shRNA, 5′-GGAATCTCATTCGATGCATAC-3′.
RT-qPCR
After treatment with zaltoprofen for 24 h, H-EMC-SS cells were lysed with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and total RNA was extracted. The RNA samples (100 ng/sample) were quantified using a spectrophotometer (NanoDrop Lite, Thermo Scientific K.K., Tokyo, Japan) and reverse-transcribed to cDNA on a T100 Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, USA) using an AffinityScript cDNA Synthesis Kit (Agilent Technologies, Santa Clara, CA, USA). RT-qPCR was performed with 10 ng cDNA per well on a StepOne™ Real-time PCR System (Applied Biosystems, Foster City, CA, USA) using the QuantiTect SYBR Green PCR Kit (Qiagen). Primers for PPARγ, CCAAT/enhancer binding protein (C/EBP) α, C/EBPß, C/EBPδ, and Krox20 were purchased from Qiagen (QuantiTect Primer Assay, Hs_PPARG_1_SG, Hs_CEBPA_1_SG, Hs_CEBPB_1_SG, Hs_CEBPD_1_SG, and Hs_EGR2_1_SG). The primer sequences for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were: forward, 5′-TGCACCACCAACTGCTTAGC-3′; reverse, 5′-GGCATGGACTGTGGTCATGAG-3′.
Western blot analyses
After treatment with various concentrations of zaltoprofen (0, 50, 100, 200, and 400 µmol/L) for 24 h, H-EMC-SS cells were lysed in radioimmunoprecipitation assay buffer (1% sodium deoxycholate, 150 mM NaCl, 50 mM sodium fluoride, 100 μM sodium orthovanadate, 0.1% sodium dodecyl sulfate, 1% Triton X-100, 10 mM Tris, pH 7.2, [FUJIFILM Wako Pure Chemical]) containing a protease inhibitor cocktail (Sigma-Aldrich Japan, Tokyo, Japan). Total protein (100 µg/sample) was boiled (2 min at 95°C) and resolved by 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis on an e-PAGEL (ATTO, Tokyo, Japan), and transferred onto a polyvinylidene fluoride membrane (Merck Millipore, Bedford, Germany). After overnight incubation in blocking solution (Odyssey Blocking Buffer PBS, LI-COR, Lincoln, NE, USA), the membranes were incubated with the following antibodies at 4°C for 60 min: anti-PPARγ (Santa Cruz Biotechnology), anti-p21 (Cell Signaling Technology, Danvers, MA, USA), anti-p27 (Santa Cruz Biotechnology), anti-p53 (Santa Cruz Biotechnology), and anti-GAPDH (Cell Signaling Technology). The membranes were washed three times with phosphate-buffered saline containing 10% Tween 20 (Sigma-Aldrich Japan) (T-PBS) and incubated with the secondary antibody (IRDye 680RD goat anti-IgG, LI-COR) for 30 min. After washing three times with T-PBS, the bands were visualized and scanned with an Odyssey Infrared Imaging system (LI-COR).
Water-soluble tetrazolium salt-8 (WST-8) assay
The viability of cells at designated time points after zaltoprofen treatment was determined using Cell Counting Kit-8 (Dojindo Laboratory, Kumamoto, Japan). After H-EMC-SS cells (5 × 103 cells/well) were treated with the indicated concentrations of zaltoprofen (0, 50, 100, 200, and 400 µmol/L), the absorbance at 450 nm was measured using an iMark microplate reader (Bio-Rad Laboratories).
5-ethynyl-2-deoxyuridine (EdU) proliferation assay
To investigate the proliferation of cells after zaltoprofen treatment, EdU assay was performed as follows. After incubating H-EMC-SSshPPARγ and H-EMC-SSshNT cells on slide chambers for 24 h, they were further incubated with 1% DMSO or 400 µmol/L zaltoprofen for 24 h. The cells were then treated with 10 mmol/L EdU stock solution (Click-iT® EdU Alexa Fluor® 555 Imaging Kit, Invitrogen) for 60 min, fixed with 4% paraformaldehyde (FUJIFILM Wako Pure Chemical), washed twice with 3% bovine serum albumin (FUJIFILM Wako Pure Chemical), permeabilized with 0.5% Triton X-100 (FUJIFILM Wako Pure Chemical), incubated with the Click-iT reaction buffer (Click-iT® EdU Alexa Fluor® 555 Imaging Kit, Invitrogen) for 30 min, and washed once with bovine serum albumin. Cell nuclei were stained with Hoechst 33,342 (NucBlue Live ReadyProbes Reagent, Invitrogen). Cells positive for EdU-stained nuclei were visualized with a BZ-9000 fluorescence microscope (Keyence, Osaka, Japan). EdU- and Hoechst 33,342-positive cells were counted using ImageJ software (National Institute of Health, Bethesda, MD, USA).
Animal experiments
To investigate the antiproliferation effect of zaltoprofen in vivo, four-week-old BALB/c female nude mice (Charles River Laboratories Japan, Kanagawa, Japan) were adapted to the laboratory conditions for a week before the experiment, and H-EMC-SS cells (4 × 10 6/100 µL) suspended in Matrigel (BD Japan, Tokyo, Japan) were implanted subcutaneously on the flanks of mice. The mice were divided randomly into zaltoprofen (30 mg/kg, oral gavage, twice a day, 42 days)-treated (n = 6) and vehicle (0.2% CMC sodium, 30 mL/kg, oral gavage twice a day for 42 days)-treated (n = 6) groups. Tumor diameters and body weights of mice were measured once a week using digital calipers and digital scales, respectively. Tumor volume (mm3) was calculated using the following formula: tumor volume (mm3) = largest diameter (mm) × smallest diameter (mm) × smallest diameter (mm) × 0.5 [Citation4]. The animals were observed on a daily basis and humanely sacrificed if they met the following endpoint criteria: severe tumor burden (more than 20 mm in diameter), prostration, significant body weight loss, difficulty breathing, rotational motion and body temperature drop. All treatment groups were housed without cage separation to minimize potential confounders.
After 42 days of treatment, the mice were sacrificed humanely, scanned by computed tomography, and tumors were resected for histopathological evaluation. The resected tumors were fixed in 10% formalin (Sigma-Aldrich Japan) and embedded in paraffin. Tumor sections were deparaffinized and rehydrated in xylene and ethanol, then stained with hematoxylin and eosin or immunostained with antibodies against Ki-67 (Abcam, Cambridge, UK), a proliferation marker, in combination with diaminobenzidine (Dako Japan, Kyoto, Japan). To observe apoptosis in the tumor sections, the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay was performed using a cell death kit (Roche) according to the manufacturer’s instructions. The TUNEL-stained area was measured and Ki-67-positive cells were counted using ImageJ software. To analyze the drug toxicity, kidney, intestine, and blood serum samples were collected from mice and evaluated pathologically and biochemically for side effects by an outsourced inspection facility (Safety Research Institute for Chemical Compounds, Sapporo, Japan).
Statistical analyses
Easy R (Saitama Medical Center, Jichi Medical University, Saitama, Japan), a modified version of R commander (The R Foundation for Statistical Computing, Vienna, Austria), which includes statistical functions for biostatistics, was used for all statistical analyses [Citation5]. Data were compared using Student’s t-test or a one-way analysis of variance with Tukey’s post-hoc test. Each experiment was repeated at least three times. All data are expressed as means ± standard error of the mean. All p-values are two-sided.
Results
Induction of PPARγ by zaltoprofen
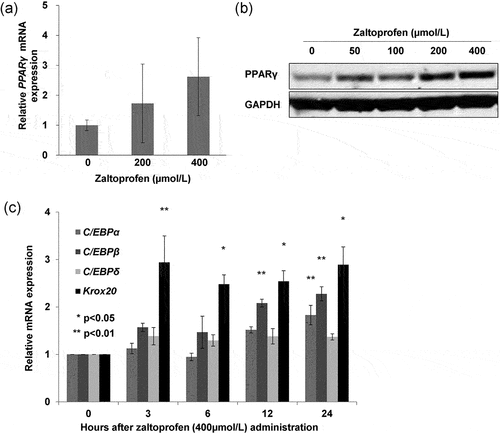
Zaltoprofen treatment upregulated the mRNA and protein levels of PPARγ in H-EMC-SS cells as evidenced by qRT-PCR and western blot analyses, respectively. This demonstrated that zaltoprofen can induce PPARγ in H-EMC-SS cells (). Next, we investigated the induction of Krox20 and C/EBPs, which regulate PPARγ in normal adipose tissue, in zaltoprofen (400 µmol/L)-treated H-EMC-SS cells. Krox20 was significantly upregulated from 3 to 24 h after the initiation of treatment, followed by a significant upregulation of C/EBPβ starting 12 h and of C/EBPα at 24 h (), demonstrating the signal pathway of PPARγ induction by zaltoprofen in H-EMC-SS cells.
Figure 1. Effect of zaltoprofen on the expression of PPARγ in H-EMC-SS cells at the mRNA (a) and protein (b) levels, as determined by qRT-PCR and western blot analyses, respectively. GAPDH was used as the housekeeping factor. (c) Effect of zaltoprofen on the expression of C/EBPα, C/EBPβ, C/EBPδ, and Krox20 mRNAs in H-EMC-SS cells, as determined by qRT-PCR. *p < 0.05, **p < 0.01. Error bars: ± SEM.
Inhibition of cell proliferation by zaltoprofen
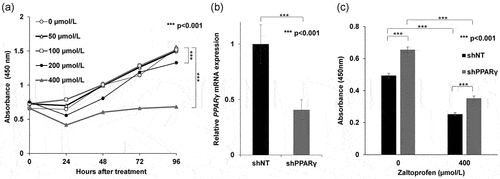
The inhibitory effect of zaltoprofen on the proliferation of H-EMC-SS cells in vitro was examined by the WST-8 and EdU assays. After treatment of H-EMC-SS cells with zaltoprofen (0–400 µmol/L) for 96 h, their growth was significantly reduced in concentration- and time-dependent manners (). To investigate PPARγ-dependency, cell growth was analyzed after downregulation of PPARγ using shRNA. The knockdown of PPARγ was confirmed using qRT-PCR (). The relative expression of PPARγ mRNA was significantly decreased in H-EMC-SSshPPARγ cells compared to that in H-EMC-SSshNT cells (p < 0.001). The growth of H-EMC-SSshPPARγ cells was significantly increased compared to that of H-EMC-SSshNT cells (, p < 0.001). Zaltoprofen (400 µmol/L) treatment significantly inhibited the growth of both H-EMC-SSshNT cells (p < 0.001) and H-EMC-SSshPPARγ cells (p < 0.001), suggesting that zaltoprofen also exerts PPARγ-independent effects on the growth of cells ().
Figure 2. (a) Effects of zaltoprofen on the viability of H-EMC-SS cells as detected by the WST-8 assay. Cells were treated with various concentrations of zaltoprofen and the absorbance was measured at each time point. (b) Confirmation of knockdown efficacy of PPARγ by qRT-PCR. PPARγ mRNA expression was analyzed in H-EMC-SSshNT and H-EMC-SSshPPARγ cells (c) PPARγ dependency of the effect of zaltoprofen on cell viability. H-EMC-SSshNT and H-EMC-SSshPPARγ cells were treated with or without zaltoprofen for 96 h before the WST-8 assay. ***p < 0.001. Error bars: ± SEM.
The EdU-positive cell ratio (EdU-incorporated cells/Hoechst 33,342-positive nuclei) was calculated, and the PPARγ-dependency of cell proliferation was confirmed by silencing PPARγ in the cells (). The EdU-positive cell ratio in H-EMC-SSshPPARγ cells was significantly higher than that in H-EMC-SSshNT cells. Zaltoprofen (200 and 400 µmol/L) significantly and concentration-dependently decreased the EdU-positive cell ratio in H-EMC-SSshNT cells. Similarly, zaltoprofen (200 and 400 µmol/L) treatment significantly reduced the EdU-positive ratio of H-EMC-SSshPPARγ cells in a concentration-dependent manner. These data suggest that proliferation of EMC cells is mediated by PPARγ and the antiproliferative effect of zaltoprofen on EMC cells is both PPARγ-dependent and -independent.
Figure 3. (a) Representative photographs of H-EMC-SSshNT and H-EMC-SSshPPARγ cells with/without zaltoprofen treatment for 24 h in the EdU proliferation assay. Scale bar = 100 µm. (b) the ratio of EdU-positive cells (the number of red nuclei [EdU-positive] per number of blue nuclei [Hoechst 33,342-positive]). *p < 0.05, **p < 0.01, ***p < 0.001. Error bars: ± SEM.
![Figure 3. (a) Representative photographs of H-EMC-SSshNT and H-EMC-SSshPPARγ cells with/without zaltoprofen treatment for 24 h in the EdU proliferation assay. Scale bar = 100 µm. (b) the ratio of EdU-positive cells (the number of red nuclei [EdU-positive] per number of blue nuclei [Hoechst 33,342-positive]). *p < 0.05, **p < 0.01, ***p < 0.001. Error bars: ± SEM.](/cms/asset/9775a6bf-c984-4a9a-b57f-2405333ed94b/kccy_a_2166195_f0003_c.jpg)
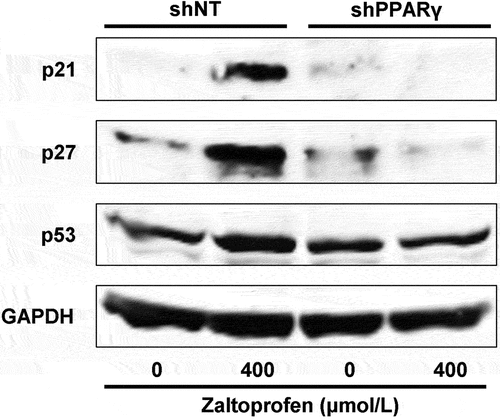
Expression of cell cycle checkpoint proteins (p21, p27, and p53) that regulate abnormal cycling of cancer cells was measured by western blot analyses in both H-EMC-SSshNT and H-EMC-SSshPPARγ cells (). Zaltoprofen (400 µmol/L) treatment induced the expression of p21, p27, and p53 in H-EMC-SSshNT cells but not in H-EMC-SSshPPARγ cells, demonstrating that zaltoprofen induced cell-cycle checkpoint proteins in a PPARγ-dependent manner in H-EMC-SS cells.
Figure 4. Effects of zaltoprofen on the expression of p21, p27, and p53 in H-EMC-SSshNT and H-EMC-SSshPPARγ cells, as detected by western blot analyses. GAPDH was used as the housekeeping protein.
Inhibitory effects of zaltoprofen on xenograft tumors in mice
The effect of zaltoprofen on xenograft tumors and its safety were tested using a xenograft mouse model of subcutaneously implanted H-EMC-SS cells. We determined the effect of 30 mg/kg zaltoprofen, which is equivalent to 400 µmol/L in vitro (the effective concentration). Zaltoprofen treatment significantly inhibited tumor growth at all time points compared with that in the vehicle group ( (a), p = 0.032). Computed tomography images of tumors and photographs of resected tumors from zaltoprofen-treated mice at the treatment end point showed decreased tumor size and increased calcification inside the tumor ().
Figure 5. (a) Tumor growth curves for the EMC xenograft model with or without zaltoprofen treatment. Line graphs indicate the tumor volume at each time point after the initiation of treatment. The tumor growth was significantly inhibited by zaltoprofen (30 mg/kg) treatment *p < 0.05, Error bars: ± SEM.(b) Photographs of the excised tumors. Scale bar = 10 mm.
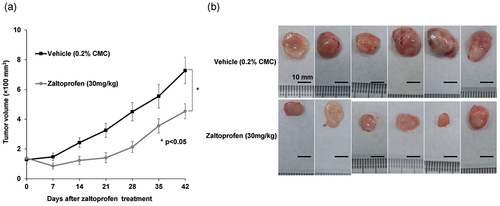
Figure 6. (a, b) Representative computed tomography images of the mice on the final day of treatment (42 days from treatment initiation). Arrows show the boundary of the tumors. (a) Vehicle (0.2% CMC, 30 mg/kg) treated group. (b) Zaltoprofen (30 mg/kg) treated group. Scale bar = 10 mm. Hematoxylin and eosin staining of the tumors in the (c) vehicle-treated and (d) zaltoprofen-treated groups. There were more necrotic scars in the zaltoprofen-treated tumor section. Scale bar = 100 µm.
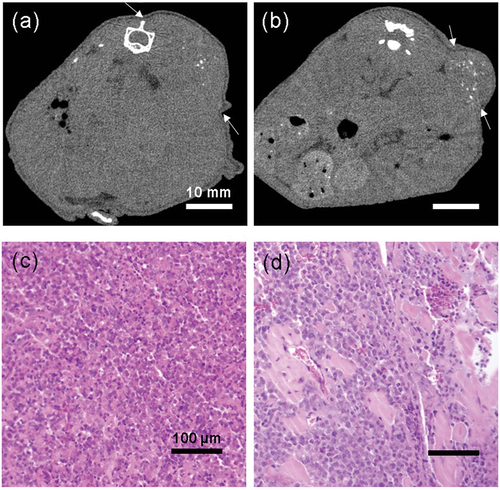
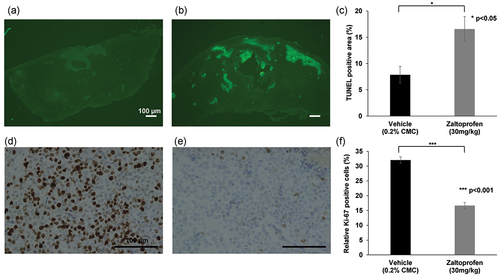
Histopathological analyses were conducted on the excised tumors. Hematoxylin and eosin-stained sections showed that tumors in the vehicle group consisted of viable highly dense tumor cells, whereas zaltoprofen-treated tumors had scattered scar areas (). There were significantly more TUNEL-positive tumor cells in the zaltoprofen-treated group than in the vehicle group (), p = 0.024). This demonstrated that zaltoprofen (30 mg/kg) induced tumor apoptosis in the H-EMC-SS xenograft model. In contrast, tumors in the zaltoprofen-treated group had significantly fewer Ki-67-positive cells compared with the vehicle group (), p < 0.001), demonstrating that zaltoprofen (30 mg/kg) inhibited the cell cycle in xenograft tumors.
Figure 7. (a, b) Representative photographs of TUNEL-stained tumors. (a) Vehicle (0.2% CMC, 30 mg/kg) group. (b) Zaltoprofen (30 mg/kg) group. Scale bar = 100 µm. (c) Bar graphs indicate the average TUNEL-positive area (percentage of fluorescent area in the tumor region). TUNEL stained areas, showing apoptotic tissue, were significantly larger in the zaltoprofen treated tumors. *p < 0.05. Error bars: ± SEM. (d, e) Representative photographs of tumors immunohistochemically stained for Ki-67 in the (d) vehicle (0.2% CMC, 30 mg/kg) and (e) zaltoprofen (30 mg/kg) groups. Scale bar = 100 µm. (f) Bar graphs indicate the percentage of Ki-67-positive cells. Ki-67-positive cells were significantly lower in the zaltoprofen treated tumors. ***p < 0.001. Error bars: ± SEM.
The body weights in both vehicle- and zaltoprofen-treated mice post-administration were higher than the pre-administration weights (p = 0.005 and p = 0.014, respectively; , suggesting natural body weight increase over 6 weeks. Generally, long-term administration of NSAIDs can cause kidney or gastrointestinal mucosa injury. However, there were no gastrointestinal damages evident upon gross observation, no abnormal renal function as evident from the absence of abnormalities in the blood levels of urea nitrogen (BUN) and serum creatinine (), and no pathological abnormalities based on renal autopsy results in both groups. In addition, no hepatotoxicity was detected in the analysis of serum total bilirubin (T-Bil) and alanine aminotransferase (ALT) in either of the groups (). There were no other observable side effects or deaths in either of the groups.
Figure 8. Changes in the pre- and post-treatment (42 days from treatment initiation) body weights of mice. Body weights of the mice significantly increased after 42 days in both groups. *p < 0.05, **p < 0.01. Error bars: ± SEM.
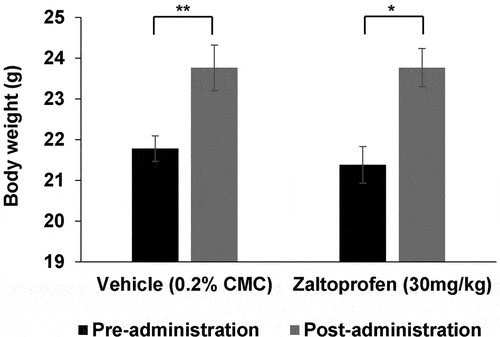
Table 1. Results from blood biochemistry tests in vehicle- or zaltoprofen-treated groups.
Discussion
We found that zaltoprofen induced the expression of PPARγ in human H-EMC-SS cells in a concentration-dependent manner by inducing KROX20, C/EBPβ, and C/EBPα in that order (). Zaltoprofen (400 µmol/L) treatment also induced expression of the cell cycle checkpoint proteins, p21, p27, and p53, and significantly inhibited the proliferation of H-EMC-SS cells (). PPARγ knockdown did not completely cancel the antitumor effect of zaltoprofen (), suggesting that zaltoprofen affects cell proliferation in both PPARγ-dependent and -independent manners. The antiproliferative effects of zaltoprofen on H-EMC-SS cells were also demonstrated in vivo for the first time () without observable side effects ( and .
The role of PPARγ in malignant tumor biology has been studied, and PPARγ ligands are known to elicit antitumor effects in cancer cells [Citation3,Citation6–8]. Several drugs, including antidiabetic and antihypertensive agents, and NSAIDs, are potential PPARγ ligands [Citation3,Citation6–8]. Among NSAIDs, indomethacin, oxaprozin, diclofenac, and zaltoprofen are potential PPARγ ligands [Citation9–11]. In the present study, we demonstrate, for the first time, that zaltoprofen induces PPARγ and elicits antitumor effects against musculoskeletal tumors, both in vitro and in vivo.
PPARγ expression has been reported to correlate with malignancy and prognosis in several cancer types [Citation12]. Downregulation of PPARγ stimulates cell proliferation in cancer cells such as esophageal carcinoma or hepatocellular carcinoma cells, suggesting that PPARγ possesses a possible protective role in cancer development [Citation13,Citation14]. In the present study, the knockdown of PPARγ by shRNA in H-EMC-SS cells significantly increased cell viability and proliferation, suggesting that PPARγ is involved in the process of malignancy in EMC. However, on the contrary, downregulation of PPARγ has been reported to inhibit cell proliferation in prostate and bladder cancers [Citation15,Citation16]. Further studies are required to elucidate the exact role of PPARγ in the oncogenesis and malignancy of cancer including EMC.
C/EBPs form a family of six homologous transcription factors that are involved in regulating cellular proliferation and differentiation, metabolism, and immunity in various organs [Citation17,Citation18]. C/EBPβ and C/EBPδ are induced early during adipogenesis and are involved in the induction of PPARγ [Citation19,Citation20]. C/EBPα is induced at a later stage of adipogenesis, which is almost the same stage at which PPARγ is induced, and is activated in mature adipocytes [Citation21,Citation22]. C/EBPα promotes adipogenesis both with and without PPARγ induction [Citation21,Citation22]. Krox20, also referred to as early growth response protein 2, a transcription regulatory factor, is an upstream regulator of C/EBPs and one of the earliest factors to be induced during adipogenesis [Citation23,Citation24]. In normal tissues such as fat tissue, some extrinsic and intrinsic factors such as dexamethasone, insulin, or 3-isobutyl-1-methylxanthine, a phosphodiesterase inhibitor which stimulates intracellular cyclic adenosine monophosphate (cAMP), have been reported to induce PPARγ via inducing Krox20 and C/EBP β and α [Citation25]. We believe that similar signal pathways may be responsible for PPARγ induction by zaltoprofen in H-EMC-SS cells. Herein, we demonstrate that, in H-EMC-SS cells, zaltoprofen induces Krox20 at the earliest stage, followed by C/EBPβ and C/EBPα, but not C/EBPδ. This pathway could be one of the mechanisms for the induction of PPARγ by zaltoprofen in H-EMC-SS cells. However, more detailed analysis to understand the relationship among these factors, including their protein expression, is needed to clarify how zaltoprofen up-regulates these proteins in tumor cells.
It has been reported that Krox20 could directly affect the apoptotic pathway mediated by p53 [Citation26]. The expression of endogenous Krox20 is significantly low in hepatocellular cancers, and the proliferation of cancer cells is suppressed by Krox20 induction [Citation26]. C/EBP α can directly inhibit cyclin-dependent kinase (CDK) 2/CDK 4 activity, and its reduced expression is correlated with poor survival in hepatocellular cancer patients [Citation17]. Biological properties of C/EBP β resemble those of C/EBP α [Citation17]. C/EBP β has been reported to be an independent predictor of overall survival in breast cancer patients [Citation27]. Whether zaltoprofen exerts its antitumor effect not only via PPARγ but also via C/EBPs and Krox20, or how these factors interact is a topic for future studies.
From the results of WST-8 assay and EdU assay in the present study, cell proliferation of H-EMC-SS cells were significantly inhibited by zaltoprofen treatment, suggesting that zaltoprofen could inhibit the cell cycle. In addition, zaltoprofen treatment up-regulated p21, p27, and p53 in a PPARγ-dependent manner demonstrated by western blot analysis, which could result in the suppression of proliferation in H-EMC-SS cells. The cyclin-dependent kinase inhibitors, p21 and p27, are essential tumor suppressor proteins that play significant roles in inhibiting the growth of cancer cells [Citation28]. It has been reported that activation of PPARγ induces cell cycle arrest through the upregulation of p21 and p27 in various cancer cells, such as hepatic cell carcinoma [Citation29–31], melanoma [Citation32], and pancreatic carcinoma [Citation33]. The tumor suppressor protein, p53, also regulates the cell cycle by repairing DNA, inducing apoptosis, and inducing p21 and p27; thus, p21, p27, and p53 interact in a complex manner to suppress the cell cycle [Citation34]. PPARγ has been reported to directly target p53, contributing to cell-cycle arrest and/or apoptosis of cancer cells [Citation35,Citation36]. Nuclear factor κB (NFκB), which is a modulator of p53, can be an intermediary between p53 and PPARγ; upregulation of PPARγ downregulates NFκB, resulting in the upregulation of p53 [Citation37]. Whether the upregulation of p53 by zaltoprofen is modulated by PPARγ directly or indirectly through the other factor, as well as the relationship between PPARγ and p53 needs to be further analyzed.
In the present study, p21, p27, and p53 were induced in H-EMC-SS cells upon zaltoprofen treatment in a PPARγ-dependent manner and inhibited their proliferation. Similarly, it has been reported that a PPARγ agonist, ciglitazone induced cell-cycle arrest in bladder cancer via upregulation of p21, p27, and p53 [Citation38]. More detailed analysis is needed to clarify the interaction between zaltoprofen-induced PPARγ expression and p21, p27, p53 expression and examination of the involvement of other mechanism in tumor cells. Flow cytometric analysis is required to elucidate the detailed antiproliferative effects of zaltoprofen on the cell cycle. These include identifying which phases of the cell cycle are involved in zaltoprofen-treated cells. In addition, the expression of the other cell-cycle related proteins that correspond to p21 and p27 such as CDK 2, CDK 4/6, and cyclin A, D and E should be studied in future. Whether zaltoprofen induces p21 or p27 in a p53-dependent or -independent manner, and PPARγ-independent antitumor effects of zaltoprofen, as evident in the WST-8 and EdU assays in the present study, require confirmation in future studies. A recent study has demonstrated that flurbiprofen and zaltoprofen platinum complexes exerted remarkable DNA damage and apoptosis with upregulation of p53 in cancer cells both in vitro and in vivo [Citation39]. In this series, inflammation in tumor tissues is restrained by the suppression of inflammasomes including cyclooxygenase (COX)-2, matrix metalloproteinase (MMP)-9, NLR family pyrin domain containing 3 (NLRP3) and caspase-1 which have been reported to promote cancer development [Citation39]. These inflammasomes may have also influenced the zaltoprofen-treated tumors in the present study.
In the present study, we demonstrated the in vivo antiproliferative effect of zaltoprofen on musculoskeletal tumor cells for the first time. The downregulation of Ki-67, a proliferation marker expressed during the active cell-cycle phases, in zaltoprofen-treated tumors indicates inhibition of the cell cycle in vivo by zaltoprofen, consistent with the in vitro results. There were more TUNEL-stained cells in zaltoprofen-treated tumors, indicating that zaltoprofen could induce apoptosis of tumor cells. The good tolerability of zaltoprofen, which was revealed by natural increase in body weight over 6 weeks and the absence of major complications including body weight loss, nephrotoxicity, and hepatotoxicity, supports further analysis on the clinical efficacy of this drug against musculoskeletal tumors. Further studies are required to determine the efficacy of zaltoprofen on normal cells and to evaluate its anti-tumorigenic effect and cell toxicity.
In conclusion, we demonstrated that zaltoprofen induces Krox20, C/EBPβ, C/EBPα, and PPARγ, and subsequently p21, p27, and p53 in H-EMC-SS cells, and contributes to the inhibition of cell proliferation. The antiproliferative effect of zaltoprofen was also demonstrated in vivo with good tolerability.
Consent to publish
Consent for publication was obtained from the participants.
Ethics approval
All research was performed in accordance with relevant guidelines and regulations. The study protocol was approved by the Kanazawa University Advanced Science Research Center (Kanazawa, Japan). All animal study procedures were performed in accordance with the Japanese national guidelines, and the protocol was approved by the Institute for Experimental Animals, Kanazawa University Advanced Science Research Center.
Author contributions
T.H. designed the research, conducted experiments, analyzed and interpreted data, and wrote the manuscript. A.T. conceived and designed the research, and approved the manuscript. S.M. and A.H. supported the experiments, interpreted data, and approved the manuscript. N.Y. and K.H. supported the experiments and approved the manuscript. Y.Y. interpreted data, and reviewed and approved the manuscript. H.T. conceived the research and approved the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article.
Additional information
Funding
References
- Paioli A, Stacchiotti S, Campanacci D, et al. Extraskeletal myxoid chondrosarcoma with molecularly confirmed diagnosis: a multicenter retrospective study within the Italian sarcoma group. Ann Surg Oncol. 2021;28(2):1142–1150.
- Kawaguchi S, Wada T, Nagoya S, et al. Extraskeletal myxoid chondrosarcoma: a multi-institutional study of 42 cases in Japan. Cancer. 2003;97(5):1285–1292.
- Higuchi T, Takeuchi A, Munesue S, et al. Anti-tumor effects of a nonsteroidal anti-inflammatory drug zaltoprofen on chondrosarcoma via activating peroxisome proliferator-activated receptor gamma and suppressing matrix metalloproteinase-2 expression. Cancer Med. 2018;7(5):1944–1954.
- Faustino-Rocha A, Oliveira PA, Pinho-Oliveira J, et al. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab Anim. 2013;42(6):217–224.
- Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013;48(3):452–458.
- Augimeri G, Gelsomino L, Plastina P, et al. Natural and synthetic PPARγ ligands in tumor microenvironment: a new potential strategy against breast cancer. Int J Mol Sci. 2020;21(24):9721.
- Ishay-Ronen D, Christofori G. Targeting cancer cell metastasis by converting cancer cells into fat. Cancer Res. 2019;79:5471–5475.
- Vallée A, Lecarpentier Y, Vallée JN. Targeting the canonical WNT/β-catenin pathway in cancer treatment using non-steroidal anti-inflammatory drugs. Cells. 2019;8:726.
- Carvalho MV, Gonçalves-de-Albuquerque CF, Silva AR. PPAR gamma: from definition to molecular targets and therapy of lung diseases. Int J Mol Sci. 2021;22:805.
- Matheson J, Le Foll B. Therapeutic potential of peroxisome proliferator-activated receptor (PPAR) agonists in substance use disorders: a synthesis of preclinical and human evidence. Cells. 2020;9:1196.
- Yamazaki R, Kusunoki N, Matsuzaki T, et al. Nonsteroidal anti-inflammatory drugs induce apoptosis in association with activation of peroxisome proliferator-activated receptor gamma in rheumatoid synovial cells. J Pharmacol Exp Ther. 2002;302:18–25.
- Ogino S, Shima K, Baba Y, et al. Colorectal cancer expression of peroxisome proliferator-activated receptor gamma (PPARG, PPARgamma) is associated with good prognosis. Gastroenterology. 2009;136:1242–1250.
- Wu K, Yang Y, Liu D, et al. Activation of PPARγ suppresses proliferation and induces apoptosis of esophageal cancer cells by inhibiting TLR4-dependent MAPK pathway. Oncotarget. 2016;7:44572–44582.
- Hsu HT, Sung MT, Lee CC, et al. Peroxisome proliferator-activated receptor γ expression is inversely associated with macroscopic vascular invasion in human hepatocellular carcinoma. Int J Mol Sci. 2016;17:1226.
- Galbraith LCA, Mui E, Nixon C, et al. PPAR-gamma induced AKT3 expression increases levels of mitochondrial biogenesis driving prostate cancer. Oncogene. 2021;40:2355–2366.
- Sanchez DJ, Missiaen R, Skuli N, et al. Cell-intrinsic tumorigenic functions of PPARγ in bladder urothelial carcinoma. Mol Cancer Res. 2021 Apr;19:598–611.
- Tolomeo M, Grimaudo S. The “Janus” role of C/EBPs family members in cancer progression. Int J Mol Sci. 2020;21:4308.
- Wang W, Xia X, Mao L, et al. The CCAAT/enhancer-binding protein family: its roles in MDSC expansion and function. Front Immunol. 2019;10:1804.
- Farmer SR. Regulation of PPARgamma activity during adipogenesis. Int J Obes (Lond). 2005;29(Suppl 1):S13–16.
- Lee JE, Cho YW, Deng CX, et al. MLL3/MLL4-associated PAGR1 regulates adipogenesis by controlling induction of C/EBPβ and C/EBPδ. Mol Cell Biol. 2020;40:e00209–20.
- Madsen MS, Siersbæk R, Boergesen M, et al. Peroxisome proliferator-activated receptor γ and C/EBPα synergistically activate key metabolic adipocyte genes by assisted loading. Mol Cell Biol. 2014;34:939–954.
- Rosen ED, Hsu C-H, Wang X, et al. C/Ebpalpha induces adipogenesis through PPARγ: a unified pathway. Genes Dev. 2002;16:22–26.
- Chen Z, Torrens JI, Anand A, et al. Krox20 stimulates adipogenesis via C/EBPbeta-dependent and -independent mechanisms. Cell Metab. 2005;1:93–106.
- Park YK, Wang L, Giampietro A, et al. Distinct roles of transcription factors KLF4, Krox20, and peroxisome proliferator-activated receptor γ in adipogenesis. Mol Cell Biol. 2017;37:e00554.
- Lee JE, Schmidt H, Lai B, et al. Transcriptional and epigenomic regulation of adipogenesis. Mol Cell Biol. 2019;39:e00601–00618.
- Zeng T, Wang D, Chen J, et al. LncRNA-AF113014 promotes the expression of Egr2 by interaction with miR-20a to inhibit proliferation of hepatocellular carcinoma cells. PLoS ONE. 2017;12:e0177843.
- Kurzejamska E, Johansson J, Jirström K, et al. C/EBPβ expression is an independent predictor of overall survival in breast cancer patients by MHCII/CD4-dependent mechanism of metastasis formation. Oncogenesis. 2014;3:e125.
- Abukhdeir AM, Park BH. P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev Mol Med. 2008;10:e19.
- Motomura W, Takahashi N, Nagamine M, et al. Growth arrest by troglitazone is mediated by p27kip1 accumulation, which results from dual inhibition of proteasome activity and Skp2 expression in human hepatocellular carcinoma cells. Int J Cancer. 2004;108:41–46.
- Rumi MA, Sato H, Ishihara S, et al. Peroxisome proliferator-activated receptor gamma ligand-induced growth inhibition of human hepatocellular carcinoma. Br J Cancer. 2001;84:1640–1647.
- Cheung K-F, Zhao J, Hao Y, et al. CITED2 is a novel direct effector of peroxisome proliferator-activated receptor γ in suppressing hepatocellular carcinoma cell growth. Cancer. 2013;119:1217–1226.
- Flori E, Rosati E, Cardinali G, et al. The α-melanocyte stimulating hormone/peroxisome proliferator activated receptor-γ pathway down-regulates proliferation in melanoma cell lines. J Exp Clin Cancer Res. 2017;36:142.
- Vitale G, Zappavigna S, Marra M, et al. The PPAR-γ agonist troglitazone antagonizes survival pathways induced by STAT-3 in recombinant interferon-β treated pancreatic cancer cells. Biotechnol Adv. 2012;30:169–184.
- Philipp-Staheli J, Kim KH, Liggitt D, et al. Distinct roles for p53, p27kip1, and p21cip1 during tumor development. Oncogene. 2004;23:905–913.
- Doonan F, Wallace DM, O’Driscoll C, et al. Rosiglitazone acts as a neuroprotectant in retinal cells via up-regulation of sestrin-1 and SOD-2. J Neurochem. 2009;109:631–643.
- Nagamine M, Okumura T, Tanno S, et al. PPAR gamma ligand-induced apoptosis through a p53-dependent mechanism in human gastric cancer cells. Cancer Sci. 2003;94:338–343.
- Mahmoud AM, Abdella EM, El-Derby AM, et al. Protective effects of turbinaria ornata and padina pavonia against azoxymethane-induced colon carcinogenesis through modulation of PPAR gamma, NF-κB and oxidative stress. Phytother Res. 2015;29:737–748.
- Plissonnier ML, Fauconnet S, Bittard H, et al. The antidiabetic drug ciglitazone induces high grade bladder cancer cells apoptosis through the up-regulation of TRAIL. PLoS ONE. 2011;6:e28354.
- Li Z, Li L, Zhao W, et al. Development of a series of flurbiprofen and zaltoprofen platinum(iv) complexes with anti-metastasis competence targeting COX-2, PD-L1 and DNA. Dalton Trans. 2022;51:12604–12619.