A GENETIC POPULATION SCREENING OF ALPHA 1 ANTITRYPSIN DEFICIENCY IN A HIGH PREVALENCE AREA OF NORTHERN ITALY
Enrica Bertella,1 Daniela Medicina,2 Giuseppe Emanuele La Piana,1 Luciano Corda,1 and Bruno Balbi3 1Centro di Riferimento Regionale per il Deficit di Alfa1-Antitripsina (Prima Divisione di Medicina Interna, Spedali Civili, Brescia—Cattedra di Malattie dell'Apparato Respiratorio, Università di Brescia), Italy; 2Prima Anatomia Patologica, Spedali Civili, Brescia and Cattedra di Istituzioni di Anatomia Patologica, Università di Brescia, Italy; 3Divisione di Pneumologia, Fondazione Maugeri Gussago (Brescia) and Veruno (Novara), Italy
We used sensitive genetic techniques to ascertain the prevalence of AAT-D in a population of a geographic area where its presence was suspected to be high. We identified all living residents of Pezzaze, Italy, as of April 1, 2005. We obtained serum and DNA from residents who consented to participate. Nephelometric serum dosage of AAT, and PCR analysis using restriction enzyme for Z and S AAT-D mutations were performed. DNA from individuals with inconsistent results of PCR-genotype and AAT levels underwent further genetic analyses as DHPLC and gene-sequencing. Serum samples were obtained from 817 of 1353 enumerated residents 18 years of age or older (60.4%). 67(8%) subjects had low (< 90 mg/dl) serum levels of AAT. By PCR, AAT-D was identified in 102 individuals. ZZ resulted 1 person, SS resulted 1 person, MZ resulted 48 persons while MS resulted 52 persons. By further DNA analysis we identified additional genotypes:2 ZMBrescia (previously classified as MZ), 9 MMBrescia, 4 MMWurzburg, 2 MI and 1 MMLowell (all previously classified as normal MM).Thus, a total of 118 individuals, 14.5% of the screened subjects were carriers of AAT-D genes. This prevalence is far beyond the expected in that region of northern Italy according to the evaluation of Hardy-Weinberg analysis (p < 0.001, Chi square test). By this adult population screening performed in a high risk area of northern Italy using molecular genetic analysis techniques, we were able to identify a higher than expected number of individuals who were carriers of AAT-D genes, including a substantial proportion of subjects with rare alleles.
AGE AT WHICH PHYSIOLOGICAL, HEALTH STATUS AND CT DENSITOMETRY SCORES BECOME ABNORMAL IN COPD RELATED TO ALPHA 1-ANTITRYPSIN DEFICIENCY
J. Holme, MB BS,1,2 J. Stockley,1 and R. A. Stockley, MD DSc FRCP1,2 1University Hospital Birmingham NHS Foundation Trust, Birmingham, England; 2University of Birmingham, Birmingham, England
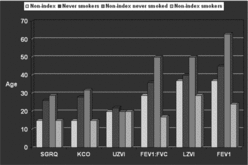
Several physiological and radiological tests are useful for diagnosing and monitoring progression in COPD related to alpha-1 antitrypsin deficiency (AATD)1,2. However, the age that these tests become abnormal, and the order in which this occurs is not known. We assessed the age that these parameters became abnormal in AATD to determine the age that screening should start and the most appropriate screening test. A total of 591 PiZ patients from the UK AATD registry were studied. A mathematical model was constructed by logistic regression with age, gender, smoking and index status as co-variates, to determine the probability of FEV1, FEV1:FVC, KCO (all % predicted), St Georges Respiratory Questionnaire (SGRQ) score and upper (UZVI) and lower zone (LZVI) CT densitometry scores being worse than the mean of the general population. The age at which this probability exceeded 50% (as expected in the normal population) was determined. For non-index and never smokers, health status, KCO and UZVI become abnormal first (20s), followed by spirometry and LZVI in 30s to 40s. [Figure 1]. All parameters were abnormal in index subjects before presentation making models inaccurate in this group due to data extrapolation. Health status and KCO are abnormal earlier than spirometry in AATD challenging current definitions and monitoring guidance.
Funded by: Talecris Biotherapeutics.
CELLULAR HANDLING OF THE NEW α1-ANTITRYPSIN BRESCIA VARIANT IN CELL TRANSFECTANTS
N. Montani, D. Medicina, L. Corda, A. Fra, L. Tiberio, C. Tantucci, F. Facchetti, and L. Schiaffonati Department of Biomedical Sciences and Biotechnology, General Pathology Unit, School of Medicine, University of Brescia, Brescia, Italy
Deficiency of the serine proteinase inhibitor α1-antitrypsin (α1-AT) is a genetic disorder associated with high risk of emphysema. Some mutations, particularly the PI Z, are also associated with liver disease. Indeed these mutations cause a perturbation in the protein structure with consequent polymerization, decreased secretion into the plasma and accumulation in the endoplasmic reticulum of hepatocytes. We identified a new defective α1-AT allele, named PI Brescia (G225A). The defective allele PI Brescia was found in heterozygous form together with the previously identified rare defective allele Mwurzburg (P369S), in a subject with reduced serum levels of α1-AT. We have characterized the cellular handling of the α1-AT Brescia variant by expressing the defective PI Brescia allele in Hepa1.6 cell line. Synthesis, secretion and intracellular accumulation of α1-AT were studied by pulse-chase experiments comparing the behaviour of the PI Brescia variant with the normal PI M protein and the PI Z variant. These experiments demonstrate that Brescia α 1-AT is secreted at lower extent compared to wild-type α1-AT but at higher extent compared to Z α1-AT. About 20% of Brescia α1-AT is degraded, but most of it accumulates in the cell in a EndoH-sensitive form, showing that it is retained in the endoplasmic reticulum (ER). A fraction of the Brescia α1-AT retained in the ER is found in NP40-insoluble aggregates resembling those described for Z α 1-AT. These results show that the PI Brescia variant may form aggregates in hepatocytes and may have a pathogenetic potential for liver disease.
DIFFERENCES IN NEUTROPHIL MIGRATORY BEHAVIOUR IN COPD COMPARED WITH AGE-MATCHED HEALTHY CONTROLS AND DISEASE-MATCHED PATIENTS WITH ALPHA 1-ANTITRYPSIN DEFICIENCY
E. Sapey, A. Ahmad, D. Bayley, R. H. Insall, and R. A. Stockley University of Birmingham, Edgbaston, Birmingham, B15 2TT
Neutrophils from patients with A1ATD are similar in their migratory behaviour to healthy controls, while neutrophils from patients with COPD appear more aggressive and demonstrate an overall enhanced migration towards common chemoattractants. However, studies have been unable to describe accurately these migratory differences. Aims were to identify differences in the speed, velocity, directional persistence and the overall accuracy of migrating neutrophils from patients with Chronic Obstructive Pulmonary Disease, age and gender matched healthy controls and patients with A1ATD matched for burden of lung disease. Phase time-lapse recordings were made using a Zeiss Axiovert 100 inverted microscope. Neutrophils from patients with moderate to severe COPD (as defined by GOLD criteria), age- and gender-matched healthy controls and patients with A1ATD (matched for FEV1 and smoking status) migrated through shallow gradients of IL-8 (at three concentrations with positive and negative controls) using a modified Dunn Chamber. Migration was assessed by speed of movement, velocity, directional persistence (a measure of cell orientation from slide to slide) and chemotactic index (overall movement towards the chemoattractant). Neutrophils from patients with A1ATD were comparable in behaviour to those from healthy controls. Neutrophils from patients with COPD moved with significantly greater speed than neutrophils from both the age-matched healthy controls and their disease-matched peers with A1ATD (p < 0.0001 for both). Neutrophils from patients with COPD also had significantly greater directional persistence than either their age matched healthy controls or their disease matched peers with A1ATD (p = 0.012 and p < 0.0001, respectively) and had a significant reduction in chemotactic index compared with the age-matched controls (p = 0.012). These data confirm that neutrophils from patients with A1ATD are comparable in behaviour to those from healthy controls, which is in keeping with previous work. Neutrophils from COPD patients move with greater speed than either their age-matched or disease-matched peers, and with insufficient attention to chemotactic stimuli which should change their direction. These findings suggest that neutrophils from patients with COPD are intrinsically different from cells from other populations and are in keeping with current theories of neutrophil-related damage that have been described in the lung.
GENETIC INFLUENCES UPON RESPIRATORY PHENOTYPE IN AATD
A. M. Wood, S. C. Gough, and R. A. Stockley University of Birmingham, Edgbaston, Birmingham, B15 2TT
AATD is associated with variable development of COPD phenotypes. We hypothesised that polymorphisms in candidate genes influencing usual COPD drive phenotypic variation in AATD. A total of 424 unrelated PiZZ subjects from the UK AATD Registry were phenotyped using clinical history, lung function, chest HRCT, sputum and plasma samples. Tag SNPs chosen using LD data for Caucasians (r2 > 0.8, MAF > 0.05) to cover 4 candidate genes and their surroundings were genotyped using TaqMan® genotyping technologies. Their influence upon quantitative and qualitative phenotypes in AATD was assessed using multiple regression accounting for all relevant covariates; 480 healthy controls were also genotyped. Phenotypic associations differed between genes, as shown in the table.
Subjects with the A allele of rs361525 tended to have higher plasma TNFα (p = 0.1), those with the G allele of rs2070741 were more likely to exhibit stable state bacterial colonisation (p = 0.04), and those with the TT genotype of rs1800469 had lower FEF25-75 than the wild-type (p = 0.03). Similar genetic associations exist in AATD to usual COPD. Detailed characterisation of patients may help to differentiate genetic effects and direct further functional work.
GENETIC VARIATION OF ALPHA 1-ANTITRYPSIN IN EUROPEAN AND AFRICAN POPULATIONS AND ORIGINS OF COMMON DEFICIENCY VARIANTS S,Z AND MMALTON
Sally Chappell, Tamar Guetta-Baranes, Maurizio Luisetti, Vanessa Hayes, and Noor Kalsheker The University of Nottingham, Queens Medical Centre, Nottingham NG7 2UH, UK
We have previously reported on genetic variation of the alpha1-antitrypsin (SERPINA1) gene in Caucasians and in African subjects from South Africa. We have undertaken a comparative analysis of the variation in the two populations (n = 685 and 110) and a Caucasian population from Australia (n = 118). Whilst the populations share many single nucleotide polymorphisms (SNPs) with no significant differences observed in the two Caucasian populations, there are some significant differences in the frequencies of some common variants as well as potentially unique variants in the African population. We have also compared our findings with information available from databases such as HapMap. There are gaps in HapMap as some of the SNPs we have studied are not on HapMap. We have also analysed the genetic background of 3 deficiency variants, Protease inhibitor (Pi) Z, S and Mmalton. By analysing combinations of SNPs in the form of haplotypes to determine the chromosomal background, each of these deficiency variants has a distinct origin. At this point in time we do not have sufficient data to understand the evolutionary history of these deficiency variants but this work is ongoing. The information we have gathered should be of value for genetic studies of alpha1-Antitrypsin in different population groups.
HIGH EFFICIENCY OF PROLASTIN® INHALATION IN PATIENTS WITH ALPHA 1-ANTITRYPSIN (AAT) DEFICIENCY AND CYSTIC FIBROSIS (CF) COMPARED TO HEALTHY VOLUNTEERS
M. Wencker,1 M. Schulte,2 J. Seitz,2 T. H. Meyer,2 and K. Hanna3 1Talecris Biotherapeutics GmbH, Frankfurt, Germany; 2Activaero GmbH, Gauteng, Germany; 3Talecris Biotherapeutics Inc, Raleigh, NC, USA
Patients with AAT deficiency and CF have an impaired protease-antiprotease balance, and inhalation of AAT may restore protective levels in the lungs. Previous inhalers required long inhalation times and resulted in variable deposition. We investigated the delivery efficiency of the AKITA2® with the APIXNEB® nebulizer, and the influence of lung function impairment on the distribution pattern after a single inhalation of 2 ml Prolastin® containing 70 mg of active 99mTc-labelled AAT in 7 patients with AAT deficiency, 7 patients with CF, and 6 healthy volunteers (mean ages for the 3 groups were 52, 29, and 33 years, respectively, and mean FEV1 % predicted were 51, 62, and 106%, respectively). There was no correlation between the amount of deposited Prolastin® and the severity of lung function impairment. Inhalation times were reasonable and not significantly different between the groups: mean inhalation time was 6.9 minutes for healthy volunteers, 13 minutes for patients with AAT deficiency, and 8.3 minutes for CF patients. Although this difference was not significant between the groups, the FEV1 correlated negatively with the inhalation time (r = −0.47, p = 0.042). There was also a negative correlation between FEV1 and the exhaled amount of drug (r = −0.46, p = 0.048). Inhalation of AAT with the AKITA2® APIXNEB® inhalation system in patients with AAT deficiency and CF results in very high delivery of Prolastin® in a short time, similar to that in normal volunteers. Funded by Talecris Biotherapeutics.
INHALED CORTICOSTEROIDS AS ADDITIONAL TREATMENT IN ALPHA1-ANTITRYPSIN DEFICIENCY-RELATED COPD
Giuseppe Emanuele La Piana, Enrica Bertella, Luciano Corda, and Claudio Tantucci Centro di Riferimento Regionale per il Deficit di Alfa1-Antitripsina (Prima Divisione di Medicina Interna, Spedali Civili, Brescia, Cattedra di Malattie dell'Apparato Respiratorio, Università di Brescia), Italy
No data are available regarding the effect of ICS in AATD-related COPD. Recent data report inflammatory effects of polymers of AAT on the peripheral lung. The aim of this study was to assess the effectiveness of an extra-fine ICS, hydrofluoroalkane-134a-bechlometasone (HFA-BDP) with median-mass-aerodynamic-diameter (MMAD) = 1.1 μm, when added to long-acting bronchodilators(BDs) on lung function and exercise tolerance in COPD patients with AATD. After one week wash-out, 8 steroid naíve COPD patients with AATD (ZZ genotype) were assigned in a double-blind randomized cross-over-study to the following 16-week treatments: 1) HFA-BDP 400 mcg t.i.d., salmeterol 50 mcg b.i.d. and bromide oxitropium 200 mcg t.i.d., or 2) placebo, salmeterol 50 mcg b.i.d. and bromide oxitropium 200 mcg t.i.d; after 2 week wash-out period they underwent to other treatment. At week1, week17, week19 and week35 patients performed spirometry (breathing air and Heliox) and Shuttle Walking Test(SWT) with the dyspnea assessment by Borg Scale. Significant improvement was found for FEV1, FVC, IC and distance covered and dyspnea perceived during SWT among treatments (p < 0.05; Friedman's test). Compared with baseline, however, only BDs + ICS showed significant increase in FEV1, FVC, IC, and Δ MEF50% and distance during SWT with reduction of maximum isostep exertional dyspnea (p < 0.05; Wilcoxon test). A greater distance was walked down at the end of SWT with ICS plus BDs as compared to BDs alone (301 ± 105 vs 270 ± 112 m; p < 0.05). In AATD-related COPD patients(ZZ genotype)the addition of extra-fine ICS to BDs decreases airway narrowing, mostly in the small airways, further reducing dynamic hyperinflation with a more marked improvement in exercise tolerance and dyspnea, suggesting a peripheral inflammatory process as a contributor of airflow obstruction in these patients.
RELATIONSHIP OF ALPHA 1-ANTITRYPSIN SERUM LEVELS AND GENETIC VARIANTS IN PATIENTS WITH PHENOTYPIC ALPHA 1-ANTITRYPSIN DEFICIENCY SAROJINI PANDEY AND DIMITRIS GRAMMATOPOULOS
Division of Clinical Sciences, Warwick Medical School and Molecular Diagnostics Laboratory, Hospital of St. Cross, UHCW NHS Trust, UK
Alpha 1-antitrypsin (α 1AT) deficiency is an autosomal hereditary disorder associated with respiratory and liver disease. Presence of abnormal alleles (S and Z) is suspected in individuals with lower than normal circulating levels of α 1AT protein in serum. At present, there is no evidence-based consensus opinion about the clinically relevant limit of α 1AT protein level required to further investigate and identify individuals carrying one or more abnormal α 1AT gene alleles. The aim of this study was to correlate α 1AT genotyping data with α 1AT protein concentrations. Patients (no = 859), with serum α 1AT concentration less than 1.4g/l were included in the study. An RT-PCR method was used to investigate the mutant alleles S and Z of the α 1AT gene. The S and Z allele frequencies for our selected patient population were found to be 0.075 and 0.067 for the S and Z allele, respectively. These were 1.5- and 3.35-fold greater than the allele frequencies reported for the general UK population. Statistical analysis showed that 100% and 85% detection rate for the Z and S allele respectively can be achieved at a cut-off level of < 1.4g/l serum α 1AT. Interestingly, significant correlation was found in the incidence of non alcoholic fatty liver disease (NAFLD) and presence of S allele (odds ratio = 2.354, CI = 1.513–3.664; p < 0.0001). In order to detect presence of Z and S alleles, genotyping of patients with α 1AT of < 1.4g/L is clinically-appropriate. Presence of the S allele might be associated with increased incidence of NAFLD.
SERPINA1 GENE VARIANTS IN SUBJECTS FROM GENERAL POPULATION WITH REDUCED ALPHA1-ANTITRYPSIN LEVEL
Michele Zorzetto,1 Erich Russi,2 Oliver Senn,2,3 Medea Imboden,3,4 Ilaria Ferrarotti,1 Ilaria Campo,1 Stefania Ottaviani,1 Roberta Scabini,1 Maurizio Luisetti,1 Nicole Probst-Hensch,3 and the SAPALDIA Team 1Center for Diagnosis of Severe Alpha1-antitrypsin Deficiency, Laboratory of Biochemistry & Genetics, Insitute for Respiratory Disease & Unit of Statistics and Biometry, Fondazione IRCCS Policlinico San Matteo, University of Pavia, Italy; 2Pulmonary Division, University Hospital of Zurich, Switzerland; 3Molecular Epidemiology/Cancer Registry, Institutes of Social and Preventive Medicine & Clinical Pathology, University of Zurich, Switzerland; 4Institute of Clinical Chemistry, University Hospital of Zürich, Switzerland
Subjects with severe deficiency of serum alpha1-antitrypsin (AAT) levels are at high risk for developing COPD, whereas individuals carrying the PI*MZ genotype are at moderately increased risk. Testing appropriate subgroups of the population for AAT deficiency (AATD) is therefore an important aspect of COPD prevention and timely treatment. No previous studies have extensively investigated at gene level subjects with moderately reduced serum AAT level, also referred to as intermediate AATD. Our work was aimed at an exhaustive identification of SERPINA1 variants in the general population, using a previously identified cut-off for serum AAT level. A total of 1,399 subjects enrolled in SAPALDIA with serum AAT level < 113 mg/dL were genotyped for the Z and S alleles; we submitted 498 of these samples to complete exon 2 → 5 sequencing. We found that 900/1,399 samples (64%), carried the normal PI*MM genotype, whereas 499 samples (36%) carried at least one SERPINA1 deficiency variant. In the samples in which AAT level ranged from 113 to 103, and from 103 to 93 mg/dL, PI*MM individuals represented the majority (86.5 and 53.8%, respectively). PI*MS subjects were predominant (54.9%) in the AAT range of 93–83 mg/dL, whereas PI*MZ represented 76.4% in the AAT range of 83–73 mg/dL. This analysis provided a detailed molecular definition of intermediate AATD, and in turn of PI*MZ individuals. This may be helpful in a diagnostic setting.
SERPINA1 IS A POTENT INHIBITOR OF IL-8-INDUCED HEMATOPOIETIC STEM CELL MOBILIZATION
Melissa van Pel,1* Ronald van Os,2* Gerjo A. Velders,1 Henny Hagoort,1 Peter M. H. Heegaard,3 Ivan J. D. Lindley,4 Roel Willemze,1 and Willem E. Fibbe1 1Laboratory of Experimental Hematology, Department of Hematology, Leiden University Medical Center, Leiden; 2Department of Cell Biology, Division of Stem Cell Biology, Groningen University Medical Center, Groningen, The Netherlands; 3Department of Veterinary Diagnostics and Research, Danish Institute of Food and Veterinary Research Copenhagen, Denmark; 4Novartis Institute for Biomedical Research, Vienna, Austria
Here we report that cytokine-induced (G-CSF, IL-8) hematopoietic stem and progenitor cell (HSC/HPC) mobilization is completely inhibited following low dose (0.5 Gy) total body irradiation (TBI). Since neutrophil granular proteases are regulatory mediators in cytokine-induced HSC/HPC mobilization, we considered a possible role for protease inhibitors in the induction of HSC/HPC mobilization. Bone marrow (BM) extracellular extracts that were obtained from murine femurs following 0.5 Gy TBI contained an inhibitor of elastase. In addition, following low-dose TBI, both serpina1 mRNA and protein concentrations were increased in BM extracts, compared to extracts obtained from controls. The inhibitory activity in BM extracts of irradiated mice was reversed by addition of an antibody directed against serpina1. To further study a possible in-vivo role of serpina1 in HSC/HPC mobilization, we administered serpina1 prior to IL-8 injection. This resulted in an almost complete inhibition of HSC/HPC mobilization, while heat-inactivated serpina1 had no effect. These results indicate that low dose TBI inhibits cytokine-induced HSC/HPC mobilization and induces serpina1 in the BM. Since exogenous administration of serpina1 inhibits mobilization, we propose that radiation-induced serpina1 is responsible for the inhibition of HSC/HPC mobilization. Furthermore, we hypothesize that cytokine-induced HSC/HPC mobilization is determined by a critical balance between serine proteases and serine protease inhibitors.
SINGLE AND REPEATED DOSE INHALATION TOXICOLOGY STUDIES WITH ALPHA-1 PROTEINASE INHIBITOR TAL6005
Vikram Arora, Kristine Bergstrand, Jeffrey Price, Stephen R. Petteway, and Philip Scuderi Research & Development, Talecris Biotherapeutics Inc, Research Triangle Park, NC 27709, USA
Acute and 28-day repeat-dose inhalation toxicology studies were performed with TAL6005, a human plasma-derived alpha 1 proteinase inhibitor, currently in development for aerosol treatment of emphysema secondary to congenital alpha-1 antitrypsin deficiency. Studies were conducted in Sprague Dawley rats and cynomolgus monkeys in support of planned clinical trials. Controls for both the acute and 28-day studies included one saline and two placebo groups (3% lactose/1.6% polysorbate 80). Acute studies delivered a single dose (followed by a 14-day recovery period) and repeated doses (rats, 5 days; monkeys, 10 days) of nebulized TAL6005. The high dose was 31.9 mg/ kg/day for rats and 41 mg/kg/day for monkeys. All treatments were well tolerated and did not result in any treatment-related effects on clinical signs, body weights, clinical or gross pathology, organ weights, or histopathology. The 28-day studies dosed nebulized test-article for 28 consecutive days, including 28-day recovery groups. In both species, the no-observed-adverse-effect-level was the high-dose (rats, 46.6 mg/kg/day; monkeys, 38 mg/ kg/day). Antigenic responses were detected in both species, but neither exhibited treatment-related effects on clinical signs, food consumption, body weights, clinical pathology or organ weights. Minimal to mild microscopic/macroscopic pathological changes indicative of local irritation in lungs, larynx and tracheobronchial lymph nodes were observed in a few monkeys across all dose groups. These were partly reversible and not considered TAL6005-related because of their distribution, which included placebo groups. In conclusion, TAL6005 is well-tolerated and does not cause dose-limiting toxicity in rodents or non-human primates at doses up to ten times those planned for patients in clinical trials.
VENTILATION INHOMOGENEITY IS INFLUENCED BY EMPHYSEMA DISTRIBUTION IN A1-ANTITRIPSIN DEFICIENCY (AATD)
L. Fregonese,1 A. Llado,1 B. C. Stoel,2 and J. Stolk1 1Pulmonology, Leiden University Medical Centre, Netherlands; 2Radiology, Leiden University Medical Centre, Netherlands
The slope of phase III (dN2) of the single breath nitrogen wash-out test (sbN2-test) measures ventilation inhomogeneity. Increased ventilation inhomogeneity has been suggested to reflect parenchymal damage in AATD-emphysema (AATDe). Computed tomography (CT) densitometry allows quantitative assessment of emphysema extent and progression. Emphysema severity and distribution at CT influences lung function outcomes such as Fev1 and Kco in AATDe. Aim:to investigate: (a) the correlation of dN2, closing volume (CV/%VC) and closing capacity (CC%TLC) with severity of emphysema as assessed by CT; (b) the influence of emphysema distribution on the same parameters. Methods: 50 subjects, ex-smokers, with AATDe were investigated cross-sectionally. Post-bronchodilator CT, spirometry, gas transfer and sbN2-test were performed. CT density was assessed in the whole lung, and separately in the lower and the upper lung zones. Results: 92% of the patients had predominantly basal emphysema, with mean total density −954.9 HU[SE1.85], lower lung density −962.1[SE1.9], upper lung density −943.5[SE2.3]. All patients had moderate-severe emphysema, with FEV1% pred 47.7[SE2.7], Kco% pred 51.2[SE1.8]. Mean values of dN2 were 4.9[SE0.3]. Analysis showed a correlation of dN2, CV/%VC and CC/%TLC with total and lower lung density (Table 1). Conclusions: ventilation inhomogeneity is related to emphysema severity at CT and the correlation is determined by lower lung disease. Future prospective studies may reveal if sbN2-test is an additional functional parameter to detect treatment effects on the progression of emphysema.
A POLYMORPHISM OF THE ALPHA1-ANTITRYPSIN GENE REPRESENTS A RISK FACTOR FOR LIVER DISEASE
Sally Chappell,1 Nedim Hadzic,2 Robert Stockley,3 Tamar Guetta-Baranes,1 Kevin Morgan,1 and Noor Kalsheker1 1The University of Nottingham, Queens Medical Centre, Nottingham NG7 2UH, England; 2University of Birmingham, Birmingham, England
Alpha1-antitrypsin deficiency (AATD) due to homozygosity of the protease inhibitor (Pi) Z variant predisposes to childhood liver disease and pulmonary emphysema. About 10% of all neonates with AATD develop liver disease, and about 3% overall progress to severe disease. AATD is a principal genetic indication for liver transplantation in children. The liver pathology is associated with accumulation of abnormally folded protein in hepatocytes, the principal producers of circulating alpha1-antitrypsin (AAT). It is currently unknown why only a small proportion of Pi ZZ individuals progress to clinically significant cirrhosis. The AAT gene shows significant variation, and we hypothesized that cryptic genetic variants within the AAT gene may contribute to susceptibility to liver disease. In a case-control study consisting of 42 patients with established moderate-to-severe liver disease and 335 homozygous Pi ZZ patients who mostly presented with chronic obstructive pulmonary disease (n = 322: 242 index cases and 80 unaffected sibs) or were asymptomatic (n = 13) with no evidence of liver disease, we identified a single nucleotide polymorphism (SNP) that conferred a significant risk for liver disease (p = 0.007). The frequency of the SNP was no different in 242 Pi ZZ cases with chronic obstructive pulmonary disease compared with 80 nonindex cases. The SNP therefore appears to confer susceptibility to liver disease, although reporter gene assays failed to show any functional differences between alleles. This is the first description of a genetic modifier of liver disease in homozygous ZZ children.
REFERENCES
- AJRCCM 2003; 168: 818
- ERJ 2001; 17: 1097