Abstract
The objective was to assess the cost-effectiveness of population screening for alpha-1 antitrypsin (AAT) deficiency. The design was a Markov-based decision analytic model. Hypothetical cohorts were analyzed from birth and followed over time until death using Monte Carlo simulation. The following strategies were compared: 1) screen all newborns, 2) screen all 10-year-old children, and 3) do not screen. Screenees found to have PI*ZZ AAT deficiency received the benefits of lower smoking rates and were offered augmentation therapy. In keeping with reported experience, most (96%) non-screened AAT deficient individuals remained undiagnosed and, therefore, missed these benefits. Under base conditions, screening all newborns cost nearly $422,000 per quality-adjusted life-year (QALY) gained; this estimate fell to $92,135 per QALY when the cost of screening was minimized to $6 in the model. Delaying screening until age 10 decreased the incremental cost-effectiveness ratio (ICER) to nearly $317,000. In sensitivity analysis, when the prevalence of PI*ZZ individuals increased from a baseline of 1.96 to 16 per 10,000, the ICER for newborn screening decreased below $100,000 per QALY. When the cost of screening and augmentation therapy were decreased simultaneously with increasing PI*ZZ prevalence, there were many scenarios in which the ICER decreased below $50,000. While population-based screening for AAT deficiency is not cost-effective under current conditions, cost-effectiveness criteria could be satisfied when case-finding in a high prevalence population is undertaken.
INTRODUCTION
In the era of the Human Genome Project and as the possibility of population screening for multiple health-threatening genetic conditions approaches, the advisability of such testing becomes a pressing scientific and societal issue [Citation[1]]. Indeed, studies to date of the impact of population screening for various genetic conditions have shown mixed results, with some studies supporting the health benefits of screening (e.g., better anthropometric characteristics of children who were identified with cystic fibrosis at birth) [Citation[2]] and others suggesting deleterious effects (e.g., psychologic strain on families and an invitation to genetic discrimination against screenees found to have the condition being studied) [Citation[3], Citation[4], Citation[5]]. In the specific instance of alpha-1 antitrypsin deficiency, two large-scale population-based screening studies have been conducted [Citation[3], Citation[6], Citation[7], Citation[8]], again with mixed results. On the one hand, individuals identified with severe (PI*ZZ) deficiency at birth were less likely to ever try smoking or to continue smoking if tried than their age-matched adolescent peers [Citation[9]]. On the other hand, as screening was conducted in Sweden, identification of individuals with PI*ZZ deficiency was associated with adverse psychological effects, especially regarding family dynamics [Citation[10]].
Importantly, unlike in cystic fibrosis [Citation[11]], none of the available screening studies in alpha-1 antitrypsin deficiency was designed as a randomized trial and all preceded the availability of specific augmentation therapy, which consists of regular intravenous infusion of pooled human plasma alpha-1 antiprotease to restore serum levels above a “protective threshold” value. As such, available analyses of the impact of population screening for alpha-1 antitrypsin deficiency have neither considered the possible benefits of earlier augmentation therapy (which has been associated with improved survival and a slower rate of forced expiratory volume in one second (FEV1) decline in available observational studies) [Citation[12], Citation[13]] nor the expense of augmentation therapy in the full analysis of population screening [Citation[14]]. In the context that full analysis of the impact of population screening for alpha-1 antitrypsin deficiency will require multi-decade longitudinal follow-up studies, attempts to elucidate the full impact of screening (i.e., with smoking avoidance counseling and augmentation therapy for alpha-1 antitrypsin deficient screenees who develop progressive airflow obstruction) invite modeling techniques.
To examine whether population screening for alpha-1 antitrypsin deficiency is cost-effective, we conducted a decision analysis that addressed 3 questions:
What is the cost-effectiveness of newborn population screening for AAT deficiency? i.e., the cost per quality-adjusted life-year of population screening when current augmentation therapy is used.
What is the cost-effectiveness of population screening for AAT deficiency in school-aged children (before the age of smoking initiation)?
What, if any, conditions must be satisfied in order for population screening to be deemed cost-effective using current, conventional criteria for cost-effectiveness?
METHODS
Decision model and natural history data ()
We performed a cost-effectiveness analysis of screening for AAT deficiency from the health care system perspective. A Markov-based decision analytic model was developed to assess three screening strategies: 1) screen all newborns for AAT deficiency with heelstick blood test; 2) screen all 10-year-old children; and 3) do not screen for AAT deficiency. The model explicitly accounted for effects of a screening program such as the cost of screening, the lower frequency of smoking in individuals with knowledge of their PI*ZZ-positive status, and the costs and benefits of treatment with pooled human plasma alpha-1-proteinase inhibitor in patients who are aware of disease.
Table 1. Summary of important natural history variables
A cohort of 50,000 individuals, weighted according to the prevalence of AAT deficiency, was evaluated in a Monte Carlo simulation. Based on results from the largest population-based newborn screening program in the United States, the frequency of AAT deficiency was estimated to be 1.96 in 10,000 newborns [Citation[6]]. Consistent with the autosomal inheritance of AAT deficiency, the study population was 50% female.
Persons in the model entered a Markov process with annual transition cycles, during which they could transition to another health state or remain in their current one (). Newborns were designated as AAT deficient (PI*ZZ) positive or AAT replete, based on the estimated prevalence of the disease (1.96 in 10,000 newborns). Alpha-1 antitrypsin deficient newborns transitioned to a health state representing the first two decades of life, during which there was assumed to be no lung dysfunction [Citation[15]]. The PI*ZZ individuals who survived the first 20 years of life then transitioned into a cascade of worsening lung function, leading to possible lung transplantation. All individuals were followed in the model until death. Population-based life table annual mortality rates were used for individuals without AAT deficiency.
Figure 1 Markov model of screening newborns for severe AAT deficiency.
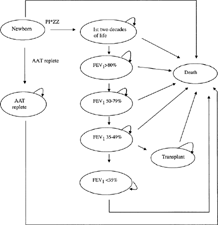
Impaired survival in individuals with AAT deficiency in the first 20 years of life was ascribed to hepatic dysfunction. Individuals who were screened had a higher likelihood of being diagnosed and, based on follow-up results from the two large population-based screening studies to date, a lower likelihood of both starting and continuing smoking [Citation[8], Citation[15]]. Alpha-1 antitrypsin deficiency smokers experienced an accelerated rate of yearly decline in lung function. In keeping with the reported natural history of lung dysfunction, the model reflects the possibility that lung dysfunction begins in the third decade of life [Citation[12]].
As in an earlier analysis of the cost-effectiveness of intravenous augmentation therapy [Citation[16]], the states of lung dysfunction used in this model were based on the American Thoracic Society COPD staging system [Citation[17]] (FEV1 > 80% predicted, Stage I 50–79%, Stage II 35–49%, and Stage III < 35% predicted). All surviving individuals started year 20 with full lung function (i.e., FEV1 = 100% predicted). As in our prior analysis [Citation[16]], the rate of annual decline of lung function and the associated increase in mortality were based on rates from the NHLBI Registry for Individuals with Severe Deficiency of Alpha-1 Antitrypsin [Citation[12], Citation[18], Citation[19]]. As lung function declined over time, individuals passed through stages of more severe airflow obstruction [Citation[17]] and increased risk of mortality. The annual decrement in lung function was determined independently for each cycle by sampling from the distribution of FEV1 decline for the patient's current health state based on NHLBI Registry findings [Citation[12]]. For AAT deficient smokers, this decline was more rapid. Lung function-dependent annual mortality rates were also based on observations from the NHLBI Registry [Citation[12]]. Alpha-1 antitrypsin-replete smokers were assumed to have a 13.85 year reduction in life span compared with non-smokers [Citation[20]]. As in our earlier analysis [Citation[16]], an estimated 19% of patients with severely compromised lung function (i.e., FEV1 < 15% predicted) received lung transplantation.
DATA version 4.0 by TreeAge Software, Inc. (Williamstown, MA) and Microsoft Excel (Seattle, WA) were used for all analyses.
Cost data ()
Direct medical costs related to screening and treatment of AAT deficiency were considered in the analysis. The cost of screening for AAT deficiency was based on a large-scale cost projection analysis that includes fixed and variable costs of the initial and confirmatory genotyping tests [Citation[21]]. The costs associated with treating different stages of AAT deficiency-related lung dysfunction were based on a cost analysis of COPD which assessed the cost of medications, oxygen therapy, laboratory and diagnostic tests, clinic and emergency department visits, and hospitalizations associated with COPD, stratified by American Thoracic Society COPD stages [Citation[22]]. The costs of lung transplantation and post-transplantation care were based on previously published pharmacoeconomic analyses [Citation[23], Citation[24]].
Table 2. Summary of important cost and quality-of-life variables
For individuals who were treated with augmentation therapy, therapy costs were based on the actual wholesale price ($0.22 per mg) plus 2003 Medicare reimbursement rates for weekly 1-hour infusions ($113) of Prolastin (Talecris BioTherapeutics, Research Triangle Park, North Carolina). The base case analysis assumed a 70 kg individual who receives weekly 1-hour augmentation therapy infusions of 60 mg/kg (i.e., 4200 mg once weekly). To account for medication wastage owing to the 500 mg vial size, the cost calculation for augmentation therapy was conducted assuming nine 500 mg vials were combined to make up the 4,200 mg dose.
All costs were adjusted to year 2003 U.S. dollars (USD$) using the medical services component of the Consumer Price Index.
Quality-of-life data ()
To account for the wide range in quality of life between the health states in our model, we explicitly incorporated quality-of-life into our model using quality-adjusted life-years (QALYs). A QALY is a health outcome measure that adjusts each unit of time by a quality factor called a utility. Utility weights range from 0 (for health states equivalent to death) to 1 (for perfect health) and are multiplied by the unit of time to calculate a QALY. As in a prior study [Citation[16]], estimates of utility weights for each health state in the model were based on expert opinion from pulmonologists experienced in treating AAT deficiency using the Health Utilities Index (Mark III) [Citation[25]].
Cost-effectiveness analysis
Screening strategies were compared based on their incremental cost-effectiveness ratio (ICER). The ICER is calculated by dividing the incremental (additional) mean cost, of the more costly strategy by the incremental effectiveness of that strategy. The effectiveness of each treatment strategy in this model was calculated by summing all QALYs for patients accumulated during their lifetime. An annual discount rate of 3% was applied to both costs and effectiveness, as recommended by the Panel for Cost Effectiveness [Citation[26], Citation[27]].
A cost-effectiveness analysis was conducted to evaluate the main goals of our project: to assess population screening of newborns and to assess screening of adolescents. Both strategies were compared to the strategy of not screening. We considered strategies costing less than $50,000 per QALY gained to be cost effective; and less than $100,000 per QALY to be possibly cost effective.
Model assumptions
Our model assumes the following conditions: 1) Based on available evidence regarding the clinical efficacy of augmentation therapy, all PI*ZZ individuals who are screened receive augmentation therapy when their FEV1 predicted falls below 50% predicted; 2) All patients, including those who do not receive augmentation therapy, receive standard medical care for COPD; 3) Lung transplantation is considered only when the FEV1 is below 30% predicted; 4) The annual probability of death for lung transplant recipients is the same as for those with FEV1 less than 50% predicted; 5) In keeping with reported rates of detecting individuals with severe AAT deficiency in clinical practice [Citation[28]], 4% of non-screened PI*ZZ individuals are diagnosed with AAT deficiency. We assumed the diagnosis occurred in the first two decades of life; 6) Eight percent of screened and 21% of non-screened PI*ZZ individuals smoke, respectively.
Sensitivity analyses
To assess the impact of parameter estimate uncertainty on the outcome of our analysis, we conducted a series of sensitivity analyses. Sensitivity ranges used in the analysis are listed in and . One-way sensitivity analysis was performed by adjusting all model estimates individually to the limits of their sensitivity ranges. Sensitivity ranges for annual FEV1 decline and probability of death were determined by 95% confidence intervals. Costs and utility weights were halved and doubled. The probability of receiving treatment with augmentation therapy if diagnosed with severe deficiency of AAT was varied between 1 and 0.5. The annual discount rate was varied from 0% to 6%. The annual probability of receiving a lung transplant if the FEV1 declines below 30% expected was varied between 5% and 30%. The prevalence of smoking was set to values between 4% and 16% for screened individuals; and 10.5% and 42% for unscreened individuals. To assess the impact of changing the time and likelihood of diagnosis in unscreened individuals, the probability of diagnosis in unscreened individuals was set to zero. In order to express outcomes in terms of life expectancy (i.e., in life-years), the effect of setting the discount rate to 0% and assigning utility weights equal to 1 for all health states was also analyzed.
Other multi-way sensitivity analyses were conducted to assess the impact of adjusting more than one variable simultaneously. The following groups of variables were adjusted simultaneously in the same direction to the upper and lower limits of their sensitivity ranges: the rate of lung function decline for all lung function states, the utility of all lung function states, and the costs of screening, of augmentation therapy, and of treating different stages of COPD. Also, the mortality rate for those treated with augmentation therapy was set low while simultaneously setting the non-treated individuals' mortality high. This same pattern was repeated for the rates of lung function decline. The smoking rate for screened PI*ZZ individuals was halved while simultaneously doubling the smoking rate for unscreened PI*ZZ individuals. For the adolescent screening analysis, the age when screening occurs was varied between ages 10 and 15 years. Also, the smoking rate for screened and unscreened adolescent PI*ZZ individuals was halved and doubled, respectively.
To begin to explore the conditions under which population screening of newborns might be cost-effective, a series of threshold analyses were conducted. In these analyses, the cost of screening, the cost and effectiveness of augmentation therapy, and the prevalence of disease in the screened population were varied individually to determine the point at which the ICER of screening fell below $100,000 and $50,000 per QALY gained, respectively. We also explored the effects of simultaneously reducing the cost of screening and augmentation therapy. These analyses were repeated for individuals screened as adolescents.
Next, guided by the findings from the one-way threshold analysis, we conducted a series of analyses in which more than one variable was varied simultaneously. For these multi-way analyses, the sensitivity ranges were greatly expanded. This was done to accommodate future changes in the cost and effectiveness of screening and therapeutic modalities. This analysis could be used to guide future efforts to improve the efficiency of population screening. In this analysis, the cost of screening was reduced from current cost ($57) to 5% of the current cost (i.e., to $3). The cost of treatment with augmentation therapy was similarly adjusted (range: $2,954–$59,086 per year). The effectiveness of augmentation therapy to slow lung function decline was raised from current levels to 190% of the rate observed in the NHLBI Registry. Finally, the prevalence of disease in the screened population was varied between 1.96 per 10,000 (0.0196%) and 250 per 10,000 (2.5%).
For all sensitivity analyses, a variable was considered potentially influential and was analyzed in further detail if it could push the ICER below $100,000 per QALY gained.
RESULTS
Under base case conditions, the incremental cost-effectiveness of newborn population screening for AAT deficiency was $421,954 per QALY gained (). Delaying screening until age 10 decreased the ICER to $316,635 per QALY, still well above the $50,000 criterion often considered an upper limit for cost-effectiveness.
Table 3. Incremental cost-effectiveness analysis of screening strategies
In one-way sensitivity analysis, the prevalence of PI*ZZ individuals in the screened population had a great impact on the ICER of newborn population screening (). Starting from a baseline prevalence of 1.96 per 10,000, small increases in the prevalence of PI*ZZ individuals in the screened population caused large decreases in the ICER of screening. The ICER of screening newborns decreased below $100,000 per QALY when the prevalence increased above 16 per 10,000. At a prevalence of 1%, the ICER of screening is approximately $60,000 per QALY gained. In other one-way sensitivity analyses, the ICER for screening decreased below $100,000 per QALY when the cost of screening was reduced to $7. The ICER did not decrease below $100,000 in any other one-way sensitivity analyses. Because of the sensitivity of the ICER to prevalence of PI*ZZ individuals in the screened population, we performed a series of multi-way sensitivity analyses using prevalence as one factor.
Figure 2 Relationship between prevalence of the PI*ZZ individuals in the population being tested and the incremental cost-effectiveness ratio (ICER) for screening.
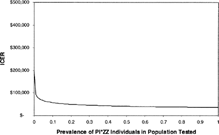
As the cost of screening and prevalence of PI*ZZ individuals were varied together, each factor had an effect on the ICER of population screening. However, as prevalence within the screened population increased, the impact of decreasing the cost of screening diminished. For example, at baseline prevalence (1.96 per 10,000) when the cost of screening was reduced from $57 to $6, the ICER decreased from $421,954 to $92,135 per QALY. In comparison, when the cost of screening was reduced from $57 to $6 in a population with a higher prevalence of PI*ZZ individuals (5 per 1,000), the ICER decreased from $69,785 to $56,918. There was no combination of prevalence and cost of screening for which the ICER was less than $50,000 per QALY gained.
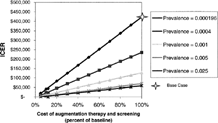
When the cost of screening, the cost of augmentation therapy, and prevalence of PI*ZZ individuals in the screened population were varied together, there were many scenarios under which the ICER of screening satisfied the criterion of less than $50,000 per QALY gained (). At the baseline prevalence observed in Oregon (1.96 per 10,000) [Citation[6]], if the cost of screening and augmentation therapy were simultaneously reduced to 12.5% of their baseline values ($7,386 and $7, respectively), the ICER of screening decreased below $50,000 per QALY. As the prevalence of PI*ZZ individuals increased in the screened population, the percent reduction in the cost of screening and augmentation therapy to achieve an ICER below $50,000 per QALY decreased. For example, at a prevalence of 1 per 1,000, the cost of augmentation and screening needed to be reduced to 40% of their initial value for the ICER to decrease below $50,000 per QALY. In comparison, when the prevalence increased to 5 PI*ZZ individuals per 1,000 population, the costs needed to be reduced to 72% of their initial value. At the highest prevalence assessed, 25 PI*ZZ individuals per 1,000 population, the costs needed to be reduced to 87% of their initial values for the ICER for population screening to decrease below $50,000 per QALY. Improving the effectiveness of augmentation did not appreciably change these results.
The effect of variation in the cost of screening, cost of augmentation therapy, and prevalence of PI*ZZ individuals in the target population on the incremental cost effectiveness ratio (ICER) for population screening for AAT deficiency. This figure demonstrates the effect of screening in populations with higher prevalence rates than a population-based sample. It simultaneously demonstrates the impact of reducing the cost of screening and augmentation therapy at each prevalence rate that was assessed. The range of prevalence rates of PI*ZZ individuals and costs assessed in this analysis was determined by making liberal assumptions regarding selection of high-risk populations and potential cost reductions. Our aim was to provide a very extensive analysis. The Figure shows that for population screening to be considered cost-effective, screening should occur in a population with much higher prevalence rates than current population-based estimates, costs of screening and augmentation therapy must be dramatically lowered from current levels, or both. On the other hand, the prevalence estimates to satisfy criteria for cost-effectiveness can be satisfied by testing a population in which alpha-1 antitrypsin deficiency is suspected with reasonable frequency (e.g., in a population of patients with known COPD).
The adolescent screening strategy was similarly affected by changes in model parameters. When the prevalence of PI*ZZ individuals in the screened population was increased to 11.5 per 10,000 (0.115%), the ICER of screening at 10 years of age was approximately $100,000. The ICER for screening adolescents at 15 years of age was $296,000. When the cost of screening and augmentation were reduced simultaneously to 17% of their baseline values ($7 and $10,045, respectively), the ICER for screening 10 year old children decreased below $50,000 per QALY.
No other multi-way sensitivity analyses, including halving the smoking rate in screened individuals while simultaneously doubling the base rate of smoking in unscreened individuals, revealed conditions where population screening was associated with an ICER below $100,000 per QALY. For example, the ICER for the multi-way smoking rate sensitivity analysis was $329,000.
To assess the impact of discounting and quality of life adjustment on life expectancy (i.e., to report raw, unadjusted life-years gained by population screening), we simultaneously set the discount rates to 0% and utilities for all health states to 1. In this analysis, the estimated the average undiscounted increase in lifespan for screened PI*ZZ individuals to be 4.9 years. Therefore, assuming a prevalence of 1.96 per 10,000, population-based screening results in an unadjusted incremental gain of 9.6 years per cohort of 10,000 screenees.
Model validation
To validate the model and detect programming errors, the latest United States Life Table data (1999) estimates of life expectancy for all newborns (76.7 undiscounted years) was compared and found similar to the undiscounted life expectancy for non-PI*ZZ individuals in our model (76.6 years) [Citation[29]].
Also, median life expectancy estimates for AAT deficient smokers and non-smokers in our model (48 and 66 years) closely resembled estimates from 2 prior studies [Citation[12], Citation[13]] of AAT deficient smokers (40 and 49 years) and non-smokers (65 and 69 years).
DISCUSSION
The main finding of this analysis is that, under the conditions of currently available augmentation therapy (i.e., cost, treatment effect, etc.), true population screening for severe AAT deficiency is not cost effective, while case-finding in populations of sufficient prevalence has potential to be cost-effective if costs for augmentation therapy and screening are reduced. Specifically, in the base analysis, the ICER of screening for and treating severe AAT deficiency exceeded $400,000 for the strategy of screening all newborns. At the same time, these results were very sensitive to the prevalence of PI*ZZ individuals in the population tested and to the cost of screening. Therefore, large-scale testing for AAT deficiency would satisfy current criteria for cost-effectiveness (i.e., an ICER < $50,000) in many scenarios as these prevalence and cost factors are varied (). For example, when screening a population with a prevalence of 10 PI*ZZ individuals per 10,000 population (prevalence = 0.001), the ICER of screening is less than $50,000 per QALY if the cost of screening and augmentation therapy are each reduced to 40% of their baseline values. As the prevalence of the screened population increases, less cost reduction in screening and augmentation therapy is needed to satisfy the criterion for costeffectiveness.
There were also numerous scenarios under which population screening met our criteria as possibly cost-effective (i.e., ICER < $100,000 per QALY):
The cost of screening was less ≤ $7, or
The prevalence of PI*ZZ individuals in the population tested was at least 16 per 10,000 (0.16%).
When conditions of prevalence and the cost of screening are varied together, there are many scenarios in which the ICER reached less than $100,000, as long as the prevalence of PI*ZZ individuals in the screened population was at least 10 per 10,000. Interestingly, varying the cost and efficacy of augmentation therapy independently of prevalence had little impact of the ICER of screening. This is, in part, a dilutional effect due to the fact that even in the highest prevalence group analyzed (25 PI*ZZ individuals per 1,000 tested), very few of those screened would ultimately receive augmentation therapy.
In analyzing the model under current conditions of cost and treatment effectiveness, a number of factors conspire against the cost-effectiveness of population screening for severe alpha-1 antitrypsin deficiency: the relatively low prevalence of disease, the cost of screening, and the relatively long life span of the patients in both groups (i.e., if there were the same 10 year raw difference in life span but deaths occurred much earlier, the impact of discounting future life-years would be lessened). The cost of screening deserves comment in that, although seemingly low for a single individual ($57), this cost is multiplied many times to detect one individual with AAT deficiency.
Screening adolescents at 10 or 15 years of age, while not meeting the criteria of being cost-effective, did produce more favorable ICERs than newborn screening because the cost of screening was deferred, while the benefits were preserved (assuming screening occurs before smoking behavior begins). Deferring costs for 10–15 years ensures that the ICER for delayed screening will always be less than newborn screening, all other factors held equal. However, the ICER for adolescent screening is still relatively high (approximately $300,000) and the difficulty of administering an adolescent screening program would likely detract from the appeal of such a strategy.
In support of the current analysis, the model we used considered many factors deemed critical in assessing screening, including: the prevalence of severe AAT deficiency, the clinical impact of the disease, the cost and accuracy of genetic testing, the efficacy of augmentation therapy, and the likelihood that knowledge of genetic predisposition would change smoking behavior. At the same time, several important limitations of our analysis warrant comment. First, we did not account for disutility associated with negative psychological or social effects of genetic testing. Indeed, available results regarding the psychosocial impact of screening are mixed. For example, Sveger et al. observed no excess psychosomatic complaints among PI*ZZ screenees than among AAT replete controls [Citation[30]]. Also, Helton et al. observed no excess adverse psychologic effects among parents of children found to have cystic fibrosis (CF) with screening compared to parents of traditionally diagnosed CF children [Citation[31]]. At the same time, McNeil et al. observed long-term adverse effects on mother-child interactions in families of PI*ZZ screenees [Citation[32]]. The impact of not explicitly accounting for this potential disutility is minimized in that it would only detract from the cost effectiveness of screening, which was not cost effective under base conditions. However if this analysis was extended to a population of higher prevalence where screening might be cost effective, the issue of accounting for disutility of knowledge of positive screen would need to be revisited. We believe that the current analysis serves as an important springboard to this discussion.
A second potential limitation of the current study is that we did not formally assess other detection strategies, such as case-finding among asymptomatic individuals known to have an elevated risk for AAT deficiency or targeted testing for AAT deficiency among individuals with fixed airflow obstruction. At the same time, our finding that the ICER was strongly influenced by the prevalence of severe AAT deficiency in the target population suggests that more directed approaches such as these are more cost-effective than newborn or school-age population screening. This finding suggests the current program of state wide targeted case-finding (e.g., that being conducted under the auspices of the Alpha-1 Foundation in Florida) has potential to be cost-effective under the right set of conditions [Citation[33]]. (e.g., the prevalence of PI*ZZ individuals in the population is 2.5%).
In conclusion, this analysis addresses an important but heretofore unanswered question: Is population screening for alpha-1 antitrypsin deficiency cost-effective? In the absence of long-term available longitudinal studies in which the cost of population screening was compared with usual diagnostic testing, inclusive decision analyses such as this offer the best available strategy to address this question. Our analysis suggests that strategies of screening newborns or school-age children for alpha-1 antitrypsin deficiency are not cost-effective under conditions of currently available treatment. However, our model identifies conditions under which testing for AAT deficiency would satisfy currently conventional criteria for a cost-effective intervention and suggests that large scale testing in a suitably enriched target group could satisfy cost-effectiveness criteria.
GLOSSARY
AAT = alpha-1 antitrypsin
CF = cystic fibrosis
COPD = chronic obstructive pulmonary disease
FEV1 = forced expiratory volume in 1 second
ICER = incremental cost-effectiveness ratio
NHLBI = National Heart, Lung, and Blood Institute
QALY = quality-adjusted life year
This work was conducted at The Cleveland Clinic Foundation and The Johns Hopkins Hospital. Keywords: Costs and cost analysis, Genetic screening, Alpha-1 antitrypsin deficiency.
REFERENCES
- Khoury M J, McCabe L L, McCabe E RB. Population screening in the age of genomic medicine. N Engl J Med 2003; 348: 50–58, [CSA]
- Farrell P M, Kosorok M R, Laxova A, Shen G, Kosaik R E, Bruns W T, Splaingard M, Mischler E H. Nutritional benefits of neonatal screening for cystic fibrosis. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. N Engl J Med 1997; 337: 963–969, [CSA]
- Sveger T, Thelin T. Four-year-old children with alpha 1-antitrypsin deficiency. Clinical follow-up and parental attitudes towards neonatal screening. Acta Paediatr Scand 1981; 70: 171–177, [CSA]
- Clayton E W. Ethical, legal, and social implications of genomic medicine. N Engl J Med 2003; 349: 562–569, [CSA]
- McCabe E R. Clinical genetics: compassion, access, science, and advocacy. Genet Med 2001; 3: 426–429, [CSA]
- O'Brien M L, Buist N R, Murphey W H. Neonatal screening for alpha1-antitrypsin deficiency. J Pediatr 1978; 92: 1006–1010, [CSA]
- Sveger T, Mazodier P. Alpha 1-antitrypsin screening of 18-year-old men. Thorax 1979; 34: 397–400, [CSA]
- Piitulainen E, Sveger T. Effect of environmental and clinical factors on lung function and respiratory symptoms in adolescents with alpha1-antitrypsin deficiency. Acta Paediatr 1998; 87: 1120–1124, [CSA]
- Sveger T, Thelin T, McNeil T F. Young adults with alpha 1-antitrypsin deficiency identified neonatally: their health, knowledge about and adaptation to the high-risk condition. Acta Paediatr 1997; 86: 37–40, [CSA]
- Thelin T, McNeil T F, Aspegren-Jansson E, Sveger T. Psychological consequences of neonatal screening for alpha 1-antitrypsin deficiency. Parental reactions to the first news of their infants' deficiency. Acta Paediatr Scand 1985; 74: 787–793, [CSA]
- Fost N, Farrell P M. A prospective randomized trial of early diagnosis and treatment of cystic fibrosis: a unique ethical dilemma. Clin Res 1989; 37: 495–500, [CSA]
- The Alpha-1-Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha 1-antitrypsin. Am J Respir Crit Care Med 1998; 158: 49–59, [CSA]
- Seersholm N, Dirksen A, Kok-Jensen A. Airways obstruction and two year survival in patients with severe alpha 1-antitrypsin deficiency. Eur Respir J 1994; 7: 1985–1987, [CSA]
- Stoller J K. Augmentation therapy for severe alpha 1-antitrypsin deficiency: is the jury still out on a trial?. Thorax 1998; 53: 1007–1009, [CSA]
- Wall M, Moe E, Eisenberg J, Powers M, Buist N, Buist A S. Long-term follow-up of a cohort of children with alpha-1-antitrypsin deficiency. J Pediatr 1990; 116: 248–251, [CSA]
- Gildea T R, Shermock K M, Singer M E, Stoller J K. Cost-effectiveness analysis of augmentation therapy for severe alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2003; 167: 1387–1392, [CSA]
- American Thoracic Society. Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1995; 152: S77–S121, [CSA]
- McElvaney N G, Stoller J K, Buist A S, Prakash U B, Brantly M L, Schluchter M D, Crystal R G. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of alpha 1-antitrypsin deficiency. Alpha 1-Antitrypsin Deficiency Registry Study Group. Chest 1997; 111: 394–403, [CSA]
- The Alpha 1-Antitrypsin Deficiency Registry Study Group. A registry of patients with severe deficiency of alpha 1-antitrypsin. Design and methods. Chest 1994; 106: 1223–1232, [CSA]
- CDC. Annual smoking-attributable mortality, years of pitential life lost, and economic costs–United States, 1995–1999. MMWR 2002; 51: 300–303, [CSA]
- Brantly M. Personal communication. 2003
- Hilleman D E, Dewan N, Malesker M, Friedman M. Pharmacoeconomic evaluation of COPD. Chest 2000; 118: 1278–1285, [CSA]
- Ramsey S D, Patrick D L, Albert R K, Lewis S, Albert R K, Raghu G. The cost-effectiveness of lung transplantation. A pilot study. University of Washington Medical Center Lung Transplant Study Group. Chest 1995; 108: 1594–1601, [CSA]
- Sharples L D, Taylor G J, Karnon J, Caine N, Buxton M, McNeil K, Wallwork J. A model for analyzing the cost of the main clinical events after lung transplantation. J Heart Lung Transplant 2001; 20: 474–482, [CSA]
- Feeny D, Furlong W, Boyle M, Torrance G W. Multi-attribute health status classification systems. Health Utilities Index. Pharmacoeconomics 1995; 7: 490–502, [CSA]
- Siegel J ES, Weinstein M CP, Russell L BP, Gold M R. Recommendations for reporting cost-effectiveness analyses. JAMA 1996; 276: 1339–1341, October 23/30[CSA]
- Gold M R, Siegel J E, Russell L B. Cost-effectiveness in health and medicine. Oxford University Press, New York 1996
- Silverman E K, Miletich J P, Pierce J A, Sherman L A, Endicott S K, Broze G J, Jr., Campbell E J. Alpha-1-antitrypsin deficiency. High prevalence in the St. Louis area determined by direct population screening. Am Rev Respir Dis 1989; 140: 961–966, [CSA]
- Anderson R, De Turk P. United States Life Tables, 1999. National Vital Statistics Reports, 2002; 50
- Sveger T, Thelin T, McNeil T F. Neonatal alpha 1-antitrypsin screening: parents' views and reactions 20 years after the identification of the deficiency state. Acta Paediatr 1999; 88: 315–318, [CSA]
- Helton J L, Harmon R J, Robinson N, Accurso F J. Parental attitudes toward newborn screening for cystic fibrosis. Pediatr Pulmonol Suppl 1991; 7: 23–28, [CSA]
- McNeil T F, Harty B, Thelin T, Aspegren-Jansson, Sveger T. Identifying children at high somatic risk: long-term effects on mother-child interaction. Acta Psychiatr Scand 1986; 74: 555–562, [CSA]
- Brantly M L, Mishra V S, Viranovskaya N. Statewide targeted screening and detection of alpha-1 antitrypsin deficient individuals. Am J Respir Crit Care Med 2003; 167: A222, [abstract][CSA]