Abstract
In alpha 1-antitrypsin deficiency in humans, inadequately regulated activity of serine protease activity is responsible for the chronic lung tissue degeneration and irreversible loss of pulmonary function seen in those individuals with emphysema. Typically, disease symptoms in this patient population are exacerbated by cigarette smoke. Here we show that inhaled recombinant alpha 1-antitrypsin (rAAT) can provide significant protection against the development of emphysema in cigarette smoke-treated mice. As has been reported previously, cigarette smoke was seen to increase significantly the recruitment of neutrophils and macrophages into the lungs of these animals, leading to concomitant alveolar airspace enlargement and emphysema. In smoking animals treated for 6 months with inhaled rAAT, effects on lavage levels of neutrophils and macrophages were only moderate when compared with untreated animals. Furthermore, neutralizing antibodies to rAAT were generated in all rAAT-treated animals. Despite this, however, reductions in airspace enlargement of up to 73% were observed. These findings demonstrate that delivery of rAAT directly to the lungs of smoke-treated mice can inhibit lung tissue damage mediated by proteases, suggesting that rAAT inhalation therapy might represent a practical approach towards treating emphysema in humans, by modifying the course of the disease.
INTRODUCTION
Inflammatory cell-derived proteases are known to be intimately involved in the development of emphysema (Citation[1], Citation[2], Citation[3], Citation[4]). For example, in individuals with the inherited form of emphysema, greatly reduced levels of circulating alpha 1-antitrypsin (AAT), the major physiological inhibitor of neutrophil elastase (NE), lead to uncontrolled proteolytic activity and the progressive loss of lung function through the degradation of lung elastin (Citation[5], Citation[6], Citation[7]). In addition to this direct deleterious activity, unregulated NE has additional upstream activities that contribute to inflammation and reduced lung function in other forms of chronic obstructive pulmonary disease (COPD) such as in chronic bronchitis (Citation[8]). Similarly, abnormally high levels of NE have been proposed to account for enhanced lung damage in cystic fibrosis (CF) (Citation[9]), and in bronchopulmonary dysplasia (BPD) in premature neonates (Citation[10]). Thus, as the most potent secretagogue known for mucin production, the major generator of neutrophil-chemotactic elastin degradation fragments, an activator of matrix metalloproteases (MMPs), and an inactivator of endogenous MMP inhibitors (Citation[11], Citation[12], Citation[13], Citation[14]), the serine protease NE, when incorrectly regulated, clearly plays a key role in the development of chronic lung diseases in humans.
Since it is well known that cigarette smoke exacerbates significantly the progression of emphysema in AAT-deficient individuals (Citation[7]), it has been proposed that NE also plays a central role in the development of emphysema in smokers who are not necessarily diagnosed as being AAT-deficient. Indeed, further support for this proposal has been established with recent findings from large-scale screening for AAT genotypes in individuals with COPD (Citation[15]). Not only are the homozygous deleterious ZZ genotypes found at an expected high frequency, but heterozygosity of the Z allele with other AAT alleles, such as the normal or M form, is also found at a greatly elevated frequency in such individuals (Citation[15]). These findings of reduced levels of AAT in a substantial proportion of individuals with COPD complement earlier hypotheses and empirical observations that cigarette smoke can reduce AAT activity in the human lung. For example, it has been shown that AAT isolated from the sputum of smokers with COPD exhibits greatly reduced activity due to both oxidation of the protein (Citation[16]) and by proteolytic degradation of endogenous AAT by MMPs (Citation[7]).
In emphysema associated with cigarette smoking, a significant role for MMPs in lung tissue degradation has also been established (Citation[17], Citation[18], Citation[19], Citation[20]). This class of protease is known to be important in the development of smoke-induced emphysema as well as other forms of COPD. Such studies have also implicated tumor necrosis factor-α (TNF-α) as a central mediator of this process (Citation[21]). Indeed, Churg et al. have shown that MMP-12 promotes smoke-induced inflammation by releasing TNF-α from macrophages, with subsequent endothelial cell activation, neutrophil influx, and proteolytic breakdown of elastin by excess NE activity (Citation[22]).
Given the above, therefore, it has been difficult to dissect the exact contributions of each class of protease in the degeneration of lung tissue caused by cigarette smoke in animal model systems and in humans. Furthermore, the relatively low fractional incidence of emphysema in even heavy smokers shows that additional, complex genetic factors are likely to be involved in human cigarette smoke-induced emphysema (Citation[23]). That notwithstanding, however, mouse model systems have been used to great effect in rationalizing key roles for both MMPs and NE in disease progression (Citation[14], Citation[24], Citation[25]). For example, transgenic knockout mice that are deficient in macrophage elastase (MMP-12) production do not develop emphysema upon prolonged exposure to cigarette smoke (Citation[20]). Similarly, marked attenuation of emphysema has been observed in wild-type mice treated orally (Citation[26]), and in guinea pigs treated subcutaneously (Citation[27]) with broad-spectrum MMP inhibitors (MMPIs).
More recently, two studies have extended the use of animal model systems by addressing practical issues related to the potential use of protease inhibitor therapy for the treatment of human emphysema. First, our groups have shown recently that the small molecule, broad-spectrum MMP inhibitor ilomastat can prevent the development of emphysema in smoking mice, as evidenced by almost total inhibition of airspace enlargement (Citation[28]). In a second model therapeutic approach, Churg et al. have extended the possibility of using AAT for the treatment of smoking-related emphysema by showing that intraperitoneally-injected plasma-derived AAT can ameliorate the development of airspace enlargement in smoking mice (Citation[29]). Indeed, the development of emphysema was inhibited by over 60% during a six-month period of daily smoke treatment. The importance of this observation stems from the fact that infused plasma-derived AAT (pAAT) has been used for almost three decades in the replacement therapy of symptomatic AAT-deficient individuals (Citation[7]). Although being an impractical approach for COPD therapy because of pAAT pricing and manufacturing constraints, the study of Churg et al. has, however, shown that concerns regarding the inactivation of AAT by cigarette smoke, reactive oxygen species, or excessive MMP activity found in the smoker's lung, may be overcome by systemic pAAT therapy, albeit at exceptionally high doses.
In this study, we have combined elements of the above two studies, and we now show that low doses of recombinant AAT (rAAT) when delivered directly to the lung by inhalation, can reduce significantly the degradation of lung tissue induced by chronic exposure to cigarette smoke. Our results demonstrate for the first time that rAAT delivered via the airways can reach sites, such as the alveolar interstitium within murine lung tissue, that are sensitive to smoke-induced proteolytic degradation, suggesting strongly that when delivered by inhalation, rAAT has the potential to address a major, if not the major underlying cause of smoking-related emphysema in humans.
MATERIALS AND METHODS
Reagents
rAAT was produced in Saccharomyces cerevisiae by methods described in detail elsewhere (Citation[30]). The recombinant protein was purified to > 99% purity by column chromatography. Purity was assessed by reverse phase high performance liquid chromatography (RP-HPLC), and activity was determined essentially as described previously (Citation[31]). The PPE substrate Suc-Ala-Ala-Ala-pNA was purchased from Bachem (Torrance, CA). PPE was purchased from Roche Diagnostics (Indianapolis, IN). The LTB4 assay was obtained from R&D Systems (Minneapolis, MN). Results were read using a Molecular Devices VERSAmax microplate reader and SOFTmaxPro software (Sunnyvale, CA).
Preparation of solutions for aerosol delivery
A stock solution of rAAT was prepared by diluting a frozen rAAT solution (50 mg/ml) in 1X phosphate buffered saline (PBS), pH 7.4 (10 ml). The resulting solution was filtered (0.45 μ m Acrodisc; Gelman Inc.), and the concentration (mg/ml) of dissolved rAAT calculated based on the absorption coefficient of rAAT, followed by confirmatory RP-HPLC. This stock solution was further diluted with sterile PBS to make solutions of 1.15 mg/ml, 5.75 mg/ml and 11.5 mg/ml rAAT. Aliquots (20 ml) of these solutions were frozen at −70°C.
Aerosol generation and delivery system characterization
Aerosols were generated using an Aeroneb Pro nebulizer (Aerogen Inc., Mountain View, CA). The nebulizer was integrated into a single chamber plenum with five ports for exposing animals to aerosol and an inlet for airflow. This system was described in more detail previously (Citation[28]). Aerosols were generated at one end of the plenum and carried to the five exposure ports by a constant airflow (2.5 L/min) into and across the plenum. Confirmation that particle sizes were in the respirable size range (< 5 μ M) was obtained for various concentrations of rAAT, and control buffer using a Spraytec laser diffraction unit (Malvern Instruments, Southborough, MA).
Test animal exposure times and drug delivery
Studies were performed on A/J female mice, 12 weeks of age at study initiation. All animal procedures were approved by the Animal Studies Committee of the Washington University School of Medicine. Then, 10 mL of sterile saline or drug was placed in the nebulizer and delivered to each group of 5 animals, as described previously (Citation[28]). Between deliveries, the nebulizer was rinsed thoroughly with warm water. Drug was delivered 6 days per week, just prior to smoking. After delivery of drug, each smoking group was subjected to smoke from 2 non-filtered cigarettes per day (Kentucky 2R4F reference cigarettes, Tobacco Research Institute, University of Kentucky), 6 days per week for 6 months. Non-smoking, age-matched littermates were used as controls.
Study groups
This study was composed of 5 groups of mice. Group 1 animals (thirty-two) were control non-smoking animals. Group 2 animals (thirty-two) were administered aerosolized PBS (10 ml/5 animals) followed by cigarette smoke. Group 3 animals (thirty two) were similarly administered low-dose nebulized rAAT (1.15 mg/ml in PBS) followed by cigarette smoke. Group 4 animals (thirty two) were administered mid-dose nebulized rAAT (5.75 mg/ml in PBS) followed by cigarette smoke. Group 5 animals (thirty-two) received high-dose nebulized rAAT (11.5 mg/ml in PBS) followed by cigarette smoke. After 1 week and 6 months, 10 animals, and 12 animals, respectively, from each group were sacrificed, and bronchoalveolar lavage (BAL) samples collected as described previously (Citation[20], Citation[28]).
Inflammatory cell analysis
BAL samples were prepared as described above and analyzed for neutrophil and macrophage content. BAL return volume was measured and 100 ul was cytospun onto slides. Slides were stained with Leukostat Stain Kit. The numbers of each cell type were identified by nuclear shape and staining pattern and quantified by counting a 100-cell field. The total cell count and differential count was calculated.
Tissue processing and morphometry
BAL samples were analyzed for neutrophil and macrophage content as described previously (Citation[20], Citation[28]). The right lung was inflated to a fixed pressure (25 cm H2O) by instilling 10% buffered formalin (for 15 minutes), and it was then ligated and removed. Inflated lungs were fixed for 48 hours and then serially dehydrated before being embedded in paraffin. Serial sagittal sections were obtained for histological analysis. Lm, an indicator of airspace size was calculated for each mouse by a single, blinded observer (SDS) from 7 random fields at X200 as described previously (Citation[20], Citation[32]).
Chemiluminescent immunoblotting with 6-month serum samples
Triplicate samples of 1 μ g of the study lot rAAT and 15 μ g of yeast host cell protein impurities from the purification process were run on a 15-lane 4–12% SDS gel. The resolved proteins were transferred to a nitrocellulose membrane (VWR, San Francisco, CA) that was rinsed with PBS, and blocked with PBS containing 1% non-fat dry milk for 30 minutes. Blots were incubated overnight at room temperature with a 1:500-fold dilution in PBS of pooled 6-month serum samples from each of the 5 animal groups. Blots were washed three times each for 10 minutes, in PBS containing 0.05% Tween-20, incubated for 1 hour in a 1:25,000 fold dilution of goat anti-mouse IgG peroxidase conjugate (Sigma, St. Louis, MO), washed three times as above, developed using SuperSignal West Dura Substrate (Pierce, Rockford, IL), and then imaged using a Kodak Image Station 440CF.
Six-month serum IgG purification
Six month serum samples were combined into five pools from each of the animal groups, and used to purify IgG Abs using a Melon Gel IgG purification kit (Pierce Biotechnology, Rockford, IL). The purified samples were quantified using UV280 spectrophotometry, as recommended by the manufacturer.
PPE inhibition assay with purified IgG antibodies
rAAT (at 52.6 mg/mL) was diluted to 2.2 μ g/mL with assay activity buffer (50 mM Tris-HCl, 500 mM NaCl, pH 8.0, 0.01% BSA). The Abs were diluted to 88 μ g/mL. Diluted Abs and rAAT were mixed in triplicate wells, 25 μ L each, in a 96-well microtiter plate (15 mins). The inhibitor control contained 25 μ L of diluted rAAT and 25 μ L of purification buffer. PPE enzyme, diluted to deliver 0.00049 Units per 200 uL reaction in 50 μ L, was added to the wells, mixed and incubated 15 minutes at 30°C. Then, 100 μ L of 6 mM PPE substrate solution in assay activity buffer was added to each well, mixed, and incubated for 5 minutes at 30°C. This gives a concentration of rAAT that inhibits the PPE activity by 50% (275 ng/mL final reaction concentration). PPE positive and substrate controls were also included. A second positive control of IgG purified neutralizing polyclonal rabbit anti-human AAT (DakoCytomation, Denmark) was included, at 44 μ g/mL.
Statistics
All graphs show mean value +/− standard error of the mean. P values were determined by Student's unpaired two-tailed t-test using Microsoft Excel. p values < 0.05 were considered significant and marked in figures with one asterisk (*). p values < 0.01 are marked with two asterisks (**).
RESULTS
To measure the effects of inhaled rAAT on the development of emphysema in a smoking mouse model, we developed a high yielding process for the production of rAAT in yeast (Citation[30]), and isolated the protease inhibitor by column chromatography to a purity level of greater than 99%. Aqueous solutions of rAAT were nebulized into the lungs of test animals for up to six months, using nebulizers and chambers that were designed and built specifically for this type of experiment, and have been described previously (Citation[28]). Our approach to optimizing the dose level required to achieve significant reductions in emphysema development in the murine lung was to administer rAAT in amounts that were equimolar to those shown previously, using a matrix metalloprotease inhibitor (MMPI), that gave virtually quantitative reductions in airspace enlargement. When using this MMPI, ilomastat, in the identical model and with the same inhalation delivery device, a 96% reduction in airspace enlargement was observed at the mid-dose (Citation[28]). Based on the known efficiencies of delivery and deposition in the mouse lung versus the human lung, we estimated that the mid-dose level of delivered drug corresponds to an approximate human dose of 100mg of rAAT per day, using a high efficiency device (75% deposition into the deep lung; data not shown), with a consequent low-dose human equivalent of 20 mg of rAAT per day.
shows the effect on neutrophil levels of administering cigarette smoke to groups of mice for time periods of 1 week and 6 months, with or without concomitant treatment with inhaled rAAT at the dose levels noted above. Lavage neutrophil levels were highly variable and no statistically significant differences between groups were observed at the one week time point. At 6 months, however, although statistical significant elevations were not reached, lavage neutrophil levels increased in the smoking group compared to the non-smoking animals. In the low- and mid-dose rAAT-treated groups, there was a moderate reduction of lavage neutrophil levels at the 6-month time point compared with the smoking group and normalization to levels found in the non-smoking control group. In the high-dose group at 6 months, neutrophil levels were also lower than those in the smoking group but were, however, higher than the low- and mid-dose levels, possibly reflecting additional inflammatory processes arising from the inhalation of a heterologous recombinant human protein, with its obligatory, albeit low level trace host cell protein impurities, into the murine lung.
Figure 1. The effects of cigarette smoke and inhaled rAAT on lavage neutrophils. Time points are (A) 1 week and (B) 6 months. The groups included control animals, age-matched cigarette smoke-treated animals (Smoke) and three groups of animals that received both smoke exposure and inhaled rAAT at three dose levels; see “Materials and Methods.” There was an increase in lavage neutrophils in the smoking group at 6 months when compared with control animals, and a reduction in neutrophil levels in the inhaled rAAT Dose 1 and Dose 2 groups when compared with smoking, untreated animals. These levels, however, did not reach statistical significance.
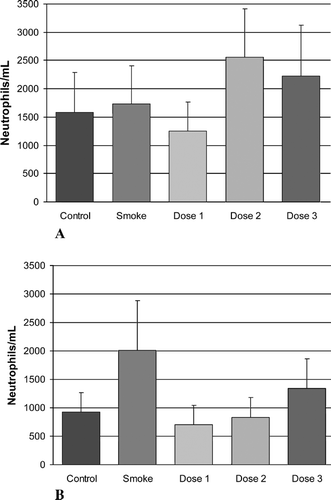
shows the effect on lavage macrophage levels of the above treatment. As has been observed previously, an increase in lavage macrophage levels was noted in the smoking group versus the control non-smoking group, at both time points, with statistical significance being reached at the 6-month time point. However, although reduced macrophage levels in the rAAT-treated groups were observed when compared with the smoking group, these reductions did not reach statistical significance.
Figure 2. The effects of cigarette smoke and inhaled rAAT on lavage macrophages. Time points are (A) 1 week and (B) 6 months. The groups included control animals, age-matched, cigarette smoke-treated animals (Smoke) and three groups of animals that received both smoke exposure and inhaled rAAT at three dose levels, see “Materials and Methods.” There was a significant increase (marked with one asterisk) in lavage macrophages in the smoking group at the 6-month time point when compared with control animals. At 6 months, there was a reduction in lavage macrophages in all three dose groups when compared with smoke-treated animals, but this did not reach statistical significance.
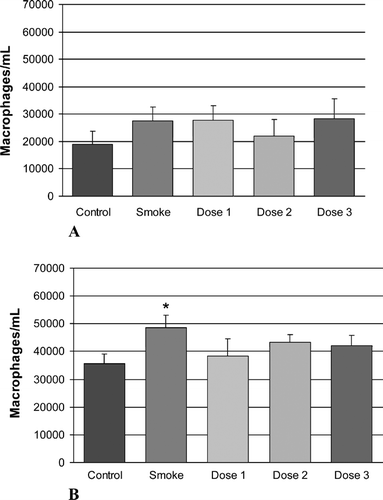
We also measured lavage levels of the inflammatory mediator LTB4 in lavage fluids at 6 months. shows that, as expected, levels of the inflammatory mediator LTB4 were elevated significantly in the smoking group at 6 months. However, although there was an amelioration of this elevation in the rAAT-treated groups, levels remained higher than in non-smoking control animals in all treated groups.
Figure 3. LTB4 measurements at the 6-month time point show a statistically significant increase in levels of this inflammatory marker in the lavage fluids of the smoke-treated group. Reduced levels of LTB4were observed in all rAAT-treated groups, but this reduction did not reach statistical significance.
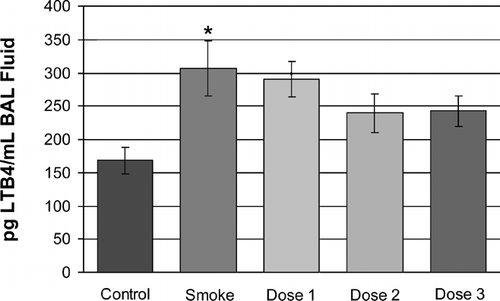
Presumably, as the sequelae of increased inflammatory cells and their proteolytic enzyme products, airspace size was increased by 15% in the smoke-exposed animals when compared with non-smoking controls (non-smoking control, 34.3± 0.74 μ m; p value = 0.0004 vs. smoker, smoke-treated, 39.5± 0.85 μ m) (). Also shown in , is the reduction in airspace enlargement (Lm) obtained by treating cigarette smoke-treated mice with inhaled rAAT at the dose levels described above. There was a striking reduction of airspace enlargement at 6 months in the low- and mid-dose groups (Dose 1 and Dose 2) of 71% and 73% respectively (35.8 ± 0.79 μ m and 35.7 ± 0.86 μ m; p values < 0.007 and < 0.008 vs. smoke-treated group, respectively). A somewhat lower reduction in airspace enlargement of 42% was noted in the high dose group (37.2 ± 0.86 μm; p value = 0.1 vs. smoke-treated group). This may be a consequence of additional inflammatory processes arising from the long-term inhalation of a heterologous recombinant human protein, with its obligatory trace host cell protein impurities, into the murine lung, which is likely correlated with the trend towards a reduced effect of our rAAT preparation on neutrophil influx in this Dose 3 study group.
Figure 4. Airspace enlargement in response to cigarette smoke exposure and rAAT treatment. Smoke-treated mice (Smoke) have significantly increased emphysema (p < 0.01, marked with two asterisks) when compared with age-matched non-smoke-exposed control mice (Control). rAAT-treated mice exhibited significantly less airspace enlargement in Dose 1 and Dose 2 groups (71% and 73% respectively, p < 0.01 vs. smoke-treated, marked with two asterisks). In the Dose 3 group there was a lower level of protection by rAAT (42%, p = 0.1 vs. smoke-treated) against the effects of chronic cigarette smoke administration, see Results.
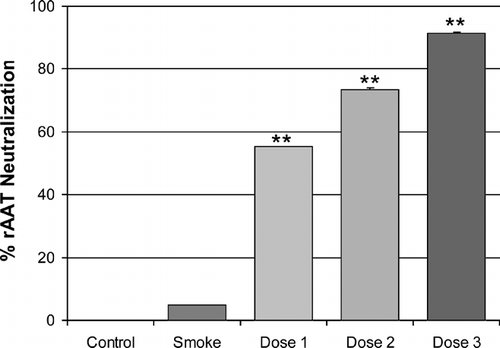
In order to measure the extent to which antibodies directed against the heterologous human rAAT protein could neutralize its activity and possibly impact its efficacy with respect to inhibiting the development of emphysema, we determined the reactivity of 6-month serum-derived IgG antibodies to rAAT by immunoblotting, and by measuring neutralization capacity in a porcine pancreatic elastase (PPE) assay. Reactivity of sera from all three treated groups was readily demonstrated by immunoblotting, with control sera from the vehicle buffer and smoke only-treated animals being negative (data not shown). Furthermore, under the conditions tested (see Materials and Methods), IgG antibodies purified from the sera of each of the rAAT-treated groups were shown to inhibit rAAT's activity against PPE in a dose-dependent manner ().
Figure 5. Human rAAT-neutralizing antibodies are generated by the inhaled protein. Murine IgG antibodies were purified from each group of animals and were used to inhibit the activity of rAAT against PPE in a microtiter plate assay. Pooled sera from animals that were not treated with rAAT (Control, Smoke) exhibited little or no rAAT-neutralizing activity. Under the conditions tested in this study (see “Materials and Methods”), purified IgG antibodies from the pooled sera of mice from the smoking groups that were treated with rAAT, inhibited rAAT's activity against PPE in a dose-dependent manner. Each data point was derived from triplicate assays with the 0% point defined as rAAT inhibition alone with no antibody, and the 100% point defined as PPE alone with no rAAT and no antibody. As a further positive control for rAAT-neutralization, purified commercially available anti-plasma AAT rabbit IgG was shown to be capable of inhibiting rAAT's activity quantitatively (data not shown).
DISCUSSION
In this study, we have shown that a recombinant DNA-derived version of the natural NE inhibitory protein AAT, when delivered by inhalation, can reduce significantly the effects of chronic cigarette smoke on airspace enlargement in susceptible mice. That the maximal levels of protection against smoke-induced airspace enlargement were observed in the low- and mid-dose treated groups (71% and 73%, respectively) is somewhat surprising. The extent of the protection, however, correlates well with previous studies using NE knockout mice (NE-/- mice). In these transgenic mice, an approximately 60% protection at 6 months was observed (Citation[14]). Interestingly, we and others have shown that complete protection can be observed using broad spectrum MMPIs (reviewed in ref. 4, 28). It is possible, therefore, that the residual proteolytic degradation contributing to the almost 30% increase in airspace enlargement in the present study is derived from the well-established elevated MMP activities of the smoke-treated lung, although other mechanisms cannot be excluded. For example, cysteinyl proteinases may be involved, either directly through matrix proteolysis, or through mechanisms that lead to enhanced levels of alveolar cell apoptosis and consequent alveolar destruction (Citation[2], Citation[4], Citation[33]). A simplified model involving the NE and MMP pathways can be invoked to explain some of our findings (). In this model, we propose that substantial protection occurs through the direct inhibition by rAAT of NE-mediated elastin degradation. Further inhibitory effects may be obtained by protection of endogenous inhibitors of MMPs (TIMPs) against cleavage and inactivation by NE. This, in turn, leads to a diminution in the destructive effects of the MMPs through either inhibition of direct matrix degradation, or through the reduced activation and release of TNF-α, an inflammatory mediator that has been implicated strongly in the development of smoke-induced emphysema (Citation[21], Citation[22]).
Figure 6. Schematic representation of the proposed role for rAAT in interrupting the protease/protease inhibitor imbalance and resulting inflammatory cascade provoked by cigarette smoke. First and foremost, rAAT inhibits directly the degradation of elastin by NE. Further protection against matrix degradation may occur through lowering of both neutrophil and macrophage recruitment by reducing the production of chemotactic matrix breakdown products such as elastin-derived peptides (Citation[12]), and by protection of the endogenous tissue inhibitors of metalloproteases (TIMPs) against degradation by NE.
![Figure 6. Schematic representation of the proposed role for rAAT in interrupting the protease/protease inhibitor imbalance and resulting inflammatory cascade provoked by cigarette smoke. First and foremost, rAAT inhibits directly the degradation of elastin by NE. Further protection against matrix degradation may occur through lowering of both neutrophil and macrophage recruitment by reducing the production of chemotactic matrix breakdown products such as elastin-derived peptides (Citation[12]), and by protection of the endogenous tissue inhibitors of metalloproteases (TIMPs) against degradation by NE.](/cms/asset/805ebe14-12bf-4ba4-83f2-437a8b367bf4/icop_a_165098_uf0006_b.gif)
The seemingly paradoxical observation that the highest dose of rAAT gave a lower level of protection may arise from the fact that in this study we used a heterologous human protein that also contains low levels (< 1%) of yeast protein impurities. Thus, additional immunological and inflammatory processes could be contributing significantly to the eventual magnitude of the critical parameter under study, namely airspace enlargement. These could include activation of the innate immune system against rAAT or yeast cell impurities. In order to possibly dissect out the contributions of components of the drug substance, i.e., rAAT versus impurities, the mouse CD-1 strain that is transgenic for low levels of human AAT, could be used in future studies. These mice are tolerant to exogenous human pAAT (Citation[29]). Unlike pAAT, however, rAAT from yeast is not glycosylated and it is unclear what effect this would have on the immunogenicity of the nebulized rAAT protein in such a mouse strain. Furthermore, we chose to use the heterologous mouse model to determine the magnitude of the effect that neutralizing antibodies (NAbs), if generated, would have on the ability of rAAT to control alveolar destruction. This could be of high clinical significance, as NAbs have been observed in individuals undergoing long-term therapy with both native and recombinant human proteins (reviewed in 34).
Not unexpectedly, given the 6-month treatment period using a heterologous protein, we did indeed find IgG antibodies to rAAT. These antibodies could be purified and shown to neutralize rAAT's activity in an in vitro elastase assay. Although we cannot rule out other mechanisms whereby the high dose rAAT therapy led to the lowest protection against alveolar destruction, the dose-dependent neutralization of rAAT's activity represents one mechanism that would explain our observations. There is considerable debate on the role of NAbs in reducing the efficacy of human recombinant proteins (Citation[34]). Here we have shown that under the conditions tested, NAbs were generated against rAAT, but there still remained a high level of efficacy of the protease inhibitor in all three dose groups.
A number of preclinical animal model studies and human clinical studies have been performed wherein biosynthetic versions of AAT were delivered by inhalation (Citation[35], Citation[36], Citation[37]). Furthermore, an inhaled human pAAT preparation has been studied extensively in both primate models and in humans (Citation[38]). Previous studies with inhaled unglycosylated yeast-derived rAAT in humans have shown that the molecule can be delivered to the lung via nebulization, and can attain levels that appear to be capable of neutralizing excessive NE activity in human lung BAL fluids (Citation[35], Citation[37]). Furthermore, in this study in AAT-deficient individuals, yeast-derived rAAT was shown to be non-immunogenic under the conditions tested (Citation[35]). The combined data suggested that inhaled rAAT could be a viable alternative to infusion of plasma-derived AAT as a replacement therapy for this condition. Despite this, however, infused plasma-derived AAT has been the mainstay of replacement therapy for symptomatic AAT-deficiency for almost two decades. Although being the subject of much debate, several studies have shown that, at least in those AAT-deficient individuals with significantly depleted lung function, infused AAT can reduce the further loss of lung function as measured by spirometry or by high-resolution computed tomography (HRCT) (Citation[7], Citation[39]).
The high cost, limited availability and potential safety concerns of plasma-derived AAT have restricted any possibility of its use in the therapy of smoking-related emphysema. Furthermore, there has been much debate as to whether or not infused AAT could even have any effect on the course of this disease, given that most individuals suffering from this condition have normal levels of circulating AAT (Citation[7]). For example, the sustained sequelae of cigarette smoke in the lung include the up-regulation of several factors that are known to inactivate AAT. The study of Churg et al. (Citation[29]) has, however, shown that appropriate, albeit very high systemic doses of injected plasma-derived AAT can indeed modify the course of the disease in a mouse model system.
The primary purpose of this study was to evaluate whether or not lower doses of rAAT, achieved by direct administration to the lungs of smoking mice, could have similar protective effects against the destructive nature of chronic cigarette smoke and its inevitable induced inflammatory and oxidative secondary products. We sought to address two key, currently unanswered questions by way of these experiments. We first wished to show that rAAT could be delivered by nebulization to the mouse lung in an active form, and that this rAAT could inhibit alveolus-degrading elastolytic activity and normalize neutrophil recruitment, as measured in the BAL fluids of test animals. Secondly, we sought to determine whether or not nebulized rAAT could inhibit the progression of emphysema in smoke-treated animals, as measured by a reduction in airspace enlargement, and how this compared with pAAT administered to the lung by the more natural, but less efficient bloodstream route of delivery.
Our results have shown quite clearly that indeed, in this mouse model system, inhaled rAAT could fulfill each of these stated objectives. Furthermore, our findings demonstrate that significantly lower doses of nebulized rAAT can give greater protection than systemically administered pAAT against the effects of cigarette smoke. This approach, therefore, has the potential to be both a practical and an efficacious method for the treatment of individuals with cigarette smoke-associated emphysema.
We thank Ms. Rebecca Wheeler for assistance with preparation of the manuscript.
Supported by Arriva Pharmaceuticals, Inc.
REFERENCES
- Stockley R A. Neutrophils and protease/antiprotease imbalance. Am J Respir Crit Care Med 1999; 160: S49–52, [INFOTRIEVE], [CSA]
- Barnes P J. Medical progress: Chronic obstructive pulmonary disease. New Eng J Med 2000; 343: 269–280, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Barnes P J, Shapiro S D, Pauwels R A. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 2003; 22: 672–688, [INFOTRIEVE], [CSA]
- Churg A, Wright J L. Proteases and emphysema. Curr Opin Pulm Med 2005; 11: 153–159, [INFOTRIEVE], [CSA], [CROSSREF]
- Laurell C -B, Eriksson S. The electrophoretic α -1-globulin pattern of serum in α -1 antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15: 132–140, [CSA]
- Tobin M J, Cook P JL, Hutchison D CS. Alpha-1 antitrypsin deficiency: the clinical and physiological features of pulmonary emphysema in patients homozygous for Pi type Z. Brit J Dis Chest 1983; 77: 14–27, [INFOTRIEVE], [CSA]
- American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168: 818–900, [CSA], [CROSSREF]
- Stockley R A. Chronic bronchitis: the antiproteinase/proteinase balance and the effect of infection and corticosteroids. Clin Chest Med 1988; 9: 643–656, [INFOTRIEVE], [CSA]
- Birrer P, McElvaney N G, Rudeberg A, Sommer C W, Liechti-Gallati S, Kraemer R, Hubbard R, Crystal R G. Protease-antiprotease imbalance in the lungs of children with cystic fibrosis. Am J Resp Crit Care Med 1994; 150: 207–213, [INFOTRIEVE], [CSA]
- Watterberg K L, Carmichael D F, Gerdes J S, Werner S, Backstrom C, Murphy S. Secretory leukocyte protease inhibitor and lung inflammation in developing bronchopulmonary dysplasia. Pediatrics 1994; 125: 264–269, [CSA], [CROSSREF]
- Glass M. The feasibility of an outcome trial in the preventive therapy of emphysema. Ann NY Acad Sci 1991; 624: 195–208, [INFOTRIEVE], [CSA]
- Senior R M, Griffin G L, Mecham R P. Chemotactic activity of elastin-derived peptides. J Clin Invest 1980; 66: 859–862, [INFOTRIEVE], [CSA]
- Zhu Y, Liu X, Skold C M, Wang H, Kohyama T, Wen F Q, Ertl R F, Rennard S I. Collaborative interactions between neutrophil elastase and metalloproteinases in extracellular matrix degradation in three-dimensional collagen gels. Respir Res 2001; 2: 300–305, [INFOTRIEVE], [CSA], [CROSSREF]
- Shapiro S D, Goldstein N M, Houghton A M, Kobayashi D K, Kelley D, Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Path 2003; 163: 2329–2335, [INFOTRIEVE], [CSA]
- Lieberman J, Winter B, Sastre A. Alpha1-antitrypsin Pi-types in 965 COPD patients. Chest 1986; 89: 370–373, [INFOTRIEVE], [CSA]
- Carp H, Miller F, Hoidal J R, Janoff A. Potential mechanism of emphysema: alpha 1-proteinase inhibitor recovered from lungs of cigarette smokers contains oxidized methionine and has decreased elastase inhibitory capacity. Proc Natl Acad Sci USA 1982; 79: 2041–2045, [INFOTRIEVE], [CSA]
- Segura-Valdez L, Pardo A, Gaxiola M, Uhal B D, Becerril C, Selman M. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest 2000; 117: 684–694, [INFOTRIEVE], [CSA], [CROSSREF]
- Russell R E, Culpitt S V, De Matos C, Donnelly L, Smith M, Wiggins J, Barnes P J. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2002; 26: 602–609, [INFOTRIEVE], [CSA]
- Finlay G A, O'Driscoll L R, Russell K J, D'Arcy E M, Masterson J B, FitzGerald M X, O'Connor C M. Matrix metalloprotease expression and production by alveolar macrophages in emphysema. Am J Respir Crit Care Med 1997; 156: 240–247, [PUBMED], [INFOTRIEVE], [CSA]
- Hautamaki R D, Kobayashi D K, Senior R M, Shapiro S D. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 1997; 277: 2002–2004, [INFOTRIEVE], [CSA], [CROSSREF]
- Churg A, Wang R D, Tai H, Wang X, Xie C, Wright J L. Tumor necrosis factor-alpha drives 70% of cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med 2004; 170: 492–498, [INFOTRIEVE], [CSA], [CROSSREF]
- Churg A, Wang R D, Tai H, Wang X, Xie C, Dai J, Shapiro S D, Wright J L. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via TNF-alpha release. Am J Respir Crit Care Med 2003; 167: 1083–1089, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Joos L. COPD and genetics—what's new?. Swiss Med Wkly 2004; 134: 437–439, [INFOTRIEVE], [CSA]
- Churg A, Dai J, Tai H, Xie C, Wright J. Tumor necrosis factor—is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am J Respir Crit Care Med 2002; 166: 849–854, [INFOTRIEVE], [CSA], [CROSSREF]
- Churg A, Zay K, Shay S, Xie C, Shapiro S D, Hendricks R, Wright J L. Acute cigarette smoke-induced connective tissue breakdown requires both neutrophils and macrophage metalloelastase in mice. Am J Respir Cell Mol Biol 2002; 27: 368–374, [INFOTRIEVE], [CSA]
- Martin R L, Shapiro S D, Tong S E, Van Wart H E. Macrophage metalloelastase inhibitors. Prog Respir Res 2001; 31 177–180, [CSA]
- Selman M, Cisneros-Lira J, Gaxiola M, Ramíerez R, Kudlaez E M, Mitchell P G, Pardo A. Matrix metalloproteinases inhibition attenuates tobacco smoke-induced emphysema in guinea pigs. Chest 2003; 123: 1633–1641, [INFOTRIEVE], [CSA], [CROSSREF]
- Pemberton P A, Cantwell J S, Kim K M, Sundin D J, Kobayashi D, Fink J B, Shapiro S D, Barr P J. An inhaled matrix metalloprotease inhibitor prevents cigarette smoke-induced emphysema in the mouse. COPD 2005; 3: 303–310, [CSA]
- Churg A, Wang R D, Xie C, Wright J L. Alpha-1-antitrypsin ameliorates cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med 2003; 168: 199–207, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Barr P J, Gibson H L. Methods of protein production in yeast. World Intellectual Property Organization. 2005, International Publication Number WO 2005/014825 A2
- Feinstein G, Kupfer A, Sokolovsky M. N-acetyl-(L-Ala)3-p-nitroanilide as a new chromogenic substrate for elastase. Biochem Biophys Res Commun 1973; 50: 1020–1026, [INFOTRIEVE], [CSA], [CROSSREF]
- Dunnill M S. Quantitative methods in the study of pulmonary pathology. Thorax 1962; 17: 320–328, [CSA]
- Petrache I, Natarajan V, Zhen L, Richter A T, Cho C, Hubbard W C, Berdyshev E V, Tuder R M. 2005. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med 2005; 11: 491–498, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Hemmer B, Stuvem O, Kieseier B, Schellekens H, Hartung H -P. Immune response to immunotherapy: the role of neutralizing antibodies to interferon beta in the treatment of multiple sclerosis. Lancet Neurol 2005; 4: 403–412, [INFOTRIEVE], [CSA], [CROSSREF]
- Hubbard R C, McElvaney N G, Sellers S E, Healy J T, Czerski D B, Crystal R G. Recombinant DNA-produced α 1-antitrypsin administered by aerosol augments lower respiratory tract antineutrophil elastase defenses in individuals with α 1-antitrypsin deficiency. J Clin Invest 1989; 84: 1349–1354, [INFOTRIEVE], [CSA]
- Cantin A M, Woods D E, Cloutier D, Heroux J, Dufour E K, Leduc R. Leukocyte elastase inhibition therapy in cystic fibrosis: role of glycosylation on the distribution of alpha-1-proteinase inhibitor in blood versus lung. J Aerosol Med 2002; 15: 141–148, [INFOTRIEVE], [CSA], [CROSSREF]
- Hubbard R C, Crystal R G. Strategies for aerosol therapy of α 1-antitrypsin deficiency by the aerosol route. Lung 1990; 168: 565–578, Suppl[INFOTRIEVE], [CSA]
- Romberg V, Dickneite G, Eldon M A, Roskos K V, Emmerling D, Weeks R, Bryant C, Brantly M, Stocks J, Benn V. Utility of primate toxicology studies as a predictor of human safety and tolerability: experience with dry-powder alpha1-antitrypsin administered to patients with AAT deficiency. Respiratory Drug Delivery VIII 2002; 1: 33–41, [CSA]
- Shaker S B, Stavngaard T, Stolk J, Stoel B, Dirksen A. Alpha1-antitrypsin deficiency.7: computed tomographic imaging in alpha1-antitrypsin deficiency. Thorax 2004; 59: 986–981, [INFOTRIEVE], [CSA], [CROSSREF]