
ABSTRACT
Epigenetics plays an important role in the malignant progression of tumors, in which DNA methylation can alter genetic performance without altering the DNA sequence. As a key regulator demethylation, thymine-DNA glycosylase (TDG) has been reported to participate in malignant progression of multiple tumors. In this study, we demonstrate that TDG is highly expressed in hepatocellular carcinoma (HCC) and its high expression is closely related to the poor prognosis of patients. Decreasing TDG expression can significantly inhibit the malignant biological behavior of HCC cells. ABL proto-oncogene 1(ABL1) was identified as a downstream gene regulated by TDG demethylation. In addition, TDG can affect the Hippo signaling pathway through ABL1 to regulate HCC cell proliferation, apoptosis, invasion and migration. Overall, our study demonstrated that TDG reduces DNA methylation of ABL1, increases ABL1 protein expression, and affects the Hippo signaling pathway to regulate the malignant progression of HCC.
1. Introduction
Hepatocellular carcinoma (HCC) is common malignant tumors [Citation1]. High metastasis is an important biological characteristic of HCC [Citation2]. Tumor invasion and metastasis and postoperative recurrence are the main causes of death in HCC patients. Fundamentally tumors are a disease of tissue growth dysregulation, and tumor metastasis is an important manifestation of this abnormal growth regulation [Citation3]. A variety of approaches, such as surgical resection of tumor mass, liver transplantation and traditional Chinese medical science treatment, have played an important role [Citation1–4]. However, the development of HCC is often regulated by multiple factors including epigenetic changes, which is one of the most common causes of gene function changes [Citation5–7]. However, the mechanism of abnormal expression of gene in the malignant progression of HCC of is still unclear.
DNA methylation is a common epigenetic modification, which changes the cytosine at the 5 “− end of CpG dinucleotide into 5” - methylcytosine under the action of DNA methyltransferases (DNMTs), and then inhibits the normal expression of genes [Citation8]. Studies have shown that DNA methylation is a dynamic process, that can be reversed by removal of 5-methylcytosine (5mC) through DNA demethylation process, leading to reduced gene DNA methylation level and subsequent gene reactivation and expression [Citation9]. DNA demethylation can be completed through two processes: passive demethylation and active demethylation, among which active demethylation of DNA is pivotal for the whole process [Citation10]. Active DNA demethylation starts with 5mC and ends with unmodified C: TET dioxygenase can oxidize 5mC and 5-hydroxymethylcytosine (5hmc) into 5-carboxycytosine (5cac), and 5cac is recognized and digested by thymine DNA glycosylase (TDG) [Citation11]. TDG expression plays an important role in gene methylation and the occurrence and development of malignant tumors [Citation12]. Therefore, exploring the role of TDG in DNA methylation and its target genes is an important for elucidating the molecular basis of HCC development.
In this study, we aimed to investigate the expression of TDG in HCC and its relationship with the prognosis of HCC patients. The role of TDG on the malignant behavior of HCC cells (proliferation, migration and invasion) was further explored to determine the potential molecular mechanism by which TDG regulates HCC development.
2. Materials and methods
2.1 Tissue samples
A total of 99 patients with HCC samples (HCC cancer specimens and adjacent para‑carcinoma tissue) were confirmed by the Department of Hepatobiliary Surgery, with surgical treatment from June 2015 to December 2018. The primary cancer tissues and the paired normal adjacent tissues were immediately snap-frozen and stored in liquid nitrogen until use for mRNA and protein assessment. The present study was approved by the Ethics Committee of the Human Trial Ethics Committee of Guizhou Provincial People’s Hospital. The present study was performed in accordance with the principles outlined in the Declaration of Helsinki. Written informed consent was obtained from the patients who provided the specimens.
2.2 Cell culture
The Human HCC cell lines Hep3B, Huh-7, MHCC-97 H, MHCC-97 L, and human normal hepatocyte THLE2 were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All cells were cultured in high glucose DMEM with 10% fetal bovine serum (FBS). Cells cultured in a closed incubator with constant temperature of 37°C and 5% CO2.
2.3 Plasmid constructs and transfection
Short hairpin RNA (shRNA) against TDG (De-TDG), ABL1 overexpression plasmid (In-ABL1), and the control vector and control shRNA were purchased from Shanghai Jima Company (Shanghai, China), Transfection of cell lines was performed with Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s instructions.
2.4 Reverse transcription‑quantitative PCR
Total RNA from HCC tissues, adjacent para‑carcinoma tissue and cell lines using TRIzol reagent (TaKaRa, Japan). PrimeScript RT reagent (TaKaRa, Japan) was used for reverse transcription of cDNA, and RT–qPCR was performed with SYBR Premix Ex Taq II (TaKaRa) using a LightCycler system (Roche). GAPDH as an internal reference and the TDG mRNA expression level was analyzed using the 2−ΔΔCt method.
2.5 Western blotting
Total protein from HCC tissues, adjacent para‑carcinoma tissue and cell lines were extracted mixed solution of RIPA buffer (Beyotime Institute of Biotechnology) and PMSF (Beyotime Institute of Biotechnology) (100:1). An equal target proteins (25 μg) was separated via SDS‑PAGE on a 12% gel and transferred to PVDF membranes. The membranes were blocked with 5% non‑fat dried milk at room temperature for 2 h. The membranes were probed at 4°C overnight with primary antibodies ABL1 (1:2000, Abcam), TDG (1:1500, Abcam), YAP (1:1000, Abcam), p-YAP (1:1000, Abcam), N-cadherin (1:2000, Abcam), Vimentin (1:2000, Abcam), Bax (1:2000, Abcam), Bcl-2 (1:2000, Abcam), E-cadherin (1:1000, Abcam), Ki-67 (1:1000, Abcam) and GAPDH (1:5000, Abcam). The membranes were then incubated with the appropriate secondary antibodies. GAPDH was used as an internal reference. Finally, protein expression was analyzed by chemiluminescence reagents (Hyperfilm ECL, USA). Image J (National Institutes of Health, USA) software was used to analyze the Western blotting results.
2.6 Immunohistochemistry
Tissue microarrays were prepared from HCC tissue samples. Tissue sections were rehydrated in xylene and alcohol followed by 3% H2O2 for 30 min at 37°C. All sections were incubated for 15 min with goat serum to block nonspecific binding, followed by incubation with TDG (1: 500, Abcam) at 4°C overnight. Then, the sections incubated with anti-rabbit secondary IgG antibody at 37°C for 30 minutes. The visualization signal was achieved using diaminobenzidine (DAB, Boster).
TDG staining classifications were as follows: 0, negative; 1, weak; 2, moderate; and 3, strong (range 0–3). The percentage of positive cells range 0–4: 0, negative or <5%; 1, 6%–25%; 2, 26%–50%; 3, 51%–75%; and 4, 76%–100%. The percentage of positive cells and the intensity to determine the final staining scores. Grades <4 were defined as low TDG expression, while grades ≥4 were defined as high TDG expression [Citation13].
2.7 Public data acquisition and analysis
TDG expression profile of HCC patients were downloaded from TCGA database with corresponding clinical information (https://portal.gdc.cancer.gov/repository). Additionally, the mRNA expression of TDG was also download from GEO database (GSE10143, GSE14520)(https://www.ncbi.nlm.nih.gov/geo/). All these data were processed using R studio software (R version 3.6.3, Boston, MA, USA). The expression of TDG was analyzed by GraphPad Prism 9.0 (GraphPad, San Diego, CA, USA). For gene set enrichment analysis (GSEA), the online website tool LinkedOmics (http://www.linkedomics.org/login.php) was used as described.
2.8 CCK-8 assay
Briefly, 100 μL of cell suspension (1 × 103 cells) was added in each well of 96 well plates. Then,10 μL of CCK-8 solution (CCK-8; Dojindo Laboratories, Kumamoto, Japan) was added to each well. The 96-well plates into the cell incubator and incubated for 1 hour. Finally, the absorbance of each well at 450 nm was detected with a microplate reader.
2.9 Flow cytometry analysis of cell apoptosis
After transfection, HCC cells were seeded into 6 well plates. Once they reached confluence, cells were collected and incubated with Annexin V‑FITC (5 μl; Biogot Technology Co., Ltd.) and propidium iodide (PI) solution (5 μl; Biogot Technology Co., Ltd.) at room temperature for 10 min according to the manufacturers’ instructions. Cells were subsequently suspended in 500 μl binding buffer.
2.10 5-Ethynyl-2′-deoxyuridine (EdU) assay
After transfection, HCC cells were seeded into 96-well plates and cultured for 24 h. Fixed in 4% formaldehyde for 30 min. Then, EdU (50 μM) for 2 h at 37°C, and permeabilized with 0.5% Triton X-100 solution for 10 min at room temperature. After two washes with PBS twice, 1× ApolloR reaction cocktail (100 μL) was added, and the reaction proceeded for 30 min at room temperature in the dark. Cell nuclei were stained by the addition 1× Hoechst 33,342 (100 μL) for 30 min. Cell proliferation was analyzed using the mean number cells in three fields for each sample.
2.11 Wound healing assay
After transfection, HCC cells were grown in six-well plates. After reaching confluence, the cell monolayer was scratched with a pipette tip (1 ml) to generate 3 scratch wounds and then rinsed twice with PBS to remove non-adherent cells. After 0 and 48 h, the distance between the wound sides was measured.
2.12 Cell invasion assays
After transfection, HCC were seeded onto the upper chamber. Transwell chambers (Corning Costar, Cambridge, MA, USA) were used to detect cell invasion. The inserts were precoated with Matrigel (1 mg/mL), and 500 ul of high-glucose DMEM containing 10% fetal bovine serum (FBS) was added to the matched lower chamber. After incubation for 48 h, the transwell chambers were fixed in methanol and stained with 0.1% crystal violet. Finally, the cells were counted under an inverted optical microscope.
2.13 Bisulfite Sanger sequencing (BSP)
500ng of genomic DNA extracted from HCC tissues (tumor tissues and adjacent para‑carcinoma tissue) was bisulfite converted using a MethylCode™ Bisulfite Conversion kit (Applied Biosystems; Thermo Fisher Scientific, Inc.). The ABL1 promoter was amplified via PCR with Taq DNA Polymerase (Invitrogen; Thermo Fisher Scientific, Inc.). The primer sequence was designed using Methyl Primer Express™ Software v1.0 (Applied Biosystems; Thermo Fisher Scientific, Inc.). The PCR products were electrophoresed, purified using Spin‑X tubes, and then cloned into a pUC ‑T vector (both from CWbiotech). Ten single products were sequenced for each sample.
2.14 DNA extraction and methylation‑specific PCR
DNA was isolated from HCC tissues (tumor and paired normal adjacent tissues) and HCC cell lines by using a DNA Isolation kit (Tiangen). Bisulfite conversion was performed using the EpiTect Bisulfite Kit (Qiagen Inc.). Methylation‑specific PCR (MSP) was performed with 2 μl of bisulfite‑modified DNA (100 ng/50 μl) and 48 μl of PCR mixture consisting of 10× PCR Buffer (Mg2+ free), 25 mM MgCl2, dNTP mixture (each 2.5 mM), sense primer (20 μM), antisense primer (20 μM), and Takara EpiTaq HS (5 U/μl; Takara). PCR amplification was conducted using 40 cycles (98°C for 10 s, 55°C for
30 s, and 72°C for 30 s). For parallel quality control, a plasmid containing a methylated ABL1 sequence and water without DNA template were used as positive and negative controls, respectively.
2.15 Tumorigenesis assay
Twelve male BALB/c nude mice (5-weeks-old) were purchased from the Shanghai Experimental Animal Center (Shanghai, China) and fed at the Animal Center of Guizhou Provincial People’s Hospital. All animal experiments were performed in accordance with institutional guidelines and were approved by the Committee on the Ethics of Animal Experiments of Guizhou Provincial People’s Hospital. BALB/c nude mice were house with free food and water under specific‑pathogen‑free conditions at 21‑25°C with 40‑70% humidity and 12/12‑h light/dark cycle. HCC cells (1 × 106 cells/ml) were subcutaneously injected into the flank of each nude mouse. Tumor volume calculated formula: volume = (width2 × length)/2. Tumor diameter was measured with Vernier callipers every 3 days after tumor emergence (10 days after inoculation) to measure tumor volume. At 27 days after injection, the mice were euthanized and tumors were collected for analysis. The mice were anaesthetized with 1% pentobarbital sodium (45 mg/kg, intraperitoneal) and subsequently euthanized by cervical dislocation (when the heart had stopped completely, the mouse was determined to be dead). Body weight loss >20% was considered a humane endpoint for euthanasia.
2.16 Statistical analyses
All data are presented as the means ± SD. Statistical data were analyzed using SPSS 20.0 software (SPSS Inc., Chicago, IL, USA) or GraphPad Prism version 6.0 (CA, USA). A paired Student’s t test was used to analyze the significant difference in TDG expression in HCC tissues. Analysis of variance (ANOVA) was used for multiple group comparisons. Tukey’s test was used as the post hoc test after ANOVA. Associations between clinicopathological parameters and TDG expression were analyzed using Pearson’s chi‑squared test. Kaplan‑Meier survival analysis and Cox regression assays were used to analyze the prognostic significance of TDG (overall survival recorded as the time from liver tumor resection as complete as possible to patient death, recurrence-free survival recorded as the time from liver tumor resection as complete as possible to liver tumor recurrence). The threshold for statistical significance was p < 0.05.
3. Results
3.1 TDG expression is upregulated in HCC
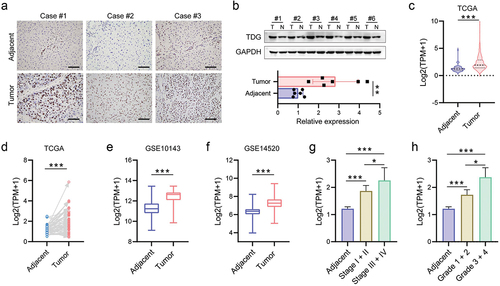
To investigate the role of TDG in HCC development, we firstly evaluated the expression of TDG via immunohistochemistry, and the results showed that TDG was highly expressed in HCC (). Next, WB results confirmed that TDG was significantly increased in HCC cancer specimens compared with adjacent tissue (). Through TCGA and GEO databases, we found that TDG was significantly increased in HCC cancer specimens ().
Figure 1. TDG expression was upregulated in HCC. (a) Immunohistochemical analysis of TDG expression in HCC. (b) TDG protein expression in HCC. (c-f) TDG expression in TCGA and GEO database. (g) Association between TDG and stage in HCC patients. (h) Association between TDG and grade in HCC patients. *P<.05, **P<.01, ***P<.001. Scale bars, 100 mm.
3.2 TDG expression is associated with poor prognosis in HCC
Additionally, according to TCGA database, the expression of TDG was higher in advanced HCC patients with higher tumor stage and grade (). Kaplan–Meier survival analysis based on TCGA data showed that HCC patients with high TDG expression had poor OS, DSS and RFS (). To further evaluate the prognostic value of TDG in HCC patients, IHC staining of TDG was carried out on TMA slides. The tissue microarray results reconfirmed that TDG is highly expressed in HCC (). HCC patient clinical data showed that TDG expression was positively correlated with TNM stage (P = 0.001) and tumor size (P = 0.004) (). Moreover, Kaplan – Meier survival analysis revealed that HCC patients with high TDG expression showed significantly shorter overall survival (). Next, univariate analysis indicated that TNM stage (P = 0.027), tumor size (P = 0.020) and TDG (P = 0.011) were significantly associated with overall survival in HCC (). Multivariate analysis suggested that TNM stage (P = 0.021), tumor size (P = 0.023) and TDG (P = 0.014) were independent prognostic factors for overall survival in HCC (). Similar results were found in recurrence-free survival of HCC patients (, ).
Figure 2. The relationship between TDG expression and prognosis in HCC. (a) Association between TDG and OS, DSS and PFS of HCC patients. (b) Immunohistochemical analysis of TDG expression in HCC. (c) Kaplan–Meier analysis of overall survival and recurrence-free survival between in HCC patients with high and low TDG expression. Scale bars, 100 mm.
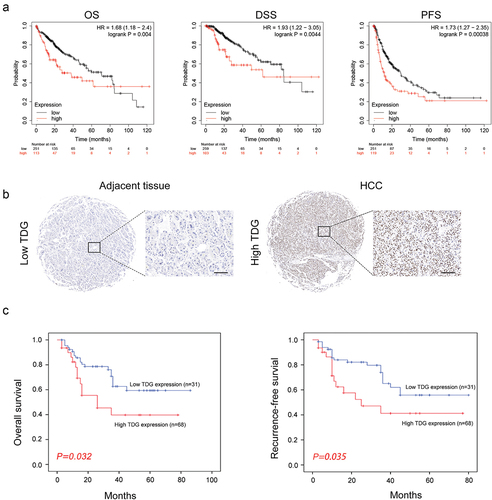
Table 1. Correlations between TDG and clinicopathological features of HCC patients.
Table 2. Univariate and multivariate analysis of different prognostic variables influencing overall survival in HCC patients.
Table 3. Univariate and multivariate analysis of different prognostic variables influencing recurrence-free survival in HCC patients.
3.3 A decrease in TDG weakens the malignant behavior of HCC cells
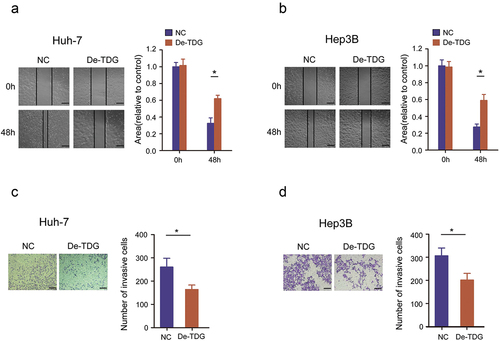
To explore the role of TDG in HCC development, gain-of-function experiments were performed in Huh-7 and Hep3B cell lines. WB results confirmed that TDG was effectively inhibited in Huh-7 and Hep3B (). CCK-8 assay results showed that decreasing the expression of TDG significantly inhibited the proliferation of Huh-7 and Hep3B (). EdU incorporation assays revealed that TDG downregulation significantly inhibited DNA replication in Huh-7 and Hep3B (). In addition, flow cytometry showed that decreased TDG expression significantly promoted apoptosis in Huh-7 and Hep3B (). Finally, wound healing assays and transwell assays revealed that cell mobility and migration of Huh7 and Hep3B cells were significantly impaired by TDG silencing ().
Figure 3. Decreased TDG significantly suppressed cell proliferation and promoted apoptosis in HCC cell lines. (a) Western blot assay showing the TDG protein expression level in TDG decreased (De-TDG) Huh-7 cells. (b) Western blot assay showing the TDG protein expression level in TDG decreased (De-TDG) Hep3B cells. (c) CCK-8 assay to detect proliferation in Huh-7 cells. (d) CCK-8 assay to detect proliferation in Hep3B cells. (e) EdU assays were used to analyze the effect of TDG on Huh-7 cell proliferation. Scale bars, 50 μm. (f) EdU assays were used to analyze the effect of TDG on Hep3B cell proliferation. Scale bars, 50 μm. (g) Flow cytometry was used to examine the effects of TDG on Huh-7 cell apoptosis. (h) Flow cytometry was used to examine the effects of TDG on Hep3B cell apoptosis. *P<.05.
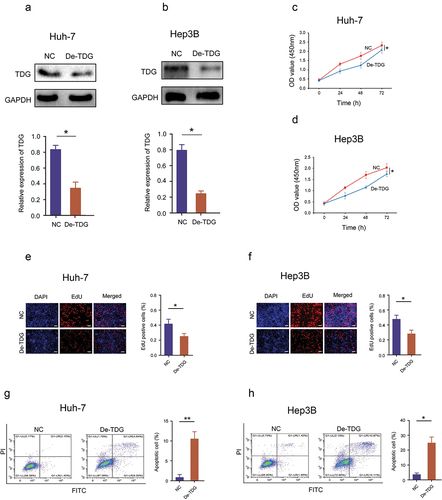
Figure 4. Decreased TDG significantly suppressed the cell invasion and migration in HCC cell lines. (a) Wound healing assays were performed to determine the effects of TDG on Huh-7 cell migration. Scale bars, 50 μm. (b) Wound healing assays were performed to determine the effects of TDG on Hep3B cell migration. Scale bars, 50 μm. (c) Transwell assays were performed to determine the effects of TDG on Huh-7 cell invasion. Scale bars, 100 μm. (d) Transwell assays were performed to determine the effects of TDG on Hep3B cell invasion. Scale bars, 100 μm. *P<.05.
3.4 Hypomethylation of ABL1 promoter CpG island contributes to high ABL1 expression in HCC
Bioinformatics correlation analysis showed a significant positive correlation between TDG and ABL1 (). ABL1 was reported to significantly promote HCC development, and was found to be regulated by DNA methylation. The methylation status of ABL1 was assayed in randomly selected HCC cancer specimens and adjacent para‑carcinoma tissue. ABL1 promoter methylation was lower HCC tissues than in adjacent para‑carcinoma tissue (). Next, bisulfite sequencing‑PCR (BSP) was used to analyze the specific methylation of CpG island of ABL1, and the results showed that ABL1 promoter methylation was lower in HCC tissues than in adjacent tissues (). In addition, analysis of THLE2, MHCC‑97 H, Hep3B MHCC‑97 L, and Huh‑7 cell lines revealed a significantly low degree of methylation in the HCC cell lines compared with normal TELE 2 cells (). Correspondingly, ABL1 was highly expressed in HCC cell lines (). Similarly, we found ABL1 was significantly increased in HCC tissues ().
Figure 5. Aberrant DNA hypermethylation of ABL1 and aberrant expression of ABL1 in HCC. (a, b) Correlation analysis between TDG and ABL1. (c) Bisulfite sequencing analysis of ABL1 promoter methylation in randomly detected in HCC samples. (d) the methylation status of ABL1 was randomly detected in HCC. (e) the methylation status of ABL1 was detected in HCC cell lines. (f) ABL1 protein expression in HCC. (g) ABL1 protein expression in HCC cell lines. *P<.01. Black dots, methylated; white dots, unmethylated.
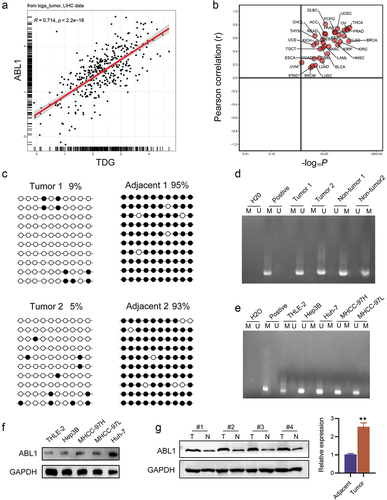
3.5 TDG changes ABL1 protein expression of by regulating the DNA methylation
Immunoblotting assay results demonstrated that TDG silencing decreased the expression of ABL1 (). Consistently, the results of cellular immunofluorescence staining revealed that the protein expression of ABL1 was dramatically reduced after TDG knockdown (). More importantly, methylation level of ABL1 was significantly elevated in TDG silenced Hep3B and Huh7 cells ().
Figure 6. TDG regulates ABL1 expression through DNA methylation. (a) ABL1 protein expression after TDG expression was decreased in Huh-7 cells. (b) ABL1 protein expression after TDG expression was decreased in Hep3B cells. (c) TDG and ABL1 protein expression was detected by immunofluorescence after TDG expression was decreased in Huh-7 cells. Scale bars, 50 µm. (d) the methylation status of ABL1 after TDG expression was decreased in Huh-7 cells. (e) TDG and ABL1 protein expression was detected by immunofluorescence after TDG expression was decreased in Hep3B cells. Scale bars, 50 µm. (f) the methylation status of ABL1 after TDG expression was decreased in Hep3B cell. *P<.05.
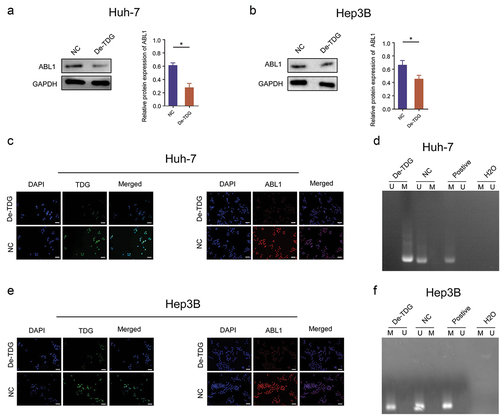
3.6 ABL1 compensates for the inhibitory effect of TDG on HCC malignant behavior in vitro and in vivo
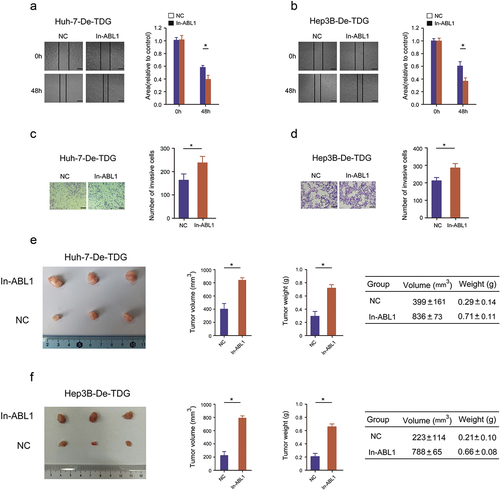
A cell function experiment was performed to confirm whether TDG affects the malignant biological behavior of HCC. ABL1 expression was increased by lentivirus infection in Huh-7-De-TDG and Hep3B- De-TDG cells (). The increase of ABL1 expression significantly abolished the inhibitory effects of TDG on proliferation in Huh-7-De-TDG and Hep3B- De-TDG (). Moreover, the inhibitory effects of TDG silencing on EdU incorporation the promoting effects on cell apoptosis were reversed after ABL1 elevation (). In addition, forced ABL1 expression significantly undermined the inhibitory effects of TDG on migration and invasion in Huh-7-De-TDG and Hep3B-De-TDG (). More importantly, forced ABL1 expression significantly abolished the inhibitory effects of TDG on tumorigenicity in Huh-7-De-TDG and Hep3B- De-TDG ().
Figure 7. Rescue experiments are performed to confirm that ABL1 ameliorates the inhibitory effect of TDG on HCC cell proliferation and apoptosis. (a) ABL1 protein expression after ABL1 expression was increased in Huh-7-De-TDG. (b) ABL1 protein expression after ABL1 expression was increased in Hep3B-De-TDG cells. (c) CCK-8 assay to detect the proliferation in Huh-7-De-TDG cells with increased ABL1 expression. (d) CCK-8 assay to detect the proliferation of Hep3B-De-TDG cells with increased ABL1 expression. (e) the uptake capacity of EdU was analyzed in Huh-7-De-TDG cells with increased ABL1 expression. Scale bars, 50 μm. (f) the uptake capacity of EdU was analyzed in Hep3B-De-TDG cells with increased ABL1 expression. Scale bars, 50 μm. (g) Flow cytometry was used to detect apoptosis in Huh-7-De-TDG cells with increased ABL1 expression. (h) Flow cytometry was used to detect apoptosis in Hep3B-De-TDG cells with increased ABL1 expression. *P<.05.
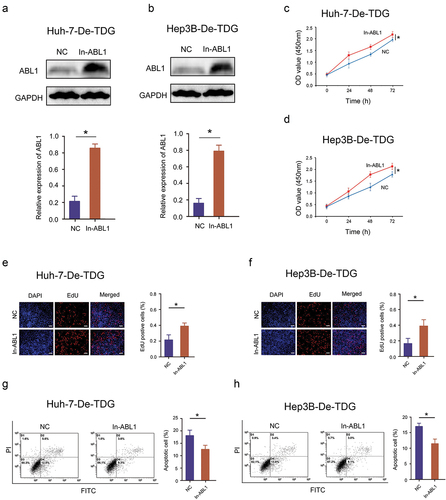
Figure 8. Rescue experiments are performed to confirm that ABL1 ameliorates the inhibitory effect of TDG on HCC tumorigenicity, invasion and migration. (a) Wound healing assays were performed to determine the migration in Huh-7-De-TDG cells with increased ABL1 expression. Scale bars, 50 μm. (b) Wound healing assays were performed to determine the migration of Hep3B-De-TDG cells with increased ABL1 expression. Scale bars, 50 μm. (c) Transwell assays were performed to determine the invasion of Huh-7-De-TDG cells with increased ABL1 expression, Scale bars, 100 μm. (d) Transwell assays were performed to determine the invasion in Hep3B-De-TDG cells with increased the expression of ABL1. Scale bars, 100 μm. (e) Animal model to verify the effect of overexpression on tumor formation induced by Huh-7-De-TDG cells. (f) Animal model to verify the effect of overexpression on tumor formation induced by Hep3B-De-TDG cells.*P<.05.
3.7 TDG regulates Hippo signaling pathway and the expression of proliferation and apoptosis-related factors
GSEA showed that genes with similar expression pattern with TDG were extensively enriched or involved in Hippo signaling pathway (). Furthermore, increase ABL1 expression significantly inhibited the expression of p-YAP, Bax and E-cadherin but significantly promoted the expression of YAP, Bcl-2, N-cadherin, Vimentin and Ki-67 in Huh-7-De-TDG and Hep3B- De-TDG (). This evidence indicates that high TDG expression promotes HCC development by regulating ABL1 expression, which further inhibits Hippo signaling pathway ().
Figure 9. TDG and ABL1 affect malignant progression by regulating EMT through Hippo signaling pathway. (a-d) Bioinformatics analysis of the relationship between TDG, ABL1 and downstream signaling pathways. (e) p-YAP, YAP, Bcl-2, Bax, E-cadherin, N-cadherin, Vimentin and Ki67 protein expression in Huh-7-De-TDG cells with increased ABL1 expression. (f) p-YAP, YAP, Bcl-2, Bax, E-cadherin, N-cadherin, Vimentin and Ki67 protein expression in Hep3B-De-TDG cells with increased ABL1 expression of. *P<.05.
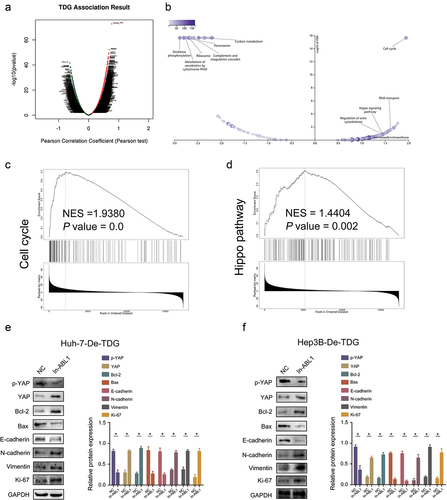
Figure 10. Schematic illustration of TDG promotes HCC development by regulating ABL1, which further inhibits Hippo signaling pathway.
4. Discussion
ABL1 is a protein kinase that regulates key processes related to cell growth and survival [Citation14]. ABL1 regulates cytoskeletal remodeling during cell differentiation, cell division and cell adhesion [Citation15]. ABL1 localizes to dynamic actin structures and can phosphorylate CRK, CRKL, DOK1, and other proteins that control cytoskeletal dynamics [Citation16]. When DNA damage is too severe to repair, ABL1 can potentially regulate DNA repair by activating proapoptotic pathways [Citation17]. In colorectal cancer, deletion of ABL1 inhibits the expression of TGF-β1 through the IRS1/PI3K/AKT pathway, thereby regulating the malignant progression of colorectal cancer [Citation15]. In addition, ABL1 expression in HCC tissues was higher than that in nontumor liver tissues, which was associated with shorter survival time of patients [Citation18]. Deletion of ABL1 reduces HCC cell proliferation and slows liver tumor growth in mice, and its molecular mechanisms are linked to MYC and NOTCH1 [Citation18]. More importantly, DNA methylation plays a significant role in regulating ABL1 expression and influences various cell activities in blood (such as cell proliferation and survival) and the transmission of cell signal molecules in chronic myeloid leukemia [Citation19]. Our findings are consistent with previous findings, and more importantly, we found that ABL1 is modified by DNA methylation regulated by TDG, and through its regulation of ABL1 protein expression, it affects the malignant progression of HCC [Citation20].
5-Methylcytosine (5mC) is a stable epigenetic modification in mammalian genomic DNA [Citation10]. Hypermethylation in the promoter region indicates the silencing of gene expression. However, recent studies have found that 5mC can be oxidized into 5hmc, 5FC and 5cac by Tet family proteins [Citation11]. 5FC and 5cac can be excised by the DNA glycosidase TDG, triggering the base excision repair pathway to complete DNA demethylation [Citation21]. Because DNA demethylation can reactivate silenced genes, oxidative demethylation of DNA by TDG has been revealed as a key step in the process of cell reprogramming. Thymine DNA glycosidase (TDG) is a DNA demethylase that is transactivated by c-myc after insulin treatment, thereby reducing the abundance of 5-carboxycytosine (5cac) in SREBP1 promoter [Citation22]. At the same time, metformin-activated AMP-activated protein kinase (AMPK) increased the activity of DNA methyltransferase 3A (DNMT3A), thereby increasing the abundance of 5-methylcytosine (5mC) in TDG promoter [Citation22]. These results suggest that TDG is an epigenetic therapeutic target for type 2 diabetes related cancer [Citation22]. By binding with DNMT3A, TDG promotes its ubiquitination and degradation and thus inhibits the migration and invasion of human colon cancer cells through the DNMT3A-TIMP2 axis [Citation23]. In addition, TDG can remove the common DNA damage substance thymidine diol caused by oxidative stress by relying on guanine [Citation24]. TDG can specifically act on DNA mismatches caused by deamination of 5-methylcytosine and oxidative coupling deamination, and regulate the transition from normal cells to tumor cells by the above means [Citation24]. H2A. Z/H3.3 dual variant nucleosomes and the pioneering transcription factor Fork Head Box A1 (FOXA1) transcription factor are both involved in the formation of chromatin landscapes during tissue specific enhancer demethylation, regulating TDG activity on chromatin in the aforementioned manner [Citation25]. Our study showed the DNA methylation of ABL1 is regulated by TDG. TDG can inhibit the malignant biological behavior of HCC by increasing ABL1 protein expression.
Epithelial mesenchymal transition (EMT) refers to the phenotypic transformation of epithelial cells to mesenchymal cells under specific physiological or pathological conditions [Citation26]. In recent years, EMT has been verified to play an important role in tumor metastasis. EMT occurs at the initial stage of tumor metastasis [Citation27]. This process leads to loss of the expression of E-cadherin, claudin, occludin and other connective molecules in epithelial cells; destroys cell polarity; promotes the expression of certain lytic enzymes involved in degradation of the extracellular matrix and basement membrane, such as matrix metalloproteinases (MMPs); destroys the histological barrier, thereby cell invasion and facilitates separation of tumor cells from the primary tumor for invasion and metastasis [Citation28]. At the same time, EMT can also promote the production of cancer stem cells (CSCs), which are considered to be the basis of tumor invasion, metastasis and invasive growth [Citation29]. In HCC, cancer promoting molecules induce EMT in tumor cells by activating Notch and Wnt signaling pathways upstream of EMT, or promote EMT by regulating the activity of EMT related transcription factors ZEB2 and snail II [Citation30–32]. At the same time, researchers found that EMT can enable HCC cells to obtain stem cell characteristics, thus promoting the production of HCC tumor stem cells and accelerating the invasion and metastasis of tumor cells [Citation33]. Hippo signaling pathway is one of the most important molecular regulatory networks composed of a series of protein kinases and transcription factors [Citation34]. Hippo signaling pathway is highly conserved from lower animals to higher animals [Citation35]. The Hippo pathway involved in inhibition cell proliferation and promotion cell apoptosis and mediates the dynamic balance between cell proliferation and apoptosis [Citation36,Citation37]. Our study found that TDG regulates ABL1 protein expression through DNA methylation and regulates EMT in HCC cells through the Hippo signaling pathway. It has been reported that TDG can be regulated by wild type P53 and c-Myc [Citation22,Citation38], and as one of the downstream targets of microRNA-29a, TDG can be suppressed or silenced by microRNA-29a [Citation39]. However, based on the TCGA public data, we didn’t find any correlation between TDG and TP53, Myc and microRNA-29a. In fact, we do not know the reason for the TDG increase, which has been listed in our future research plan. In our future research, we will focus on the reasons for the high expression of TDG in HCC, whether there are more transcription factors that regulate the expression of TDG, and whether TDG plays more roles in HCC.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Ethics statement
The study was conducted with the approval of the Ethics Committee of Guizhou Provincial People’s Hospital. Written informed consent was obtained from all patients for the use of their tissue in this study.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data sets used and analyzed during the current study are available from the corresponding author on reasonable request.
Additional information
Funding
References
- Hatzaras I, Bischof DA, Fahy B, et al. Treatment options and surveillance strategies after therapy for hepatocellular carcinoma. Ann Surg Oncol. 2014;21(3):758–766.
- Couri T, Pillai A. Goals and targets for personalized therapy for HCC. Hepatol Int. 2019;13(2):125–137.
- Tang KY, Du SL, Wang QL, et al. Traditional Chinese medicine targeting cancer stem cells as an alternative treatment for hepatocellular carcinoma. J Integr Med. 2020;18(3):196–202.
- Zhao HT, Meng YB, Zhai XF, et al. Comparable effects of Jiedu Granule, a compound Chinese herbal medicine, and sorafenib for advanced hepatocellular carcinoma: a prospective multicenter cohort study. J Integr Med. 2020;18(4):319–325.
- Wang JJ, Lei KF, Han F. Tumor microenvironment: recent advances in various cancer treatments. Eur Rev Med Pharmacol Sci. 2018;22(12):3855–3864.
- Stoletov K, Beatty PH, Lewis JD. Novel therapeutic targets for cancer metastasis. Expert Rev Anticancer Ther. 2020;20(2):97–109.
- Kang GJ, Park JH, Kim HJ, et al. PRR16/Largen induces epithelial-mesenchymal transition through the interaction with ABI2 leading to the activation of ABL1 kinase. Biomol Ther. 2020;30(4):340–347.
- Edwards JR, Yarychkivska O, Boulard M, et al. DNA methylation and DNA methyltransferases. Epigenetics Chromatin. 2017;10(1):23.
- Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 2017;18(9):517–534.
- Greenberg MVC, Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20(10):590–607.
- Bochtler M, Kolano A, Xu GL. DNA demethylation pathways: additional players and regulators. BioEssays. 2017;39(1):1–13.
- Onabote O, Hassan HM, Isovic M, et al. The role of thymine DNA glycosylase in transcription, active DNA demethylation, and cancer. Cancers (Basel). 2022;14(3):765.
- Wu H, Qiu J, Wu Z, et al. MiR-27a-3p binds to TET1 mediated DNA demethylation of ADCY6 regulates breast cancer progression via epithelial-mesenchymal transition. Front Oncol. 2022;12:957511.
- Braun TP, Eide CA, Druker BJ. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell. 2020;37(4):530–542.
- Chiaretti S, Messina M, Foà R. BCR/ABL1-like acute lymphoblastic leukemia: how to diagnose and treat? Cancer. 2019;125(2):194–204.
- Massimino M, Stella S, Tirrò E, et al. ABL1-directed inhibitors for cml: efficacy, resistance and future perspectives. Anticancer Res. 2020; 40(5):2457–2465.
- Luo J, Zheng H, Wang S, et al. ABL1 and Cofilin1 promote T-cell acute lymphoblastic leukemia cell migration. Acta Biochim Biophys Sin (Shanghai). 2021;53(10):1321–1332.
- Wang F, Hou W, Chitsike L, et al. ABL1, overexpressed in hepatocellular carcinomas, regulates expression of notch1 and promotes development of liver tumors in mice. Gastroenterology. 2020;159(1):289–305.
- Behzad MM, Shahrabi S, Jaseb K, et al. Aberrant DNA methylation in chronic myeloid leukemia: cell fate control, prognosis, and therapeutic response. Biochem Genet. 2018;56(3):149–175.
- Liu Y, Cao J, Zhu YN, et al. C1222C deletion in exon 8 of ABL1 is involved in carcinogenesis and cell cycle control of colorectal cancer through IRS1/PI3K/Akt pathway. Front Oncol. 2020;10:1385.
- Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502(7472):472–479.
- Yan JB, Lai CC, Jhu JW, et al. Insulin and metformin control cell proliferation by regulating TDG-Mediated DNA demethylation in liver and breast cancer cells. Mol Ther Oncolytics. 2020;18:282–294.
- Miao J, Zhao C, Tang K, et al. TDG suppresses the migration and invasion of human colon cancer cells via the DNMT3A/TIMP2 axis. Int J Biol Sci. 2022;18(6):2527–2539.
- Yoon JH, Iwai S, O’Connor TR, et al. Human thymine DNA glycosylase (TDG) and methyl-CpG-binding protein 4 (MBD4) excise thymine glycol (Tg) from a Tg: g mispair. Nucleic Acids Res. 2003;31(18):5399–5404.
- Deckard CE, Banerjee DR, Sczepanski JT. Chromatin structure and the pioneering transcription factor FOXA1 regulate TDG-Mediated removal of 5-formylcytosine from DNA. J Am Chem Soc. 2019;141(36):14110–14114.
- Nieto MA, Huang RY, Jackson RA, et al. Emt: 2016. Cell. 2016;166(1):21–45.
- Ye X, Weinberg RA. Epithelial–mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 2015;25(11):675–686.
- Pastushenko I, Blanpain C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019;29(3):212–226.
- De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110.
- Xiao S, Chang RM, Yang MY, et al. Actin-like 6A predicts poor prognosis of hepatocellular carcinoma and promotes metastasis and epithelial-mesenchymal transition. Hepatology. 2016;63(4):1256–1271.
- Jiang L, Yang YD, Fu L, et al. CLDN3 inhibits cancer aggressiveness via Wnt-EMT signaling and is a potential prognostic biomarker for hepatocellular carcinoma. Oncotarget. 2014;5(17):7663–7676.
- Dong T, Zhang Y, Chen Y, et al. FOXO1 inhibits the invasion and metastasis of hepatocellular carcinoma by reversing ZEB2-induced epithelial-mesenchymal transition. Oncotarget. 2017;8(1):1703–1713.
- Liu F, Kong X, Lv L, et al. TGF-beta1 acts through miR-155 to down-regulate TP53INP1 in promoting epithelial-mesenchymal transition and cancer stem cell phenotypes. Cancer Lett. 2015;359(2):288–298.
- Ma S, Meng Z, Chen R. The hippo pathway: biology and pathophysiology. Annu Rev Biochem. 2019;88:577–604.
- Moya IM, Halder G. Hippo–yap/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol. 2019;20(4):211–226.
- Zheng Y. The Hippo Signaling Pathway in Development and Disease. Dev Cell. 2019;50(3):264–282.
- Dey A, Varelas X. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat Rev Drug Discov. 2020;19(7):480–494.
- da Costa NM, Hautefeuille A, Cros MP, et al. Transcriptional regulation of thymine DNA glycosylase (TDG) by the tumor suppressor protein p53. Cell Cycle. 2012;11(24):4570–4578.
- Cheng T, Xu M, Qin B, et al. lncRNA H19 contributes to oxidative damage repair in the early age-related cataract by regulating miR-29a/TDG axis. J Cell Mol Med. 2019;23(9):6131–6139.