
ABSTRACT
Only 3% of thyroid cancers are medullary thyroid carcinomas (MTCs), the rest are follicular epithelial cell derived non-MTCs (NMTCs). A dysfunctional INK4-CDK4-RB pathway is detected in most of NMTCs. DNA repair defects and genome instability are associated with NMTC dedifferentiation and aggressiveness. Whether inactivation of the INK4-CDK4-RB pathway induces NMTCs and how differentiation of NMTC cells is controlled remain elusive. In this study, we generated p18Ink4c and Brca1 singly and doubly deficient mice as well as p16Ink4a and Brca1 singly and doubly deficient mice. By using these mice and human thyroid carcinoma cell lines, we discovered that loss of p18Ink4c, not p16Ink4a, in mice stimulated follicular cell proliferation and induced NMTCs. Depletion of Brca1 alone or both p16Ink4a and Brca1 did not induce thyroid tumor. Depletion of Brca1 in p18Ink4c null mice results in poorly differentiated and aggressive NMTCs with epithelial-mesenchymal transition (EMT) features and enhanced DNA damage. Knockdown of BRCA1 in thyroid carcinoma cells activated EMT and promoted tumorigenesis whereas overexpression of BRCA1 inhibited EMT. BRCA1 and EMT marker expression were inversely related in human thyroid cancers. Our finding, for the first time, demonstrates that inactivation of INK4-CDK4-RB pathway induces NMTCs and that Brca1 deficiency promotes dedifferentiation of NMTC cells. These results suggest that BRCA1 and p18INK4C collaboratively suppress thyroid tumorigenesis and progression and CDK4 inhibitors will be effective for treatment of INK4-inactivated or cyclin D-overexpressed thyroid carcinomas.
Introduction
Thyroid cancer is the most common endocrine tumor whose incidence has doubled and mortality has increased in the past decade [Citation1,Citation2]. The thyroid gland is composed of follicular epithelial cells and parafollicular cells. The follicular epithelial cells are responsible for iodine uptake and thyroid hormone synthesis, and the parafollicular cells produce calcium-regulating hormone calcitonin. Accordingly, thyroid tumors are mainly derived from either follicular cells or parafollicular cells. More than 95% of thyroid tumors originate from follicular cells whereas only about 3% of tumors, referred to as medullary thyroid carcinoma (MTC), are derived from parafollicular cells. Follicular-cell-derived thyroid carcinomas, i.e. non-MTCs (NMTCs), are divided into well-differentiated papillary and follicular thyroid carcinoma, as well as poorly differentiated and undifferentiated anaplastic thyroid carcinoma [Citation3,Citation4]. Poorly differentiated thyroid carcinoma (PDTC) and anaplastic thyroid carcinoma (ATC) are highly malignant and metastatic. They likely arise by way of a dedifferentiation process from pre-existing papillary thyroid carcinoma (PTC) and follicular thyroid carcinoma (FTC) [Citation3–5]. Though it has been reported that mutations in p53, RAS, BRAF, and b-catenin, as well as rearrangement of RET, NTRK1, and PPARG play an important role in promoting thyroid tumorigenesis and progression [Citation3,Citation4,Citation6,Citation7], how follicular-cell-derived NMTCs are developed, and how differentiation of NMTC cells are controlled remain largely elusive.
Exposure to ionizing radiation is the only well-established risk factor for thyroid cancer development [Citation8]. This is likely due to the high susceptibility of the follicular cells to radiation damage. Radiation induced DNA damage enhances genomic instability, which leads to follicular cell transformation and is closely associated with thyroid tumor formation and progression [Citation3,Citation8]. Though both PTCs and FTCs belong to “well-differentiated” type of thyroid carcinomas, genome is stable in PTC but instable in FTC. FTCs are more aggressive than PTCs. Interestingly, of PTCs, about half of the total cases are classical type PTCs (C-PTCs), whereas the follicular variant of PTCs (FV-PTCs) comprises 30% of all PTCs. The prognosis of the FV-PTCs falls between that of the C-PTCs and FTCs [Citation9]. Furthermore, drastically enhanced genomic instability has been discovered in almost all PDTCs and ATCs [Citation3,Citation4,Citation8]. These findings indicate the importance of DNA damage repair and genomic stability in regulating follicular cell differentiation and tumorigenesis. However, it is poorly understood how defective DNA damage repair contributes to thyroid tumorigenesis. BRCA1 is a tumor suppressor directly involved in the repair of DNA double-strand breaks and the maintenance of genomic stability. Clinically, the functional loss of BRCA1 by mutation or promoter methylation is associated with an increased susceptibility to thyroid cancer [Citation10–15]. Decreased Brca1 expression is associated with DNA repair delay in irradiated normal thyroid cells [Citation16]. However, homozygous deletion of Brca1 in mice leads to embryonic lethality and heterozygous deletion of Brca1 results in growth defects and premature senescence [Citation17] thereby preventing us from directly determining its role in thyroid tumor development. It remains elusive whether and how loss of function of BRCA1 contributes to thyroid tumor development and progression.
The RB protein, phosphorylated and inactivated by CDK4 and CDK6 (CDK4/6), controls the G1-to-S transition of the cell cycle. CDK4/6 is activated by cyclin D and inhibited by inhibitors of CDK4/6 (INK4) such as p16INK4A (p16, encoded by CDKN2A) and p18INK4C (p18, encoded by CDKN2C). Inactivation of the INK4-CDK4/6-RB pathway (i.e. loss of INK4 or RB and amplification of cyclin D or CDK4/6) is a common event in human cancers and most follicular-cell-derived thyroid carcinomas, including PTC, FTC, and ATC, carry a dysfunctional INK4-CDK4/6-RB pathway [Citation3,Citation18–24]. However, in mouse systems, knockout of p18 or heterozygous germline deletion of Rb induces MTC [Citation25–30] and knockout of other INK4 family cell cycle inhibitors including p15INK4B, p16, p19INK4D does not induce thyroid tumorigenesis [Citation25,Citation31–33]. Though we have previously reported that p18 inhibits follicular cell proliferation and collaborates with Men1 and Pten to suppress the development of NMTCs [Citation26,Citation27], whether inactivation of the INK4-CDK4/6-RB pathway induces NMTCs remain largely elusive. This question is important because most NMTCs, particularly PDTCs and ATCs, are highly proliferative and CDK4/6 inhibitors, a newly approved drug for breast cancer treatment, may also be effective for treatment of INK4 inactivated or cyclin D/CDK4 overexpressed NMTCs. Interestingly, we noticed that mouse studies investigating the role of loss of function of INK4 or Rb were performed in BL/6 or FVB background, whereas, the thyroid tumor phenotype differs drastically in distinct mouse strains [Citation34–37]. We, therefore, hypothesize that inactivation of INK4 in other mouse strains induces NMTCs which recapitulates the role of INK4-CDK4/6-RB pathway in controlling NMTCs.
We and others have reported that BRCA1 deficiency activates p16, p18, and RB [Citation17,Citation38–40] and that loss of either p16 or p18 rescues the proliferative defects and premature senescence induced by Brca1 deficiency in epithelial cells [Citation17,Citation38,Citation39]. To employ this knowledge in our study, we generated p18 and Brca1 singly and doubly deficient mice in either Balb/c or BL/6 background as well as p16 and Brca1 singly and doubly deficient mice in Balb/c enriched background. By using these mice and human thyroid carcinoma cell lines, we demonstrated that deletion of p18, not p16, in Balb/c background induces well-differentiated PTC and FTC (NMTCs). Haploid loss of Brca1 by heterozygous germline deletion in p18 null, not p16 null, mice promotes EMT and dedifferentiation of NMTC (PTC and FTC) cells leading to the increase of tumor metastasis. We confirmed that overexpression of BRCA1 in human ATC cells inhibits EMT and knockdown of BRCA1 in human PTC cells promotes EMT and tumorigenesis.
Materials and methods
Mice, histopathology, and immunostaining
The generation of p18-/- mice in BL/6 background (BL/6-p18-/-) and in Balb/c background (Balb/c-p18-/-), as well as Brca1+/- and p18-/-; Brca1+/-mice in Balb/c background has been previously described [Citation27,Citation38,Citation41,Citation42]. The generation of p16-/- and p16-/-; Brca1+/- mice has been described [Citation39]. p16−/- (in FVB background) and p16-/-;Brca1+/- (in Balb/c-FVB mixed background) mice have both been backcrossed for five generations with Balb/c mice. NOD-Prkdcem26Cd52Il2rgem26Cd22/Nju (NCG) mice was purchased from GemPharmatech (Nanjing, China). The Institutional Animal Care and Use Committee at the University of Miami and Shenzhen University approved all animal procedures. Seventy three WT, eighty p18-/-, forty six Brca1+/- and seventy six p18-/-; Brca1+/- mice in Balb/c background, nine WT, thirty two p16-/- or p16+/-, seven Brca1+/- and twenty five p16-/-; Brca1+/- or p16+/-; Brca1+/- mice in Balb/c enriched background, as well as thirty five WT and fifty eight p18-/- mice in BL/6 background were analyzed. Histopathology and immunohistochemistry (IHC) were performed as previously described [Citation17,Citation26,Citation27]. To briefly summarize, after heat-induced antigen retrieval in retrieval buffer (10 mM Tris base, 1 mM EDTA solution, 0.05% Tween 20, pH 9.0), tissue sections were blocked with normal goat serum in PBS and incubated with primary antibodies at 4°C for 12 h. Immunocomplexes were detected using the Vectastain ABC DAB kit according to the manufacturer’s instructions (Vector Laboratories) [Citation26,Citation27]. The primary antibodies used were as follows: BRCA1 (ab16780; Abcam; 1:1000), Vimentin (VIM, #5741, Cell Signaling, 1:1000), PDGFRβ (#3169, Cell Signaling, 1:1000), TWIST (#50850, Cell Signaling,1:1000), γH2AX (#2577, Cell Signaling), p-PKCα (ab180848, Abcam, 1:1000), and Calcitonin (MA5–16325, ThermoFisher, 1:200), as well as Ki67 (ZM-0166), cytokeratin 19 (CK19, ZM-0074), Galectin-3 (ZM-0143), Hector Battifora mesothelialcell (HBME1, ZM-0386), thyroid transcription factor 1 (TTF1, ZM-0270) (Aorui Dongyuan Biotechnology, Wuxi, China. Working solution). Antibodies obtained from Aorui Dongyuan Biotechnology were directly used as a working solution, and the others were diluted with 5%BSA. The immunostained sections were then examined by microscope (Nikon; ECLIPSE, E200), as previously described [Citation43].
Cell culture, transfection, and viral infection
Human thyroid cancer cell lines B-CPAP and 8505C were purchased from Fenghui Biotechnology (Hunan, China). B-CPAP cells were maintained in RPMI 1640 with 10% fetal bovine serum (FBS), and 8505C cells were maintained in DMEM with 10% FBS. For ectopic expression of BRCA1, cells were transfected with pBabe-empty or pBabe-HA-BRCA1 (gifts from Dr. Yue Xiong at the University of North Carolina at Chapel Hill), as we previously described [Citation44]. To briefly summarize, 2 × 105 cells were seeded into six-well plate and then transfected with pBabe-empty or pBabe-HA-BRCA1 (2 μg) by Lipofectamine™ 3000 (L30000015, Invitrogen). Three days after transfection, cells were lysed for Western blot and qRT-PCR analysis. For knockdown (KD) of BRCA1, cells were infected with pGIPZ-sh-Control (sh-Ctrl) or pGIPZ-sh-BRCA1 (sh-BRCA1) (Open Biosystems), selected with puromycin at 2 μg/ml for three days, and then analyzed for gene expression in vitro, as previously described [Citation17], or subcutaneously inoculated into the back of NCG mice.
Western blot, qRT-PCR, and xenograft
Western blot analysis was carried out as previously described [Citation17]. To briefly summarize, tissues or cells were lysed with RIPA buffer (ab288006, Abcam) supplemented with Protease Inhibitor Cocktail (635673, Takara). Lysates were subjected to SDS-PAGE followed by immunoblotting. Membranes were blocked in 5% skim milk and then probed with anti-BRCA1 (ab16780, Abcam), VIM (#5741, Cell Signaling), TWIST (#50850, Cell Signaling), PDGFRβ (#3169, Cell Signaling), p-PKCα (ab180848, Abcam), p-FRA1 (#3880, Cell Signaling), and GAPDH (BL006B, Biosharp) for 12 h at 4°C. Antibodies were diluted at 1:1000 with universal antibody diluent (WB500D, NCM Biotech). Membranes were washed with TBST (50 mM Tris-HCL pH 7.6, 150 mM NaCl, 0.05% Tween 20, Sigma-Aldrich). Secondary antibodies used were goat anti-rabbit IgG (BL003A, Biosharp) or goat anti-mouse IgG (BL001A, Biosharp). For qRT-PCR, total RNA was extracted using the RNeasy kit (Qiagen) according to the manufacturer’s protocol. 1 μg RNA was used to generate random hexamer-primed cDNA using the Omniscript RT Kit (Qiagen). qRT-PCR was performed as previously reported [Citation38]. The expression level of each gene was normalized with GAPDH. Primers used are listed in Table S1. For xenograft, 5 X 105 cells that were infected with pGIPZ-empty, pGIPZ-shBRCA1-B7, or pGIPZ-shBRCA1-G6 and selected in puromycin were then suspended in 100 μl 50% Matrigel/PBS and subcutaneously inoculated on the left and/or the right side of the back of four or six NCG mice at six weeks of age. Five weeks after inoculation, animals were euthanized, and tumors were dissected for histopathological, immunohistochemical, and biochemical analyses.
Meta-analysis of gene expression data sets
Gene Expression Profiling interactive Analysis (GEPIA, http://gepia.cancer-pku.cn) [Citation45] was used for the analysis of BRCA1 mRNA in human thyroid cancers and paired normal thyroid tissues. GEPIA is a resource for gene expression analysis based on tumor and normal samples from the TCGA (https://www.cancer.gov/tcga) and GTEx (https://www.gtexportal.org) databases. GEPIA thyroid cancer dataset contains 512 TCGA PTC samples and 337 matched TCGA normal and GTEx normal samples [Citation45]. Of these 512 PTC samples, 69.4% were classical-type PTC, and 21.2% were follicular-variant PTC [Citation46]. cBioPortal for Cancer Genomics (https://www.cbioportal.org) [Citation47] was used for the analysis of correlation between the mRNA expression of BRCA1 and EMT markers in 501 TCGA NMTC samples including 395 PTC and 106 FTC samples [Citation47], or in 482 TCGA PTC samples [Citation46].
Statistical analysis
All data are presented as the mean ± SD for at least three repeated individual experiments for each group. Quantitative results were analyzed by two-tailed Student’s t-test. P-value <0.05 was considered statistically significant.
Results
Loss of p18, not p16, in mice stimulates follicular cell proliferation and induces well-differentiated NMTCs
Supported by the previous findings that in human samples most follicular-cell-derived thyroid carcinomas carry a dysfunctional INK4-CDK4/6-RB pathway and mutations in or loss of expression of p18 is frequently detected in NMTCs, we hypothesize that loss of p18 in mice with other genetic backgrounds may recapitulate the function of p18 in controlling follicular-cell-derived thyroid tumors. To test this hypothesis, we generated p18 null mice in Balb/c background, in which mice are more susceptible to tumor development. We characterized these mice and surprisingly found that as early as two months of age female p18-/- mice began to form thyroid tumors. 4 of 16 (25%), 4 of 15 (27%), and 4 of 19 (21%) female p18-/- mice spontaneously developed bilateral thyroid tumors at 2–4 months, 4–12 months, and 12–18 months of age, respectively. Overall, 24% of female p18-/- mice formed thyroid tumors, whereas, only 10% of male p18-/- mice developed thyroid tumors. As a control, 1 of 29 (4%) and 1 of 16 (6%) female wild-type (WT) mice had thyroid tumor before and after 1 year of age, respectively, and none of 28 male WT mice formed thyroid tumor at similar age (). All thyroid tumor-bearing p18-/- mice, with the exception of one, were very small in size and significantly lighter than thyroid tumor-free p18-/- mice. (, ). Histopathological analysis revealed that all thyroid tumors were characterized by intermingled cribriform, papillary, and follicular architecture which are the features of FTC, C-PTC, and FV-PTC. In each tumor, the major parts were well-differentiated FTCs, and small parts were well-differentiated C-PTCs and/or FV-PTCs. Due to the pathological difficulty of differentiating FV-PTC from C-PTC mixed/hybridized with FTC, we uniformly named these tumors well-differentiated NMTC (). No distant metastasis was detected in p18-/- thyroid tumors. To confirm the thyroid epithelial origin and differentiation status of tumors, we performed IHC analysis with antibodies against the markers of thyroid differentiation. The expression of the thyroid master regulator TTF-1, as well as CK19, Galectin-3, and HBME1 was readily detected in p18-/- thyroid tumors (). In contrast, less than 1% of tumor cells were positively stained with calcitonin which is a marker of MTC (). As controls, we detected calcitonin-positive parafollicular cells in tumor-free thyroid (, inset) and calcitonin-positive MTC tumor cells in BL/6-p18-/- mice (Fig. S1). These data not only confirm thyrocytes as the cells of origin of p18-/- thyroid tumors but also indicate the well-differentiated status of the tumors. We then examined tumor-free thyroids and found that Ki67 positive follicular cells in p18-/- mice were significantly more than those in WT counterpart, indicating that loss of p18 promotes follicular cell proliferation (). These data are in line with our previous findings derived from p18 null mice in B6 background [Citation26,Citation27]. We also characterized p16 null mice in Balb/c enriched background and found no hyperproliferative follicular cell phenotype in adult mice and no thyroid tumor formed after 11 months of age though 11 out of 32 mice developed various types of tumor in this time period (Fig. S2, Table S2). Together, these results demonstrated that loss of p18, not p16, in Balb/c background stimulates follicular cell proliferation and induces formation of NMTCs, particularly in female mice.
Figure 1. Loss of p18 stimulates follicular cell proliferation and induces bilateral papillary thyroid tumors. (a) Representative gross appearance and HE staining (inset) of the thyroid tumors from a 9-month-old p18−/− female mouse. Note the thyroid tumors in both sides of the esophagus (Eso.), Th., Tu., thyroid tumor. (b) Representative HE staining of p18−/− thyroid tumors. Note typical pathological PTC features including tumor cells containing enlarged, overlapping nuclei (blue arrows), cells with Orphan Annie nuclei (black arrows), and cells with nuclear grooves and nuclear membrane irregularities (yellow arrows). (c) Body weight analysis of p18−/− mice with or without thyroid tumors. Data represent the mean ± SD of five mice in 2–4 month females without thyroid tumor group, four mice in 2–4 month females with thyroid tumor group, six mice in 4–12 month females without thyroid tumor group, and four mice in 4–12 month females with thyroid tumor group. * p < 0.05 between the group without thyroid tumor and the group with thyroid tumor. (d) IHC analysis of thyroid tumors developed in p18−/− mice. Calcitonin-positive cells are indicated by arrows. Inset shows the representative IHC analysis of tumor-free thyroid. Note, the majority of tumor cells are negative for calcitonin, other than a few sporadically located calcitonin-positive cells. (e) IHC analysis of tumor-free thyroids from WT and p18−/− mice at the age of 2–4 months of age. The percentages of Ki67-positive cells were calculated from cells situated in clear duct/gland structures. Results represent the mean ± SD of three animals per group.
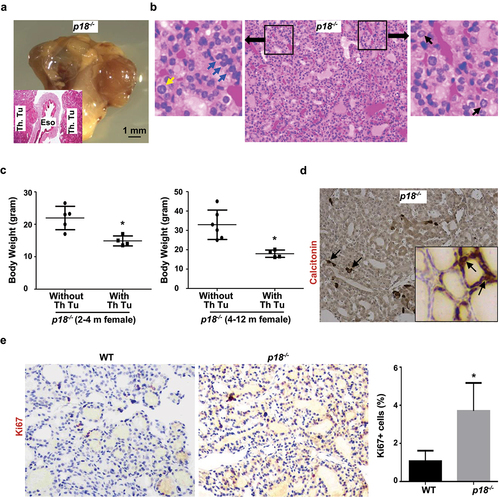
Figure 2. Heterozygous germline deletion of Brca1 in p18 null mice promotes EMT and enhances metastasis of thyroid tumors. (a) HE staining analysis of thyroid tumors developed in mutant mice. Note relative to p18−/− thyroid tumor cells, p18−/−;Brca1+/- cells are highly heterogenous and poorly differentiated. Tumor a and b show two thyroid tumors derived from two individual p18−/−;Brca1+/- mice. Picture in the most right shows lung metastasis from thyroid tumor b. (b, c) IHC analysis of thyroid tumors developed in mutant mice. Note relative to p18−/− tumor cells, more p18−/−;Brca1+/- cells expresses strong VIM and TWIST, but none or weak TTF1, CK19, Galectin-3, and HBME1. (d) IHC analysis of thyroid tumors with an antibody against Ki67. The percentages of Ki67-positive cells were calculated and plotted. Results represent the mean ± SD of three tumors per group.
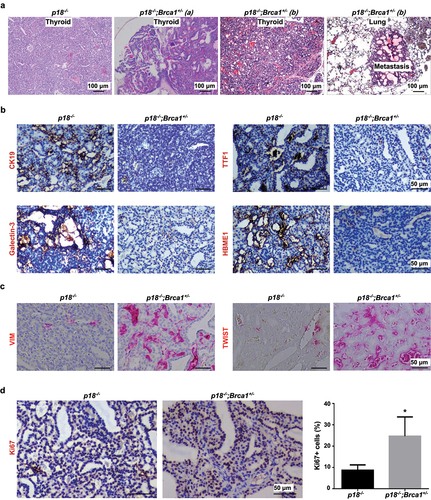
Table 1 Spontaneous thyroid tumor development in mutant mice a
Haploid loss of Brca1 promotes EMT and dedifferentiation of p18-deficient thyroid tumor cells accelerating tumor progression
To determine the effect of Brca1 deficiency in regulating thyroid cell proliferation and differentiation in an unbiased manner, we choose to use heterozygous germline Brca1+/- mutant mice. Taking advantage of our previous finding that loss of either p16 or p18 rescues the proliferative defects and the premature senescence induced by Brca1 deficiency in epithelial cells, we generated and characterized p18-/-;Brca1+/- mice in Balb/c background, which have been backcrossed for more than 10 generations with Balb/c mice, and p16-/-;Brca1+/- mice in Balb/c-enriched background, which have been backcrossed for 5 generations with Balb/c mice. Though no Brca1+/- and p16-/-;Brca1+/- mice formed thyroid tumors, 5 of 12 (42%) and 6 of 13 (46%) female p18-/-;Brca1+/- mice spontaneously developed bilateral thyroid tumors at 2–4 months and 4–12 months of age, respectively, while 6 of 18 (33%) and 5 of 14 (36%) male p18-/-;Brca1+/- mice also developed bilateral thyroid tumors at 4–12 months and 12–18 months of age, respectively (, , Table S2). Notably, due to the metabolic defects and severe malnutrition that likely resulted from thyroid tumors or thyroid tumor-induced difficulty swallowing, as well as the defects induced by the tumors simultaneously developed in other organs, no thyroid tumor-bearing female p18-/-;Brca1+/- mice lived, or was allowed by IACUC to live, beyond 11 months of age. Therefore, no assessment of thyroid tumor incidence for female p18-/-;Brca1+/- mice could be made in the 12–18 month range. Thyroid tumor incidences in p18-/-;Brca1+/- female mice at 2–4 months of age and 4–12 months of age were higher than those in p18-/- counterparts, though no statistical significance was reached, which is likely caused by the insufficient number of mice that was examined. Notably, 3 of 22 (14%) p18-/-;Brca1+/- thyroid tumors metastasized to the lungs whereas no p18-/- thyroid tumors did so (, ). Again, due to the early termination as mentioned above, we were unable to pursue the full penetrance of metastasis induced by p18-/-;Brca1+/- thyroid tumors.
Table 2. Haploid loss of Brca1 in p18-deficient mice activates EMT in thyroid tumor cells.
Histopathological analysis revealed that though p18-/-;Brca1+/- thyroid tumors retained some PTC and FTC (NMTC) features, they were more heterogeneous, invasive, metastatic, and poorly differentiated, when compared with p18-/- tumor counterparts. Consistent with p18-/- thyroid tumors, p18-/-;Brca1+/- thyroid tumors were also NMTC tumors as evidenced by sparsely scattered calcitonin-positive tumor cells (Fig. S3B, and data not shown). Notably, the expression of thyroid differentiation markers including TTF-1, CK19, Galectin-3, and HBME1 was barely detectable in p18-/-;Brca1+/- thyroid tumors, confirming that these are poorly differentiated tumors (). Inspired by the findings that NMTC thyroid tumors originate from follicular epithelial cells and haploid loss of Brca1 promotes dedifferentiation of mammary epithelia-derived tumor cells through activation of EMT [Citation17,Citation44], we examined the role of haploid loss of Brca1 in EMT activation in thyroid tumors. We observed that when compared with p18-/- tumors, significantly more p18-/-;Brca1+/- thyroid tumors expressed high level of EMT markers and EMT-transcription factors (EMT-TF) including VIM, TWIST, and p-FRA1 (, ). In addition, we also found that relative to p18-/- tumors, more p18-/-;Brca1+/- thyroid tumors expressed PDGFRβ, a EMT marker and downstream therapeutic target of BRCA1 deficiency in promoting dedifferentiation of mammary tumor cells (Fig. S3) [Citation48]. Together, these results suggest that haploid loss of Brca1 activates EMT and promotes dedifferentiation of p18- deficient thyroid tumor cells, stimulating their invasive and metastatic potential.
Furthermore, we found p18-/-;Brca1+/- thyroid tumors exhibited more Ki67 and γH2AX positive cells than p18-/- tumors (, Fig. S3). These data indicate that deficiency of p18 and Brca1 collaboratively enhances proliferation and defects in DNA damage repair of thyroid tumor cells. Interestingly, Brca1+/- thyroid epithelia displayed less Ki67 positive cells than WT thyroid epithelia, suggesting that haploid loss of Brca1 reduces thyroid epithelial cell proliferation (Fig. S2). This result is in accordance with our previous finding that depletion of Brca1 in mammary epithelial cells impairs their proliferation [Citation38]. Notably, p16-/- thyroid epithelia showed slightly more Ki67 positive cells than WT thyroid epithelia, and p16-/-;Brca1+/- thyroid epithelia exhibited significantly more Ki67 positive cells than Brca1+/- thyroid epithelia (Fig. S2). These data suggest that the loss of p16 rescues proliferative defects caused by Brca1 deficiency in thyroid epithelia; however, depletion of both p16 and Brca1 do not lead to the formation of thyroid tumor. In summary, these results suggest that Brca1 cooperates with p18, but not p16, to suppress follicular cell proliferation and dedifferentiation in thyroid tumor development and progression.
Overexpression of BRCA1 in thyroid carcinoma cells inhibits EMT and dedifferentiation
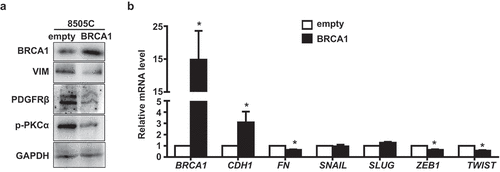
To consolidate the role of BRCA1 in regulating EMT, we ectopically overexpressed WT BRCA1 in a human undifferentiated anaplastic thyroid carcinoma cell line, 8505C. Consistent with our findings derived from poorly differentiated mammary carcinoma cells [Citation48], we observed that ectopic BRCA1 stimulated the expression of E-cad but inhibited the expression of VIM, PDGFRβ, p-PKCα, TWIST, and ZEB1 (). These data support the function of BRCA1 in suppressing EMT and dedifferentiation of thyroid tumor cells.
Figure 3. Overexpression of BRCA1 in human thyroid cancer cells inhibits EMT. 8505C cells transfected with pBabe-empty (empty) and pBabe-HA-BRCA1 (BRCA1) were analyzed by Western blot (a) or qRT-PCR (b). GAPDH was used as a loading control in Western blot.
Knockdown of BRCA1 in human thyroid cancer cells activates EMT promoting tumorigenesis
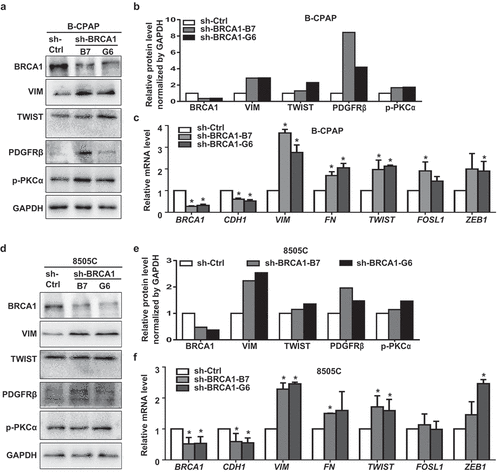
We then set out to determine whether depletion of BRCA1 in human thyroid cancer cells activates EMT and promotes dedifferentiation. To this end, we choose a human PTC cell line, B-CPAP, and an ATC cell line, 8505C. The reason we choose an ATC cell line is that ATC cells likely originate from dedifferentiation of pre-existing PTC and FTC cells and are highly heterogeneous. We found that stable knockdown of BRCA1 in both cell lines enhanced the expression of EMT markers and EMT-TFs including VIM, FN, TWIST, FRA1, ZEB1, as well as PDGFRβ and p-PKCα (), which confirm the findings derived from Brca1-deficient thyroid tumors in mice.
Figure 4. Knockdown of BRCA1 in human thyroid cancer cells promotes EMT and dedifferentiation. B-CPAP (a, b, c) and 8505C (d, e, f) cells infected with pGIPZ-sh-Control (sh-Ctrl) or pGIPZ-sh-BRCA1 targeting different sequences of human BRCA1 (sh-BRCA1-G6 and sh-BRCA1-B7) were analyzed by Western blot (a, d) or qRT-PCR (c, f). * p < 0.05 between the sh-Ctrl group and sh-BRCA1-G6 group, or between the sh-Ctrl group and sh-BRCA1-B7 group. (b, e) The protein levels of each lane in (a) and (d) were quantified, respectively, by Image-Pro Plus 6.0 and normalized by that of GAPDH.
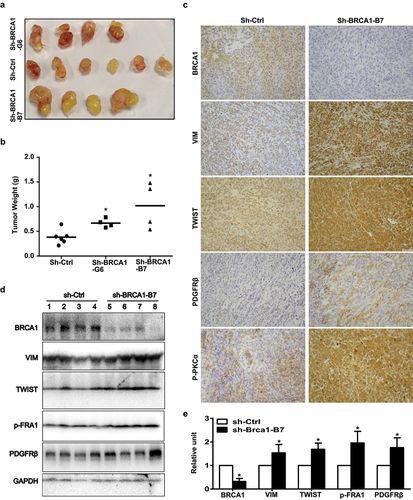
We inoculated B-CPAP-sh-control and B-CPAP-sh-BRCA1 cells into mice and found that B-CPAP-sh-BRCA1 cells generated significantly larger and heavier tumors than B-CPAP-sh-control cells (). Western blot and IHC analysis revealed that relative to B-CPAP-sh-control tumors, B-CPAP-sh-BRCA1 tumors clearly expresses less BRCA1, more VIM, TWIST, PDGFRβ, and p-PKCα (). In summary, these results demonstrate that depletion of BRCA1 in human thyroid cancer cells activates EMT promoting tumorigenesis.
Figure 5. Knockdown of BRCA1 in human thyroid cancer cells promotes EMT and tumorigenesis. (a, b) B-CPAP-sh-Ctrl and B-CPAP-sh-BRCA1-G6 cells were inoculated into the left and right back of four NSG mice, respectively, in a pairwise manner. Similarly, B-CPAP-sh-Ctrl and B-CPAP-sh-BRCA1-B7 cells were inoculated into the left and right back of two NCG mice, respectively, in a pairwise manner. In addition, the same number of B-CPAP-sh-BRCA1-B7 cells were inoculated into the left back of two individual mice. Gross appearance (a) and weight (b) of the tumors formed 5 weeks later were analyzed. Data in (b) represent the mean ± SD of four tumors in each sh-BRCA1 group and six tumors in sh-Ctrl group. * p < 0.05 between the sh-Ctrl group and sh-BRCA1-G6 group, or between the sh-Ctrl group and sh-BRCA1-B7 group. (c, d) Representative tumors derived from sh-Ctrl and sh-BRCA1-B7 cell transplants were analyzed by IHC (c) and Western blot (d). (e) The protein levels of each lane in (d) were quantified by Image-Pro Plus 6.0 and normalized by that of GAPDH (right). Results represent the mean ± SD of four tumors per group.
Expression of BRCA1 is downregulated and is inversely related to that of EMT markers in human thyroid cancers
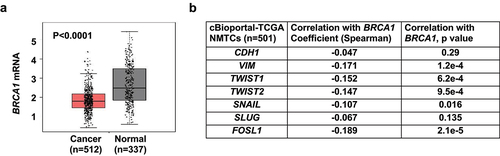
To determine whether our murine Brca1-deficient tumors model human thyroid cancers, we studied BRCA1 expression in GEPIA thyroid cancer patient sample sets [Citation45] that is a resource for gene expression analysis based on tumor and normal samples from the TCGA and GTEx databases. GEPIA thyroid cancer dataset contains 512 PTC samples including classical-type PTC and follicular-variant PTC, as well as 337 matched TCGA normal and GTEx normal samples. We found that BRCA1 mRNA was significantly lower in thyroid cancers than in normal thyroid tissues (), supporting the function of BRCA1 deficiency in promoting thyroid tumorigenesis. We then utilized cBioPortal for Cancer Genomics [Citation47] to analyze the correlation between the mRNA expression of BRCA1 and EMT markers in NMTC samples including PTC and FTC samples [Citation47], or in PTC samples [Citation46]. We detected a significant inverse correlation between mRNA expression of BRCA1 with VIM, TWIST1, TWIST2, SNAIL, SLUG, and FOSL1, all genes associated with EMT (, Table S3). Consistent with our results in mice, these findings indicate that deficiency of BRCA1 is associated with thyroid cancer development and progression.
Figure 6. Analysis of BRCA1 expression and its correlation with EMT markers in human thyroid tissues and cancers. (a) Analysis of BRCA1 mRNA expression in thyroid cancer samples (Cancer) and paired normal thyroid tissues (Normal) in GEPIA (http://gepia.cancer-pku.cn) dataset. (b) Correlation analysis of mRNA expression of BRCA1 and EMT markers in thyroid carcinoma/NMTC samples (TCGA, Firehose Legacy) by cBioportal for Cancer Genomics (https://www.cbioportal.org).
Discussion
In the present study, we showed that loss of p18, not p16, in mice stimulated follicular cell proliferation and induced well-differentiated PTC and FTC (NMTCs). We found that heterozygous germline deletion of Brca1 in mice did not result in thyroid tumor development. Though loss of p16 rescued proliferative defects caused by Brca1 deficiency in thyroid epithelia, depletion of both p16 and Brca1 did not lead to the formation of thyroid tumor. Notably, p18;Brca1 doubly deficient mice developed thyroid tumors with moderately accelerated incidence when compared with p18 singly deficient mice. The expression of thyroid differentiation markers including TTF-1, CK19, Gatectin-3, and HBME1 was readily detected in p18 singly deficient thyroid tumors, but barely detectable in p18;Brca1 doubly deficient tumors. p18;Brca1 doubly deficient thyroid tumors were more heterogeneous, invasive, and poorly differentiated, relative to p18 singly deficient tumors. Haploid loss of Brca1 activated EMT in p18-deficient thyroid tumor cells. Knockdown of BRCA1 in human thyroid carcinoma cells activated EMT and promoted tumorigenesis whereas overexpression of BRCA inhibited EMT. In addition, we observed that the expression of BRCA1 was downregulated and was inversely related to that of EMT markers in human thyroid cancers. These results demonstrate that p18 controls follicular epithelial cells proliferation and tumorigenesis, and that Brca1 suppresses dedifferentiation of NMTC cells. Furthermore, these data also indicate BRCA1 collaborates with p18, not p16, controlling thyroid tumor progression.
In clinical samples, mutations in p18 gene or loss of expression of p18 are frequently detected in human MTCs and NMTCs [Citation19–24]. Inactivation of p16 due to copy number losses, gene truncation, loss-of-function mutations, and promoter methylation is detected in 30%–50% of NMTCs [Citation3,Citation49,Citation50]. Overexpression of cyclin D1 is observed in 1/3 of NMTCs [Citation3,Citation51], and gain of function mutation of CDK4 was also detected in sporadic thyroid cancers [Citation52]. Mutations in RB gene were reported in about 7%–55% of NMTCs, and 82% of NMTCs carry either mutations in RB gene or overexpressed cyclin D1 [Citation49,Citation51]. In human thyroid cancer cell lines, the levels of cyclin D and phosphorylated RB are highly expressed in aggressive lines, and CDK4/6 inhibitor suppresses proliferation of these cell lines [Citation53]. These data suggest that in humans, inactivation of INK4-CDK-RB pathway promotes the development of NMTCs. Perplexedly, in mice deficiency of p18 or Rb induces MTCs in B6 background [Citation25–30] and deletion of other INK4s including p16, p15, and p19 has no significant effect in promoting the development of thyroid tumors [Citation25,Citation31–33]. In addition, no thyroid tumor was detected in mice harboring Cdk4R24C mutant that is functionally similar with CDK4R24L mutant, a gain of function mutation of CDK4 in human NMTCs [Citation52,Citation54]. These findings make one doubt whether INK4-CDK-RB pathway plays a role in controlling follicular cell proliferation and tumorigenesis. Our results demonstrate that loss of p18 in mice with Balb/c background promotes development of NMTCs, which provides the first genetic evidence confirming the role of INK4-CDK-RB pathway in controlling NMTCs. In addition, this study also indicates that there exist unknown genetic modifiers in mice with B6 background that suppress the development of NMTCs when INK4-CDK-RB pathway is inactivated.
An interesting discovery is that mice lacking p16 in Balb/c enriched background do not develop thyroid tumor, though inactivation of p16 is detected in 30%–50% of human NMTCs, particularly in ATC and PDTC [Citation3,Citation49,Citation50,Citation55]. The discrepant findings related with the role of p16 loss in thyroid tumorigenesis in humans and mice are likely resulted from two reasons. First, one third of p16 null mice developed sarcoma and lymphoma, which prevent from further investigating the development of thyroid tumors in the same group of mice at a later age; Second, p16 expression is weakly detected in young organs and is gradually induced during the aging process. Loss of p16 results in significant hyper-proliferative phenotype in old, but not in young organs and tissues [Citation18]. In the present study, we examined tumor phenotype of p16-deficient mice mostly between 12–16 months of age. We, therefore, cannot exclude the possibility that p16-deficient mice develop thyroid tumor after 16 months of age.
It has long been known that thyroid follicular cells are highly susceptible to ionizing radiation, which induces DNA damage, enhances genomic instability, and eventually leads to the transformation of follicular cells. Furthermore, genomic instability is closely associated with thyroid tumor formation [Citation3,Citation8]. Functional loss of many genes involved in DNA damage repair is associated with an increased susceptibility to thyroid cancer [Citation3,Citation13]. However, it is poorly understood how functional loss of genes involved in DNA damage repair contributes to thyroid tumorigenesis. Many DNA damage repair genes including BRCA1 are essential for development and the knockout of these genes causes embryonic lethality. Heterozygous germline deletion of these genes in mice induces apoptosis and cell cycle arrest in multiple organs and/or cell lineages. It is practically difficult to directly use these mutant mice to investigate the role of these genes in thyroid tumorigenesis. We previously demonstrated that Brca1 deficiency activates p18, and that loss of p18 rescues the proliferative defects and premature senescence induced by Brca1 deficiency in epithelial cells [Citation17,Citation38,Citation39]. In the present study, we found that heterozygous germline deletion of Brca1 in p18 null mice led to the formation of thyroid tumor with significantly enhanced DNA damage and cell proliferation though haploid loss of Brca1 alone had no effect in the induction of thyroid tumors. These results demonstrate that overcoming growth defects is a necessary step for the development of thyroid tumors initiated by loss of function of genes involved in DNA damage repair, like BRCA1. Importantly, our findings provide genetic evidence suggesting that CDK4/6 inhibitors will be effective for the treatment of INK4 inactivated or cyclin D/CDK4 overexpressed ATCs and PDTCs.
Between well-differentiated thyroid carcinomas, FTCs carry less stable genome and are less differentiated and more aggressive than PTCs. Relative to PTCs and FTCs, undifferentiated and poorly differentiated PDTCs and ATCs carry significantly more instable genome and are highly metastatic [Citation3,Citation4,Citation8]. These findings suggest that the level of genomic instability is closely associated with the aggressiveness and differentiation status of thyroid tumors. Notably, PDTC and ATC cells coexist with well differentiated PTC and FTC cells in thyroid carcinomas, and PDTCs and ATCs likely arise from pre-existing well-differentiated carcinomas through dedifferentiation driven by the gaining of genetic abnormalities [Citation3,Citation4,Citation8,Citation56]. For example, mutations in p53, a gene that controls DNA damage repair and genome instability, occur with increasing frequency in PDTCs and ATCs [Citation3,Citation4,Citation57] and loss of function of p53 in RET/PTC3p transgenic or BRAFV600E mice activates EMT and promotes dedifferentiation of well differentiated NMTCs leading to the development of metastatic PDTCs and ATCs [Citation58,Citation59]. In this study, we found that 18-/- thyroid tumors are typical well-differentiated carcinomas, whereas p18-/-;Brca1+/- tumors are poorly differentiated NMTCs with barely detectable thyroid differentiation markers and significantly enhanced EMT features. We confirmed that functional loss of Brca1 activates EMT and stimulates dedifferentiation of thyroid tumor cells promoting their aggressiveness and metastasis. These results further indicate that, in addition to p53 loss, functional loss of BRCA1, another gene involved in DNA damage repair, promotes dedifferentiation of thyroid carcinomas.
The molecular mechanisms underlying the role of BRCA1 in maintaining the differentiation of carcinoma cells have been investigated in mammary tumors. We have previously reported that loss of function of Brca1 activates EMT and induces dedifferentiation of carcinoma cells promoting mammary tumor aggressiveness and metastasis [Citation17,Citation43,Citation44,Citation48]. It has been demonstrated that loss of function of BRCA1 in breast cancer cells enhances the expression of several EMT inducing transcription factors (EMT-TFs) including SLUG [Citation60], TWIST [Citation17], FOXC1/C2 [Citation61]. We recently discovered that loss of function stimulates the transcription of PDGFRβ and activates the PDGFRβ-PKCα signaling pathway, an essential pathway that controls the EMT of mammary carcinoma cells [Citation62], and that inhibition of PDGFRβ and its downstream target PKCα in Brca1-deficient tumor cells suppresses EMT and tumor development and progression [Citation48]. In the present study, we demonstrate that Brca1 deficiency also activates EMT and enhances the expression of PDGFRβ in thyroid tumors, which allows us to propose that PDGFRβ pathway may also be a therapeutic target for Brca1-deficient thyroid tumors.
Ethics Statement
The Institutional Animal Care and Use Committee at the University of Miami and Shenzhen University approved all animal procedures.
Author contribution statement
FB and XHP designed the research studies. FB, XL, XZ, ZM, HW, JM, and XHP conducted experiments and analyzed data. FB and XHP wrote the manuscript. FB and XHP provided financial support. XHP supervised the project. All authors made comments on the manuscript.
Supplemental Material
Download PDF (639.7 KB)Acknowledgements
We thank Drs. Beverly Koller and Norman Sharpless for Brca1 mutant and p16 null mice, Alexandria Scott, Emely Pimentel, and Ho Lam Chan for technical support, the DVR core facility for animal husbandry.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
All data generated or analyzed during this study are included in this published article and its supplementary information files. All data generated/analyzed during the current study are available from the corresponding auther upon request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384101.2023.2225938
Additional information
Funding
References
- Du L, Zhao Z, Zheng R, et al. Epidemiology of Thyroid Cancer: incidence and Mortality in China, 2015. Front Oncol. 2020;10:1702. doi:10.3389/fonc.2020.01702
- Lim H, Devesa SS, Sosa JA, et al. Trends in thyroid cancer incidence and mortality in the United States, 1974-2013. JAMA. 2017;317(13):1338–1348. DOI:10.1001/jama.2017.2719
- Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer. 2006;6(4):292–306. doi:10.1038/nrc1836.
- Taccaliti A, Silvetti F, Palmonella G, et al. Anaplastic thyroid carcinoma. Front Endocrinol. 2012;3:84. doi:10.3389/fendo.2012.00084
- Zane M, Scavo E, Catalano V, et al. Normal vs cancer thyroid stem cells: the road to transformation. Oncogene. 2016;35(7):805–815. DOI:10.1038/onc.2015.138
- Bhaijee F, Nikiforov YE. Molecular analysis of thyroid tumors. Endocr Pathol. 2011;22(3):126–133. doi:10.1007/s12022-011-9170-y.
- Agrawal N, Akbani R, Aksoy B&, et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159(3):676–690. DOI:10.1016/j.cell.2014.09.050
- Ron E, Lubin JH, Shore RE, et al. Thyroid cancer after exposure to external radiation: a pooled analysis of seven studies. Radiat Res. 1995;141(3):259–277. DOI:10.2307/3579003
- Daniels GH. Follicular variant of papillary thyroid carcinoma: hybrid or mixture? Thyroid: Offic J Am Thyroid Association. 2016;26(7):872–874. doi:10.1089/thy.2016.0244.
- Shih HA, Nathanson KL, Seal S, et al. BRCA1 and BRCA2 mutations in breast cancer families with multiple primary cancers. Clin Cancer Res. 2000;6(11):4259–4264.
- Xu L, Doan PC, Wei Q, et al. Association of BRCA1 functional single nucleotide polymorphisms with risk of differentiated thyroid carcinoma. Thyroid: Offic J Am Thyroid Association. 2012;22(1):35–43. DOI:10.1089/thy.2011.0117
- Streff H, Profato J, Ye Y, et al. Cancer incidence in first- and second-degree relatives of BRCA1 and BRCA2 mutation carriers. Oncology. 2016;21(7):869–874. DOI:10.1634/theoncologist.2015-0354
- Hincza K, Kowalik A, Kowalska A. Current knowledge of germline genetic risk factors for the development of non-medullary thyroid cancer. Genes (Basel). 2019;10(7):482. doi:10.3390/genes10070482.
- Wojcicka A, Czetwertynska M, Swierniak M, et al. Variants in the ATM-CHEK2-BRCA1 axis determine genetic predisposition and clinical presentation of papillary thyroid carcinoma. Genes Chromosomes Cancer. 2014;53(6):516–523. DOI:10.1002/gcc.22162
- Galetzka D, Hansmann T, El Hajj N, et al. Monozygotic twins discordant for constitutive BRCA1 promoter methylation, childhood cancer and secondary cancer. Epigenetics. 2012;7(1):47–54. DOI:10.4161/epi.7.1.18814
- Penha RCC, Lima SCS, Boroni M, et al. Intrinsic LINE-1 hypomethylation and decreased Brca1 expression are associated with DNA repair delay in irradiated thyroid cells. Radiat Res. 2017;188(2):144–155. DOI:10.1667/RR14532.1
- Bai F, Chan HL, Scott A, et al. BRCA1 suppresses epithelial-to-mesenchymal transition and stem cell dedifferentiation during mammary and tumor development. Cancer Res. 2014;74(21):6161–6172. DOI:10.1158/0008-5472.CAN-14-1119
- Pei XH, Xiong Y. Biochemical and cellular mechanisms of mammalian CDK inhibitors: a few unresolved issues. Oncogene. 2005;24(17):2787–2795. doi:10.1038/sj.onc.1208611.
- Kunstman JW, Juhlin CC, Goh G, et al. Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum Mol Genet. 2015;24(8):2318–2329. DOI:10.1093/hmg/ddu749
- Grubbs EG, Williams MD, Scheet P, et al. Role of CDKN2C copy number in sporadic medullary thyroid carcinoma. Thyroid: Offic J Am Thyroid Association. 2016;26(11):1553–1562. DOI:10.1089/thy.2016.0224
- Pita JM, Figueiredo IF, Moura MM, et al. Cell cycle deregulation and TP53 and RAS mutations are major events in poorly differentiated and undifferentiated thyroid carcinomas. J Clin Endocrinol Metab. 2014;99(3):E497–507. DOI:10.1210/jc.2013-1512
- Neta G, Brenner AV, Sturgis EM, et al. Common genetic variants related to genomic integrity and risk of papillary thyroid cancer. Carcinogenesis. 2011;32(8):1231–1237. DOI:10.1093/carcin/bgr100
- van Veelen W, Klompmaker R, Gloerich M, et al. P18 is a tumor suppressor gene involved in human medullary thyroid carcinoma and pheochromocytoma development. Int J Cancer. 2009;124(2):339–345. DOI:10.1002/ijc.23977
- Maxwell JE, Gule-Monroe MK, Subbiah V, et al. Novel use of a Clinical Laboratory Improvements Amendments (CLIA)-certified Cyclin-Dependent Kinase N2C (CDKN2C) loss assay in sporadic medullary thyroid carcinoma. Surgery. 2020;167(1):80–86. DOI:10.1016/j.surg.2019.03.041
- Franklin DS, Godfrey VL, O’Brien DA, et al. Functional collaboration between different cyclin-dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Mol Cell Biol. 2000;20(16):6147–6158. DOI:10.1128/MCB.20.16.6147-6158.2000
- Bai F, Pei XH, Nishikawa T, et al. P18ink4c, but not p27Kip1, collaborates with Men1 to suppress neuroendocrine organ tumors. Mol Cell Biol. 2007;27(4):1495–1504. doi:10.1128/MCB.01764-06.
- Bai F, Pei XH, Pandolfi PP, et al. P18 Ink4c and Pten constrain a positive regulatory loop between cell growth and cell cycle control. Mol Cell Biol. 2006;26(12):4564–4576. doi:10.1128/MCB.00266-06.
- Nikitin AY, Juarez-Perez MI, Li S, et al. RB-mediated suppression of spontaneous multiple neuroendocrine neoplasia and lung metastases in Rb± mice. Proc Natl Acad Sci U S A. 1999;96(7):3916–3921. doi:10.1073/pnas.96.7.3916.
- Williams BO, Remington L, Albert DM, et al. Cooperative tumorigenic effects of germline mitations in Rb and p53. Nature Genet. 1994;7:480–484. doi: 10.1038/ng0894-480
- Loffler KA, Biondi CA, Gartside MG, et al. Lack of augmentation of tumor spectrum or severity in dual heterozygous Men1 and Rb1 knockout mice. Oncogene. 2007;26(27):4009–4017. DOI:10.1038/sj.onc.1210163
- Sharpless NE, Bardeesy N, Lee KH, et al. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413(6851):86–91. DOI:10.1038/35092592
- Bai F, Chan HL, Smith MD, et al. P19ink4d is a tumor suppressor and controls pituitary anterior lobe cell proliferation. Mol Cell Biol. 2014;34(12):2121–2134. doi:10.1128/MCB.01363-13.
- Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2(2):103–112. doi:10.1016/S1535-6108(02)00102-2.
- Cranston AN, Ponder BA. Modulation of medullary thyroid carcinoma penetrance suggests the presence of modifier genes in a RET transgenic mouse model. Cancer Res. 2003;63(16):4777–4780.
- Tiozzo C, Danopoulos S, Lavarreda-Pearce M, et al. Embryonic epithelial Pten deletion through Nkx2.1-cre leads to thyroid tumorigenesis in a strain-dependent manner. Endocr Relat Cancer. 2012;19(2):111–122. DOI:10.1530/ERC-10-0327
- Frith CH, Heath JE. Morphological classification and incidence of thyroid tumors in untreated aged mice. J Gerontol. 1984;39(1):7–10. doi:10.1093/geronj/39.1.7.
- Freeman D, Lesche R, Kertesz N, et al. Genetic background controls tumor development in PTEN-deficient mice. Cancer Res. 2006;66(13):6492–6496. DOI:10.1158/0008-5472.CAN-05-4143
- Bai F, Smith MD, Chan HL, et al. Germline mutation of Brca1 alters the fate of mammary luminal cells and causes luminal-to-basal mammary tumor transformation. Oncogene. 2013;32(22):2715–2725. DOI:10.1038/onc.2012.293
- Scott A, Bai F, Chan HL, et al. P16ink4a suppresses BRCA1-deficient mammary tumorigenesis. Oncotarget. 2016;7(51):84496–84507. DOI:10.18632/oncotarget.13015
- Sedic M, Skibinski A, Brown N, et al. Haploinsufficiency for BRCA1 leads to cell-type-specific genomic instability and premature senescence. Nat Commun. 2015;6(1):7505. DOI:10.1038/ncomms8505
- Franklin DS, Godfrey VL, Lee H, et al. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. 1998;12(18):2899–2911. DOI:10.1101/gad.12.18.2899
- Pei XH, Bai F, Smith MD, et al. CDK inhibitor p18(INK4c) is a downstream target of GATA3 and restrains mammary luminal progenitor cell proliferation and tumorigenesis. Cancer Cell. 2009;15(5):389–401. DOI:10.1016/j.ccr.2009.03.004
- Bai F, Wang C, Liu X, et al. Loss of function of BRCA1 promotes EMT in mammary tumors through activation of TGFbetaR2 signaling pathway. Cell Death Dis. 2022;13(3):195. DOI:10.1038/s41419-022-04646-7
- Bai F, Zhang LH, Liu X, et al. GATA3 functions downstream of BRCA1 to suppress EMT in breast cancer. Theranostics. 2021;11(17):8218–8233. DOI:10.7150/thno.59280
- Tang Z, Li C, Kang B, et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–w102. DOI:10.1093/nar/gkx247
- TCGAR N. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159(3):676–690. doi:10.1016/j.cell.2014.09.050.
- Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. DOI:10.1158/2159-8290.CD-12-0095
- Bai F, Liu S, Liu X, et al. Pdgfrbeta is an essential therapeutic target for BRCA1-deficient mammary tumors. Breast Cancer Res. 2021;23(1):10. DOI:10.1186/s13058-021-01387-x
- Pozdeyev N, Gay LM, Sokol ES, et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin Cancer Res. 2018;24(13):3059–3068. DOI:10.1158/1078-0432.CCR-18-0373
- Yoo SK, Song YS, Lee EK, et al. Integrative analysis of genomic and transcriptomic characteristics associated with progression of aggressive thyroid cancer. Nat Commun. 2019;10. doi:10.1038/s41467-019-10680-5.
- Zou MJ, Shi YF, Farid NR, et al. Inverse association between cyclin D1 overexpression and retinoblastoma gene mutation in thyroid carcinomas. Endocrine. 1998;8(1):61–64. doi:10.1385/ENDO:8:1:61.
- Kim JH, Jeong JY, Seo AN, et al. Genomic profiling of aggressive thyroid cancer in association with its clinicopathological characteristics. In Vivo. 2022;36(1):111–120. doi:10.21873/invivo.12682.
- Lee HJ, Lee WK, Kang CW, et al. A selective cyclin-dependent kinase 4, 6 dual inhibitor, Ribociclib (LEE011) inhibits cell proliferation and induces apoptosis in aggressive thyroid cancer. Cancer Lett. 2018;417:131–140. doi:10.1016/j.canlet.2017.12.037
- Sotillo R, Dubus P, Martin J, et al. Wide spectrum of tumors in knock-in mice carrying a Cdk4 protein insensitive to INK4 inhibitors. Embo J. 2001;20(23):6637–6647. DOI:10.1093/emboj/20.23.6637
- Schagdarsurengin U, Gimm O, Hoang-Vu C, et al. Frequent epigenetic silencing of the CpG island promoter of RASSF1A in thyroid carcinoma. Cancer Res. 2002;62(13):3698–3701.
- Ragazzi M, Torricelli F, Donati B, et al. Coexisting well-differentiated and anaplastic thyroid carcinoma in the same primary resection specimen: immunophenotypic and genetic comparison of the two components in a consecutive series of 13 cases and a review of the literature. Virchows Arch. 2021;478(2):265–281. DOI:10.1007/s00428-020-02891-9
- Volante M, Lam AK, Papotti M, et al. Molecular pathology of poorly differentiated and anaplastic thyroid cancer: what do pathologists need to know. Endocr Pathol. 2021;32(1):63–76. doi:10.1007/s12022-021-09665-2.
- McFadden DG, Vernon A, Santiago PM, et al. P53 constrains progression to anaplastic thyroid carcinoma in a Braf-mutant mouse model of papillary thyroid cancer. P Natl Acad Sci USA. 2014;111(16):E1600–E9. DOI:10.1073/pnas.1404357111
- Powell DJ, Russell JP, Li GQ, et al. Altered gene expression in immunogenic poorly differentiated thyroid carcinomas from RET/PTC3p53 mice. Oncogene. 2001;20(25):3235–3246. DOI:10.1038/sj.onc.1204425
- Proia TA, Keller PJ, Gupta PB, et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell. 2011;8(2):149–163. DOI:10.1016/j.stem.2010.12.007
- Tkocz D, Crawford NT, Buckley NE, et al. BRCA1 and GATA3 corepress FOXC1 to inhibit the pathogenesis of basal-like breast cancers. Oncogene. 2011;31(32):3667–3678. DOI:10.1038/onc.2011.531
- Tam WL, Lu H, Buikhuisen J, et al. Protein kinase C alpha is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell. 2013;24(3):347–364. DOI:10.1016/j.ccr.2013.08.005