Abstract
The CXC chemokines, IP-10/CXCL10 and IL-8/CXCL8, play a role in obstructive lung disease by attracting Th1/Tc1 lymphocytes and neutrophils, respectively. Inhaled corticosteroids (ICS) and long acting β 2-agonists (LABA) are widely used. However, their effect(s) on the release of IP-10 and IL-8 by airway epithelial cells are poorly understood. This study examined the effects of fluticasone, salmeterol, and agents which raise intracellular cAMP (cilomilast and db-cAMP) on the expression of IP-10 and IL-8 protein and mRNA. Studies were performed in cultured human airway epithelial cells during cytokine-stimulated IP-10 and IL-8 release. Cytokine treatment (TNF-α, IL-1β and IFN-γ) increased IP-10 and IL-8 protein and mRNA levels. Fluticasone (0.1 nM to 1 μM) increased IP-10 but reduced IL-8 protein release without changing IP-10 mRNA levels assessed by real time RT-PCR. The combination of salmeterol (1 μ M) and cilomilast (1–10 μ M) reduced IP-10 but had no effect on IL-8 protein. Salmeterol alone (1 μ M) and db-cAMP alone (1 mM) antagonised the effects of fluticasone on IP-10 but not IL-8 protein. In human airway epithelial cells, inhibition by salmeterol of fluticasone-enhanced IP-10 release may be an important therapeutic effect of the LABA/ICS combination not present when the two drugs are used separately.
Key words: :
INTRODUCTION
Inhaled corticosteroids are mainstays in the treatment of COPD and asthma and exert their anti-inflammatory effects, at least in part, by inhibiting cytokine (e.g., TNF-α, IL-1β) and chemokine (e.g., IL-8, GM-CSF, eotaxin, RANTES) production in several respiratory cell types (Citation[1], Citation[2], Citation[3], Citation[4], Citation[5], Citation[6], Citation[7]). Recent studies in COPD and asthma indicate that the combination of an inhaled corticosteroid (ICS) and a long-acting β2 agonist (LABA) achieve greater improvement in airway function; exacerbation rate; rescue use of a short-acting bronchodilator; and dyspnea than either drug alone (Citation[8], Citation[9]) or even a doubling of ICS dose (Citation[10], Citation[11], Citation[12]).
The greater improvement in COPD and asthma clinical endpoints achieved by the ICS/LABA combination is associated with greater anti-inflammatory effects, in particular, inhibition of cytokine and chemokine release. For example, the combination of fluticasone and salmeterol produced greater inhibition than fluticasone alone of IFN-γ and TNF-α release by CD4+ and CD8+ Th1-lymphocytes and IL-5 and IL-13 release by CD4+ Th2-lymphocytes (Citation[13]). Moreover, fluticasone/salmeterol augmented the release of IL-10 and induction of a T-regulatory cell phenotype to a greater extent than fluticasone alone (Citation[14]). In airway epithelial cells and smooth muscle cells, ICS/LABA combinations (formoterol/budesonide or salmeterol/fluticasone) potentiated corticosteroid-mediated inhibition of release of the granulocyte-monocyte chemokine, GM-CSF (Citation[4]), and the neutrophil chemokine, IL-8 (Citation[4], Citation[15]).
The molecular basis of the combined anti-inflammatory effects of an ICS/LABA is becoming clearer. The LABA, salmeterol, phosphorylates the glucocorticoid receptor (GCR) and enhances corticosteroid-induced translocation of the GCR into the nucleus and its binding to DNA (Citation[13], Citation[14], Citation[16], Citation[17]). Moreover, salmeterol, like glucocorticosteroids, inhibits histone acetylation, and hence, chromatin remodeling (Citation[18]). Salmeterol-induced effects on corticosteroid-mediated responses are dependent on cAMP signaling pathways (Citation[14], Citation[16], Citation[17]).
Airway epithelial cells play a prominent role in obstructive lung disease by releasing a number of cytokines and chemokines which contribute to the innate and adaptive immune responses to infectious and non-infectious agents (Citation[19], Citation[20]). For example, airway epithelial cells produce the CXC chemokines, IP-10 and IL-8, in response to viral infection (Citation[21]) and exposure to IFN-γ, TNF-α and IL-1β (Citation[1], Citation[2], Citation[22]). IP-10 recruits natural killer cells and CD4+ and CD8+ lymphocytes of the Th-1/Tc-1 phenotype into the airway; IL-8 recruits neutrophils (Citation[19]). Moreover, IP-10 promotes proliferation and enhanced IFN-γ production by CXCR3+ Th1 cells (Citation[23]).
The effects of ICS, LABA and other agents that raise intracellular cAMP, alone or in combination, on the production of IP-10, have not been well studied in airway epithelial cells. Accordingly, we examined the effects of fluticasone propionate, salmeterol, and cilomilast, a selective phosphodiesterase type 4 inhibitor which increases intracellular cAMP, on IP-10 chemokine expression in cultured human airway epithelial cells. Additional studies were performed using cell-permeable dibutyryl cAMP to examine more directly the role of cAMP-dependent pathways in the responses observed. The effects of these drugs on IP-10 were compared to IL-8, which is inhibited by ICS in a variety of respiratory cell types (Citation[2], Citation[4], Citation[5], Citation[15], Citation[24], Citation[25], Citation[26]). IP-10 and IL-8 expression were examined at the protein level by ELISA and at the mRNA level by real time RT-PCR.
MATERIALS AND METHODS
Cell culture and assay protocol
Human airway epithelial cells, the 16-HBE cell line, were cultured in 100 mm tissue culture dishes coated with type VI human placental collagen (Sigma/Aldrich, St. Louis, MO) in DMEM medium containing 10% fetal bovine serum (FBS), penicillin (100 U/mL) and streptomycin (100 mg/mL) in 5% CO2 at 37°C. Cells were grown to 80% to 100% confluence, and then serum-deprived for 24 hrs prior to treatment.
Cells were treated with a mixture of TNF-α (20 ng/mL), IL-1β (30 ng/mL) and IFN-γ (100 ng/mL), i.e., “cytomix”, to induce release of IP-10 and IL-8 (Citation[2], Citation[15], Citation[22]). These cytokines have been reported to contribute to airway inflammation in COPD (Citation[19], Citation[20], Citation[27]). Cytomix treatment was performed for 12 hr or 24 hr. Vehicle treated cells (0.1% bovine serum albumin/PBS) served as controls.
The effects of fluticasone propionate (a gift of GlaxoSmithKline Inc., Greenford, UK) was assessed over a range of concentrations (10− 10 to 10− 6 M) in cells simultaneously treated with cytomix. Cells incubated with vehicle (DMSO) served as control.
In separate experiments, salmeterol xinofoate (SALM 1 μ M) was added singly or in combination with fluticasone (10− 10 M) for 24 hr.
In additional experiments, cilomilast (1–10 μ M) (gift of GlaxoSmithKline Inc.), a type 4D phosphodiesterase inhibitor, was added singly or in combination with salmeterol in the presence of cytomix for 24 hr.
The role of cAMP-dependent mechanisms in IL-8 and IP-10 expression was examined by treating cells with dibutyryl-cAMP (db-cAMP–1 mM), or vehicle in the presence of cytomix and fluticasone (10− 10 M) for 24 hr.
In human airway epithelial cells, production of prostaglandin E2 (PGE2), which stimulates cAMP production, is inhibited by glucocorticosteroids (Citation[28], Citation[29]). In other cell types, cAMP stimulates IL-8 and inhibits IP-10 release (Citation[30], Citation[31]). Therefore, to determine if the effects of fluticasone on IL-8 or IP-10 production were secondary to inhibition of PGE2 synthesis, cells were treated with indomethacin (10 μM) or vehicle.
After all treatments, the cell-conditioned medium was collected and stored at −80°C for ELISA (see later). Cell number was assessed from measurement of DNA by the CyQuant assay (InVitrogen, Carlsbad, CA). In brief, cell cultures were washed twice with PBS, frozen overnight at −80°C, then thawed and reacted with a proprietary mixture of lysis buffer and DNA-binding fluorescent dye for 5 mins. Sample fluorescence was measured at 535 nm and cell number determined using standard curves.
ELISA
IP-10 and IL-8 protein concentrations in epithelial cell-conditioned medium were measured by sandwich ELISA in 96-well micro-titer plates using commercially available kits (Biosource International, Camarillo, CA). The antigen-antibody complex was detected using streptavidin horseradish peroxidase (HRP) followed by tetramethylbenzidine (TMB) as substrate. Color intensity was read at 450 nm by micro-titer plate reader (Bio-Rad, model 550, Hercules, CA). Concentrations were calculated by using human IP-10 and IL-8 as standards. The detection thresholds for IP-10 and IL-8 were 2.0 pg/mL and 5.0 pg/mL, respectively.
RNA preparation and real-time RT-PCR
Total cellular RNA was isolated in guanidinium isothiocyanate, phenol – chloroform (Tri Reagent, Sigma/Aldrich). RNA concentration and purity were measured at 260 and 280 nm by UV spectrophotometer (Beckman Instruments, Fullerton, CA). Samples were stored at −80°C. RT-PCR was performed as a one-tube reaction in a LightCycler (Roche Applied Science, Indianapolis, IN) using TaqMan® FAM-labeled probes/primers for IP-10 and IL-8 (Assays-on-Demand, Applied Biosystems, Foster City, CA). Each 20 μ L reaction contained 30 ng of total RNA, 0.9 μ M forward and reverse primers, 0.25 μM probe, and 1x Universal PCR Master Mix (Applied Biosystems), a proprietary pre-formulated master mix of enzymes, reaction buffer and dNTP's. Time/temperature parameters for the RT reaction were: 50°C for 5 min followed by 55°C for 30 min. Temperature/time parameters for 2-step PCR were: denaturation −95°C for 5 sec and annealing/elongation −60°C for 25 sec, 35 cycles total. RT-PCR assays included the housekeeping gene, β -actin.
IP-10 and IL-8 mRNA levels were assessed from the cycle number at which chemokine amplification exceeded threshold, i.e., the crossing point, (ct). To maximize the precision and sensitivity of the PCR data, the threshold was manually centered to the exponential phase of amplification during data analysis and the same setting was used in all experiments to obtain the crossing point. All samples were run in duplicate along with negative RT controls and H2O blanks. IP-10 and IL-8 mRNA levels were expressed as fold-change from control, where fold-change = 2(control ct - experimental ct).
Statistical analysis
Statistical analysis was performed using SigmaStat (Systat Software). Group data were expressed as mean ± 1 SEM. To normalize for differences in cell responses to various stimuli, data were expressed as a percentage of the control value. Relationship of IP-10 and IL-8 protein and mRNA to fluticasone concentration was assessed by 1-way ANOVA. Comparisons between treatment groups were carried out by 2-way ANOVA. Pair-wise comparisons were performed using Student's paired t-test. A p value of less than 0.05 was considered statistically significant.
RESULTS
Time course of cytokine-induced IP-10 and IL-8 mRNA and protein
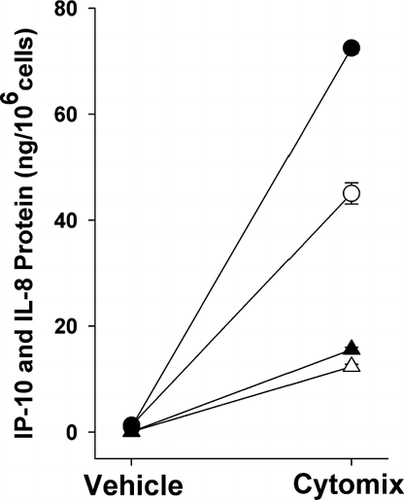
Cytomix increased IP-10 and IL-8 protein at 12 hr and 24 hr compared to vehicle-treated controls (p < 0.001 by paired t-test for all 4 conditions; n = 5) ().
Figure 1 Effect of cytokines (TNF-α, IL-1β and IFN-γ) on IP-10 and IL-8 protein release. Cytokine treatment increased IP-10 (circles) and IL-8 (triangles) protein in epithelial cell-conditioned medium at 12 hr (open symbols) and 24 hr (closed symbols) (p < 0.001 versus vehicle for both IP-10 and IL-8 at 12 and 24 hr). Mean ± SEM of 5 experiments.
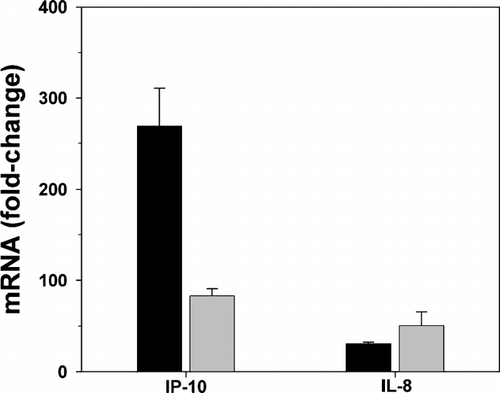
Cytomix greatly increased IP-10 and IL-8 mRNA at 12 hr and 24 hr (fold increases of > 80 for IP-10 and > 30 for IL-8; n = 2–3) ().
Figure 2 Effect of cytokines (TNF-α, IL-1β and IFN-γ) on IP-10 and IL-8 mRNA expression. Cytokine treatment increased IP-10 and IL-8 mRNA at 12 hr (black bars) and 24 hr (gray bars) as measured by real time RT-PCR. IP-10 and IL-8 mRNA are expressed as fold-change from control, where fold-change = 2 (control ct–chemokine ct). Mean ± SEM of 2–3 experiments.
Effect of fluticasone on IP-10 and IL-8 protein
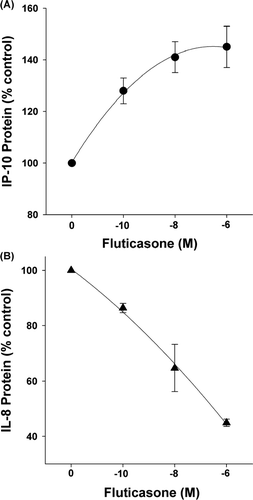
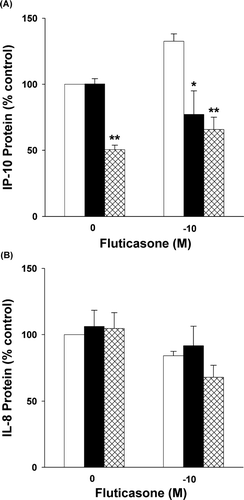
Fluticasone (10− 10–10− 6 M for 24 hr) differentially affected cytomix-induced IP-10 and IL-8 protein production at 24 hr ( and ). Fluticasone significantly enhanced IP-10 and inhibited IL-8 protein (p < 0.001 by 1-way RM ANOVA for both IP-10 and IL-8; n = 5).
Figure 3 Effect of fluticasone on cytokine-induced IP-10 and IL-8 protein. Fluticasone dose-dependently increased IP-10 (Panel A) and decreased IL-8 (Panel B) protein in cytokine-stimulated, airway epithelial cell-conditioned medium at 24 hr (p < 0.001 by 1-way RM ANOVA for both). Chemokine protein expressed as percent of control, i.e., cytokine-treated cells. Mean ± SEM of 5 experiments for each.
Effect of fluticasone on IP-10 and IL-8 mRNA
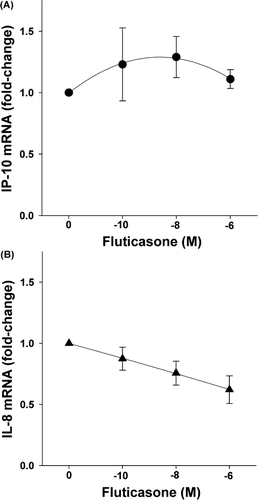
Fluticasone (10− 10–10− 6 M) reduced IL-8 mRNA slightly but significantly (p < 0.04), but had no significant effect on IP-10 mRNA at 12 hr (p = 0.54 by 1-way RM ANOVA) (n = 5) ( A, B). Similar results were obtained for fluticasone at 24 hr (data not shown).
Figure 4 Effect of fluticasone on cytokine-induced IP-10 and IL-8 mRNA. Fluticasone (12 hr) had no significant effect on IP-10 (p = 0.54) (Panel A) but reduced IL-8 mRNA (p < 0.04) (Panel B), as assessed by real-time RT-PCR. Mean ± SEM of 5 experiments.
Effect of salmeterol and cilomilast on IP-10 and IL-8 protein
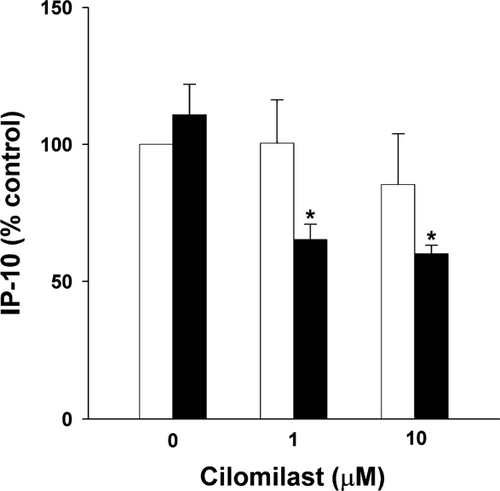
Salmeterol (1 μ M) and cilomilast (1–10 μ M) individually had no effect on cytomix-induced IP-10 release at 24 hr (p = 0.44 by paired t-test for salmeterol and p = 0.57 by 1-way RM ANOVA for cilomilast; n = 3). However, in the presence of salmeterol, cilomilast reduced IP-10 release (p = 0.013 by 1-way RM ANOVA; n = 3) (). Neither agent, alone or in combination, affected cytomix-induced IL-8 release (data not shown).
Figure 5 Effect of salmeterol and cilomilast on cytokine-induced IP-10 protein. Cells were stimulated with cytokines for 24 hr. Addition of cilomilast alone (0, 1 μ M, 10 μ M-open bars) during this time (24 hr) had no effect on IP-10. The addition of salmeterol (1 μ M for 24 hr-closed bars) to cilomilast inhibited IP-10 (* p = 0.013 by 1-way RM ANOVA). Mean ± SEM of 3 experiments.
Effect of salmeterol and fluticasone in combination on IP-10 and IL-8 protein
In the presence of fluticasone (10− 10 M), salmeterol (1 μ M) significantly inhibited cytomix-induced IP-10 at 24 hr (p = 0.013 by 2-way RM ANOVA; n = 3) (). In contrast, salmeterol did not affect fluticasone-induced reductions in IL-8 release (p = 0.97 by 2-way RM ANOVA; n = 3) ().
Figure 6 Effect of salmeterol or dibutyryl cAMP on fluticasone-induced IP-10 and IL-8 Protein. Panel A: Cells were stimulated with cytokines for 24 hr. Addition of salmeterol (1 μ M-solid bars) or db-cAMP (1 mM-cross-hatched bars) during this time (24 hr) inhibited fluticasone-induced (10− 10 M-open bars) increases in IP-10 protein (* p = 0.013 for salmeterol and ** p < 0.001 for db-cAMP by 2-way RM ANOVA). Mean ± SEM of 3 experiments. Panel B: Neither salmeterol (solid bars) nor db-cAMP (cross-hatched bars) affected fluticasone-induced (open bars) decreases in IL-8 protein (p = 0.97 for salmeterol and p = 0.10 for db-cAMP by 2-way RM ANOVA). Mean ± SEM of 3 experiments.
Effect of db-cAMP on IP-10 and IL-8 protein
Dibutyryl cAMP (1 mM) markedly inhibited fluticasone-induced increases in IP-10 (p < 0.001 by 2-way RM ANOVA; n = 3) () but had no significant effect on fluticasone-induced inhibition of IL-8 (p = 0.10; n = 3) ().
Indomethacin (10 μ M) had no significant effect on fluticasone-induced changes in IP-10 or IL-8 protein over 24 hr (n = 3) (data not shown).
DISCUSSION
The present study examined the effects of fluticasone and salmeterol on IP-10 and IL-8 expression in human airway epithelial cells under conditions in which chemokine expression was stimulated by cytokines. TNF-α, IL-1β and IFN-γ were used to up-regulate IP-10 and IL-8 since these several cytokines appear to contribute to the pro-inflammatory conditions in the airway in COPD (Citation[19], Citation[32], Citation[27]).
Our results indicate that fluticasone dose-dependently augments IP-10 protein release but inhibits cytokine-stimulated IL-8 protein release. Changes in IP-10 protein were not accompanied by statistically significant changes in mRNA as assessed by real-time RT PCR. The lack of significant changes in IP-10 mRNA suggests that fluticasone-mediated changes in protein expression may occur primarily at the translational or post-translational level.
In this study, neither salmeterol (a β 2-adrenergic agonist) nor cilomilast (a PDE inhibitor) alone affected cytokine-stimulated IL-8 or IP-10 protein production. However, the combination of these two agonists inhibited IP-10 release but had no effect on IL-8 release. Dibutyryl cAMP also markedly inhibited IP-10 release strongly suggesting that cAMP-dependent mechanisms are responsible for the effects of salmeterol and cilomilast. That the combination of salmeterol and cilomilast produced effects on IP-10 not observed with either agent alone, is likely to be explained by a greater increase in cAMP with the combination of the two agents.
Of interest, salmeterol antagonized the effects of fluticasone on IP-10 release but had no effect on fluticasone-mediated inhibition of IL-8 release. The inhibitory effect of salmeterol on IP-10 release in the presence, but not the absence of fluticasone, is likely explained by the well-described potentiation of β2-agonist responses by corticosteroids. For example, in human airway epithelial cells, corticosteroids increase β2-adrenergic receptor expression, increase β2-adrenergic receptor coupling to the stimulatory G-protein, Gαs, and facilitate protein kinase A (PKA)-mediated intracellular signaling (Citation[29]).
Finally, db-cAMP eliminated the enhancing effect of fluticasone on IP-10 release. This result suggested the possibility that fluticasone increased IP-10 by decreasing cellular cAMP levels. We previously observed in human airway epithelial cells that fluticasone inhibits a known stimulus for cAMP production, i.e., PGE2 production (Citation[28], Citation[29]). However, since the prostaglandin inhibitor, indomethacin, had no effect on IP-10 release in the present study, it seems unlikely that fluticasone-induced increases in IP-10 are PGE2 dependent.
Inhibition of IL-8 expression by glucocorticosteroids has been reported previously in bronchial epithelial cells (Citation[2], Citation[4], Citation[5], Citation[6], Citation[24], Citation[25]); airway smooth muscle (Citation[15]); the alveolar epithelial cell line, i.e., A549 cells (Citation[2], Citation[26]); and alveolar macrophages (Citation[26]). In contrast, the effect of corticosteroids on IP-10 expression by human respiratory cells is limited to semi-quantitative, immunohistochemical approaches performed in airway biopsies following treatment with methylprednisolone 40 mg/day for two weeks (Citation[33]). Our data indicating that fluticasone increases IP-10 production in cultured human airway epithelial cells represent the first quantitative assessment of the effects of glucocorticosteroids on IP-10 expression at the protein or mRNA level in any respiratory cell type. Our results are in agreement with those demonstrating an increase in IP-10 immunoreactivity in airway epithelial cells following treatment with methylprednisolone (Citation[33]).
Moreover, to our knowledge, the interacting effects of LABAs and corticosteroids on IP-10 release by human airway epithelial cells has not been studied previously. The inhibitory effects of salmeterol and db-cAMP on fluticasone-induced IP-10 release are of particular interest in view of previous studies which have described additive or synergistic interactions of LABA and ICS on a variety of cellular responses (see preceding). Our data indicating that salmeterol antagonizes the effects of fluticasone on IP-10 release are of interest since they indicate that LABAs and corticosteroids may exert opposing effects on at least some biological responses in the respiratory tract. Furthermore, the opposing effects of the two agents suggest that salmeterol-induced effects on IP-10 release in human airway epithelial cells do not involve activation of the GR-α. The observation that salmeterol produces an effect on IP-10 production opposite to that of fluticasone is not surprising, however. In other cell types (e.g., keratinocytes, microglia), agents which increase cAMP inhibit IP-10 expression, an effect which can be blocked by an adenylyl cyclase inhibitor (Citation[31], Citation[34]).
Of importance, IP-10 expression is increased in COPD and asthma (Citation[27], Citation[32], Citation[35], Citation[36], Citation[37]). In subjects with COPD, IP-10 expression is greater than normal in small airway epithelial cells (Citation[32]) and in whole lung tissue (Citation[37]). In subjects with asthma, IP-10 levels in bronchoalveolar lavage fluid are increased following allergen challenge (Citation[36]). IP-10 and other ligands of the CXCR3 chemokine receptor, recruit Th1 and Tc1 lymphocytes (and possibly macrophages) into the airway (Citation[19]). Finally, over-expression of IP-10 in a mouse model of asthma increases airway eosinophilia and airway hyperreactivity (Citation[38]). Accordingly, IP-10 is believed to be important in the pathogenesis of COPD and asthma by promoting airway inflammation and airway hyper-reactivity (Citation[19], Citation[20], Citation[27], Citation[32], Citation[36]). The results of the present study indicate that in airway epithelial cells, inhibition of fluticasone-enhanced IP-10 release by salmeterol and other agents which increase intracellular cAMP may be an important therapeutic benefit of the LABA/ICS combination not present when the two drugs are used separately.
Grant support: A grant from GlaxoSmithKline, and institutional funds.
REFERENCES
- Levine S J, Larivee P, Logun C, Angus C W, Shelhamer J H. Corticosteroids differentially regulate secretion of IL-6, IL-8, and G-CSF by a human bronchial epithelial cell line. AmJ Physiol 1993; 265: 360–368
- Kwon O J, Au B T, Collins P D, Adcock I M, Mak J C, Robbins R R, Chung K F, Barnes P J. Tumor necrosis factor-induced IL-8 expression in human airway cells. AmJ Physiol Lung Cell Mol Physiol 1994; 267: L398–405
- Stellato C, Beck L A, Gorgone G A, Proud D, Schall T J, Ono S J, Lichtenstein L M, Schleimer R P. Expression of the chemokine RANTES by a human bronchial epithelial cell line. Modulation by cytokines and gluccorticoids. J Immunol 1995; 155: 410–418
- Korn S H, Jerre A, Brattsand R. Effects of formoterol and budesonide on GM-CSF and IL-8 secretion by triggered human bronchial epithelial cells. Eur RespirJ 2001; 17: 1070–1077
- Chang M MJ, Juarez M, Hyde D M, Wu R. Mechanism of dexamethasone-mediated interleukin-8 gene suppression in cultured airway epithelial cells. AmJ Physiol Lung Cell Mol Physiol 2001; 280: 107–115
- Escotte S, Tabary O, Dusser D, Majer-Teboul C, Puchelle E, Jacquot J. Fluticasone reduces IL-6 and IL-8 production of cystic fibrosis bronchial epithelial cells via IKK-beta kinase pathway. Eur RespirJ 2003; 21: 574–581
- Necela B M, Cidlowski J A. Mechanisms of glucocorticoid receptor action in noninflammatory and inflammatory cells. Proc Am Thorac Soc 2004; 1: 239–246
- Mahler D A, Wire P, Horstman D, Chang C N, Yates J, Fischer T, Shah T. Effectiveness of fluticasone propionate and salmeterol combination delivered via the Diskus device in the treatment of chronic obstructive pulmonary disease. AmJ Respir Crit Care Med 2002; 166: 1084–1091
- Calverley P, Pauwels R, Vestbo J, Jones P, Pride N, Gulsvik A, Anderson J, Maden C. TRial of Inhaled Steroids and long-acting beta2 agonists study group. Combined salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease: a randomized controlled trial. Lancet 2003; 361: 449–456
- Woolcock A, Lundback B, Ringdal N, Jacques L A. Comparison of addition of salmeterol to inhaled steroids with doubling of the dose of inhaled steroids. AmJ Respir Crit Care Med 1996; 153: 1481–1488
- Pauwels R A, Lofdahl C G, Postma D S, Tattersfield A E, O'Byrne P, Barnes P J, Ullman A. Effect of inhaled formoterol and budesonide on exacerbations of asthma. Formoterol and Corticosteroids Establishing Therapy (FACET) International Study Group. N EnglJ Med 1997; 337: 1405–1411
- Hancox R J, Cowan C O, Flannery E M, Herbison G P, McLachlan C R, Wong C S, Taylor D R. Randomised trial of an inhaled beta2 agonist, inhaled corticosteroid and their combination in the treatment of asthma. Thorax 1999; 54: 482–487
- Goleva E, Dunlap A, Leung D YM. Differential control of TH1 versus TH2 cell responses by the combination of low-dose steroids with beta2-adrenergic agonists. J Allergy Clin Immunol 2004; 114: 183–191
- Peek E J, Richards D F, Faith A, Lavender P, Lee T H, Corrigan C J, Hawrylowicz C M. Interleukin-10-secreting “regulatory” T cells induced by glucocorticoids and beta2-agonists. AmJ Respir Cell Mol Biol 2005; 33: 105–111
- Pang L, Knox A J. Synergistic inhibition by beta(2)-agonists and corticosteroids on tumor necrosis factor-alpha-induced interleukin-8 release from cultured human airway smooth-muscle cells. AmJ Respir Cell Mol Biol 2000; 23: 79–85
- Eickelberg O, Roth M, Lorx R, Bruce V, Rudiger J, Johnson M, Block L H. Ligand-independent activation of the glucocorticoid receptor by beta2-adrenergic receptor agonists in primary human lung fibroblasts and vascular smooth muscle cells. J Biol Chem 1999; 274: 1005–1010
- Usmani O S, Ito K, Maneechotesuwan K, Ito M, Johnson M, Barnes P J, Adcock I M. Glucocorticoid receptor nuclear translocation in airway cells after inhaled combination therapy. AmJ Respir Crit Care Med 2005; 172: 704–712
- Nie M, Knox A J, Pang L. Beta2-Adrenoceptor agonists, like glucocorticoids, repress eotaxin gene transcription by selective inhibition of histone H4 acetylation. J Immunol 2005; 175: 478–486
- D'Ambrosio D, Mariani M, Panina-Bordignon P, Sinigaglia F. Chemokines and their receptors guiding T-lymphocyte recruitment in lung inflammation. AmJ Respir Crit Care Med 2001; 164: 1266–1275
- Barnes P J, Shapiro S D, Pauwels R A. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur RespirJ 2003; 22: 672–688
- Spurrell J CL, Wiehler S, Zaheer R S, Sanders S P, Proud D. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. AmJ Physiol Lung Cell Mol Physiol 2005; 289: L85–L95
- Sauty A, Dziejman M, Taha R A, Iarossi A S, Neote K, Garcia-Zepeda E A, Hamid Q, Luster A D. TheT cell-specific CXC chemokines IP-10, MIG, and I-TAC are expressed by activated human bronchial epithelial cells. J Immunol 1999; 162: 3549–3558
- Whiting D, Hsieh G, Yun J J, Banerji A, Yao W, Fishbein M C, Belperio J, Strieter R M, Bonavida B, Ardehali A. Chemokine monokine induced by IFN-gamma/CXC chemokine ligand 9 stimulates T lymphocyte proliferation and effector cytokine production. J Immunol 2004; 172: 7417–7424
- Sabatini F, Silvestri M, Sale R, Serpero L, Raynal M E, Di Blasi P, Rossi G A. Modulation of the constitutive or cytokine-induced bronchial epithelial cell functions in vitro by fluticasone propionate. Immunol Lett 2003; 89: 215–224
- Noah T L, Wortman I A, Becker S. The effect of fluticasone propionate on respiratory syncytial virus-induced chemokine release by a human bronchial epithelial cell line. Immunopharmacology 1998; 39: 193–199
- Ek A, Palmberg L, Larsson K. Influence of fluticasone and salmeterol on airway effects of inhaled organic dust; an in vivo ex vivo study. Clin Exp Immunol 2000; 121: 11–16
- Chung K F. Cytokines in chronic obstructive pulmonary disease. Eur RespirJ Suppl 2001; 34: 50s–59s
- Penn R B, Kelsen S G, Benovic J L. Regulation of beta-agonist- and prostaglandin E2-mediated adenylyl cyclase activity in human airway epithelial cells. AmJ Respir Cell Mol Biol 1994; 11: 496–505
- Aksoy M O, Mardini I A, Yang Y, Bin W, Zhou S, Kelsen S G. Glucocorticoid effects on the beta-adrenergic receptor-adenylyl cyclase system of human airway epithelium. J Allergy Clin Immunol 2002; 109: 491–497
- Yu Y, Chadee K. Prostaglandin E2 stimulates IL-8 gene expression in human colonic epithelial cells by a posttranscriptional mechanism. J Immunol 1998; 161: 3746–3752
- Kanda N, Watanabe S. 17 beta-estradiol inhibits the production of interferon-induced protein of 10 kDa by human keratinocytes. J Invest Dermatol 2003; 120: 411–419
- Saetta M, Mariani M, Panina-Bordignon P, Turato G, Buonsanti C, Baraldo S, Bellettato C M, Papi A, Corbetta L, Zuin R, Sinigaglia F, Fabbri L M. Increased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with chronic obstructive pulmonary disease. AmJ Respir Crit Care Med 2002; 165: 1404–1409
- Fukakusa M, Bergeron C, Tulic M K, Fiset P-O, Al Dewachi O, Laviolette M, Hamid Q, Chakir J. Oral corticosteroids decrease eosinophil and CC chemokine expression but increase neutrophil, IL-8, and IFN-gamma-inducible protein 10 expression in asthmatic airway mucosa. J Allergy Clin Immunol 2005; 115: 280–286
- Delgado M. Inhibition of interferon (IFN) gamma-induced Jak-STAT1 activation in microglia by vasoactive intestinal peptide: inhibitory effect on CD40, IFN-induced protein-10, and inducible nitric-oxide synthase expression. J Biol Chem 2003; 278: 27620–27629
- Keatings V M, Collins P D, Scott D M, Barnes P J. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. AmJ Respir Crit Care Med 1996; 153: 530–534
- Bochner B S, Hudson S A, Xiao H Q, Liu M C. Release of both CCR4-active and CXCR3-active chemokines during human allergic pulmonary late-phase reactions. J Allergy Clin Immunol 2003; 112: 930–934
- Hardaker E L, Bacon A M, Carlson K, Roshak A K, Foley J J, Schmidt D B, Buckley P T, Comegys M, Panettieri R A, Jr, Sarau H M, Belmonte K E. Regulation of TNF-alpha- and IFN-gamma-induced CXCL10 expression: participation of the airway smooth muscle in the pulmonary inflammatory response in chronic obstructive pulmonary disease. FASEB J 2004; 18: 191–193
- Medoff B D, Sauty A, Tager A M, Maclean J A, Smith R N, Mathew A, Dufour J H, Luster A D. IFN-gamma-inducible protein 10 (CXCL10) contributes to airway hyperreactivity and airway inflammation in a mouse model of asthma. J Immunol 2002; 168: 5278–5286