
Abstract
Current paradigms of chronic obstructive pulmonary disease (COPD) treatment suggest stratifying patients by their symptoms, utilising three main drug classes, but it is unclear if this approach will substantially alter the progression of the disease in the long term. More treatment options are needed which target the underlying pathology of the condition. Whilst many inflammatory cells are implicated in COPD, the neutrophil is by far the most abundant and has been extensively associated with disease pathogenesis. Neutrophil products are thought to be key mediators of inflammatory changes in the airways of COPD patients, causing pathological features such as emphysema and hypersecretion of mucus. High rates of bacterial colonisation and recurrent infective exacerbations of COPD, as well as evidence of neutrophil-associated host damage suggest that neutrophil functions may be impaired in COPD. This concept is supported by studies demonstrating impaired migratory accuracy and increased degranulation and reactive oxygen species release, with some evidence of altered cellular signalling pathways which might be exploitable as therapeutic targets. This review discusses our evolving understanding of neutrophil function in both health and COPD and highlights the role of this cell in disease pathogenesis, to determine whether this key inflammatory mediator represents a viable therapeutic target to prevent disease progression.
Introduction
Chronic obstructive pulmonary disease (COPD) remains a significant global health challenge. It has not seen the same improvements in morbidity and mortality as many other chronic inflammatory diseases with only one novel drug class reaching the market in the past 20 years (Citation1). After 25 years of smoking approximately 30–40% of adults would have developed COPD (Citation2) but despite most patients having this shared risk factor, COPD is heterogeneous in presentation, the age of onset and speed of decline. Whilst the disease is defined by the presence of airflow obstruction, there are a number of recognised clinical phenotypes. These include a predominance of emphysema (Citation3), obstructive bronchiolitis (Citation3, Citation4), the presence or absence of chronic bronchitis, frequent exacerbations (Citation5) and a faster decline in lung function (Citation6), and patients exhibit a spectrum of these features. Clinical phenotypes are stable within individual patients (Citation6) and cluster within families (Citation7) and thus are likely to reflect genetic traits. Indeed, genome wide association studies in COPD have identified a large number of signals for genes associated with lung development, lung parenchyma formation, and repair and epigenetic regulation such as the inositol phosphate pathway (Citation8, Citation9). These studies suggest there may be a wide range of therapeutic targets in different subsets of COPD patients, providing hope for personalised medicine. However, genes linked to lung development may not be therapeutically targetable in an adult with lung disease, drug development is costly and slow and the burden of COPD is rising fastest in low income countries (Citation10). A more common and shared targetable mechanism across disease phenotypes could provide a treatment for a larger number of patients.
The complex inflammatory milieu in COPD
Most pro-inflammatory mediators and immune cells have been shown to be raised in patients with COPD (Citation11, Citation12) and this inflammation is heightened and self-sustaining in smokers who are susceptible to COPD in contrast to those smokers who are not (Citation13, Citation14). However, while many cells and mediators have been implicated in COPD pathogenesis at some level, few have reliably demonstrated their importance as therapeutic targets in human studies. For example, despite the promising resistance to COPD-like lung damage shown by TNFα receptor (Citation15) or IL-1 receptor 1 knock-out mice (Citation16) studies targeting these individual mediators in unselected cohorts of patients with COPD have been disappointing with TNFα and IL-1 receptor 1 inhibition showing no improvements in disease endpoints (Citation17, Citation18). This might suggest that, unlike murine models, only sub groups of patients will respond to individual mediator-based therapies. In support of this concept, there is variability in inflammatory patterns between patients, even when matched for age, smoking status and disease severity (Citation19) which might reflect specific genetic traits as shown in some studies of TNFα (Citation20) and IL-1β (Citation21) polymorphisms.
To complicate matters further, there is significant intra-patient variability in the concentration of plasma and sputum mediators and cells on a day-to-day basis. Some mediators increase while others decrease suggesting the variability not only reflects dilution but also fluctuations in specific components of the inflammatory load (Citation19, Citation22). This supports an alternative explanation to the negative trial results reported to date, where there is so much compensation and overlap within the complex inflammatory storm that is established COPD (Citation23) that end-cell effects can be driven by an alternative cytokine, should one be abrogated. For example, toll-like receptors, TNFα and IL-1 signalling to NF-κB all converge on a common IκB kinase complex that phosphorylates the NF-κB inhibitory protein IκBα, despite the upstream signalling components being to a large part receptor-specific (Citation24). Despite the effects of these mediators being synergistic when they converge on the same pathway, inhibiting one has not proved efficacious enough to impact robustly on cellular inflammation or COPD disease progression. Potentially targeting the functions of the end-cell and not the intermediatory cytokine might be more effective and there is a strong rationale for targeting the neutrophil in COPD.
Classical neutrophil functions in health
Neutrophils are the most abundant leukocyte, accounting for 70% of all circulating white blood cells. They are short-lived cells (with a half-life of around eight hours), with basal production of 1–2 × 1011 neutrophils/day in health; though this can increase to 1012 during infection (Citation25). Following myeloblastic differentiation in the bone marrow, the mobilisation of terminally differentiated neutrophils into the circulation is tightly controlled by bone marrow signals and circulating growth factors. Interactions between neutrophil CXCR4 and bone marrow stromal cell CXCL12 cause cell retention or cell return to the bone marrow and increasing neutrophil CXCR2 results in neutrophil release into the circulation (Citation26, Citation27).
Neutrophils are characterised by the presence of a multi-lobed nucleus and granular cytoplasm, due to the presence of azurophillic (primary), specific (secondary) and gelatinase (tertiary) granules, as well as secretory vesicles. These granules and vesicles contain a complex and specialised arsenal of proteins which facilitate cell to cell communication and functional modification, neutrophil migration from the systemic circulation through the dense extracellular matrix to areas of inflammation, microbial killing and tissue remodelling, degradation and repair. Some of these proteins are listed in .
Table 1. The contents of human neutrophil granules and secretory vesicles.
In COPD, a significant physiological role of these granular contents is bacterial killing, achieved mainly when bacteria are ingested into phagosomes that fuse with lysosymes containing proteinases, bacteriocidal proteins such as perforin and granzyme B (the two molecules known as the cytotoxic entity of natural killer cells and of cytotoxic T lymphocytes) (Citation28) and reactive oxygen species following the formation of nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase from subunits within the cytosol. However, before this can occur, circulating neutrophils must reach the site of infection, and they do so following chemotactic signals in a gradient-dependent manner (Citation29). ATP released from chemoattractant-stimulated neutrophils amplifies the external migratory signal, controlling gradient sensing and migrational speed in an autocrine and paracrine manner, recruiting more neutrophils to the site of inflammation (Citation30). Circulating neutrophils then cross the endothelium via the Leukocyte Adhesion Cascade (LAC). Here, neutrophils interact with endothelial cells at sites of high inflammatory signalling by initially rolling on the endothelial surface by reversibly binding selectins which are found on both the neutrophil and endothelial cells. This transitions to firm adhesion by the sequential activation of integrins, followed by transendothelial migration (Citation31).
Once within inflamed tissue, the neutrophil further utilizes chemotactic gradients created by host-derived inflammatory cytokines (e.g. Interleukin-8) and bacterial products (e.g., lipopolysaccharide [LPS] and N-formyl-methionyl-leucyl-phenylalanine [fMLP]) released from the site of infection to navigate towards invading pathogens (Citation29). Activated neutrophils have a clear “front” and “back” and use this polarised form to amplify external gradients internally within the cell, allowing accurate mobilisation of internal structures (generation of pseudopods for migration, granules for phagolysis) towards the inflammatory or infectious insult. Chemotactic signals such as Leukotriene B4 (LTB4), integrin binding and neutrophil ATP release promotes the formation of neutrophil “swarms” within these damaged tissues (Citation32) sealing off the sites of infection and maximising the clearance of bacteria and cell debris by phagocytosis and netosis (Citation33). During these episodes, neutrophils far outnumber any other immune cells in the inflamed tissue.
Phagocytosis is an active, receptor-dependent process through which a phagocyte internalises material into membrane-bound vacuoles (Citation34). Phagocytosis of opsonised particles (by IgG, for example) occurs in seconds (Citation35), faster than the minutes required for macrophage phagocytosis of bacteria (Citation36). The most extensively studied opsonin receptors of neutrophils, the Fc receptors (e.g. FcγRIIA [CD32], FcγIIIb [CD16]) bind IgG-bound particles triggering a signalling cascade involving the tyrosine kinase Syk and phosphatidylinositol 3-kinase (PI3K). Complement molecules also opsonise pathogens by interacting with neutrophil complement receptors (e.g. CR1 [CD35], CR3 [CD11b/CD18]) (Citation37–40). Once bacteria are enveloped, bacterial killing occurs through the delivery of neutrophil granules and the NADPH oxidase machinery into the vacuole by membrane trafficking (Citation29, Citation41, Citation42). These processes are depicted in .
Figure 1. Phagosome maturation.
The neutrophil NADPH oxidase machinery, activated by delivery of its membrane-bound components to the phagosome, pumps electrons into the phagosomal space to generate toxic ROS. Membrane trafficking allows delivery of primary and secondary granules to the phagosomal membrane, which release a variety of microbicidal proteins into the phagosomal space. MPO released from primary granules reacts with ROS to further produce highly toxic substances. Adapted from (Citation161).
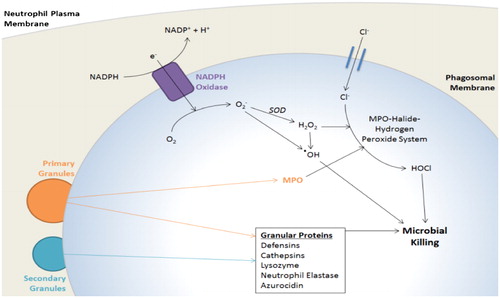
In contrast with the highly acidic pH of macrophage phagolysosomes, the neutrophil phagosome appears to undergo a transient alkalisation, which is optimal for neutrophil protease activity (Citation43). Then, with maturation and acidification of the phagolysosome, Reactive Oxygen Species (ROS) are released into phagosomes with NADPH oxidase acting as a channel for electrons from the cytosol into phagosomal vacuoles, stimulating reduction of oxygen (O2) to the superoxide anion O2 - (Citation44). Superoxide can then dismutate, a process accelerated by the enzyme superoxide dismutase (SOD), to form the highly oxidative hydrogen peroxide (H2O2), which can react further (the MPO-halide-hydrogen peroxide antibacterial system) to form strongly bactericidal hypohalous acids (e.g., HOCl) (Citation45–50). The role of ROS and NADPH oxidase in elimination of pathogens was long-believed to be a dominant killing mechanism in neutrophils, however, the exact role of these processes in neutrophil bacterial killing has come under recent scrutiny with some research supporting their role as facilitatory rather than obligatory (Citation48).
Neutrophils are also able to produce extracellular traps (NETs), consisting of a backbone of uncondensed chromatin to which bactericidal products such as cathepsins, MPO and nuclear histones are bound (Citation51). Microscopic studies suggest NETs are responsible for the killing of a wide range of pathogens, including gram-negative (Citation51) and gram-positive (Citation51, Citation52) bacteria as well as fungi (Citation53). However, NETosis occurs later in neutrophil activation than other killing processes such as phagocytosis or generation of ROS (Citation54) and this appears to be a “final act” for most neutrophils, when faced with overwhelming infection.
The crucial role of neutrophils in clearing infection is amply evidenced by the devastating outcomes seen when neutrophil numbers are reduced or functions are inhibited including during neutropenic sepsis, chronic granulomatous diseases (most commonly associated with abnormalities in cytochrome b558, preventing NADPH oxidase formation (Citation55)); Chediak–Higashi Syndrome (characterised by giant lysosomal granules which lack neutrophil elastase and cathepsin G (Citation56)) and specific granule deficiency, where neutrophil migration is comprised due to failure to upregulate surface receptors upon activation (Citation57); this is more fully reviewed in (Citation58).
The capacity for neutrophil-induced host damage and its mitigation
The same cytotoxic proteins crucial in bacterial killing also have the potential for significant host harm, as exemplified by neutrophil elastase (NE). Each neutrophil contains on average 400 elastase-positive granules (Citation59) and each granule contains approximately 67,000 molecules of NE at a mean concentration of 5.33 mMol (Citation60). Free NE is inhibited by Alpha 1 anti-trypsin (AAT) but on a one-to-one molar basis and the average concentration of AAT in healthy individuals in 30 μMol, resulting in a localised area of uninhibited and thus active NE around the cell following degranulation. Free NE can cause significant damage to host by a number of mechanisms () and degranulation can occur during migration, frustrated phagocytosis or “sloppy eating” and netosis as well as during cellular necrosis. NE is not the only protein implicated in host damage and many other granular products have been implicated including proteinase 3 (Citation61), reactive oxygen species generation (Citation62) and MPO-mediated nitrosative stress (Citation63). In stable COPD, sputum samples can contain up to 10 × 106/ml neutrophils (Citation22); therefore, the potential for lung damage is high.
Table 2. The actions of neutrophil elastase.
To protect the host, neutrophils have evolved a number of safe-guarding mechanisms. Neutrophils exist in three potential states of activation; quiescent, primed or activated, akin to a tri-coloured warning system and primed cells both enable a faster response to full activation but also permit a return to the quiescent state prior to degranulation if full activation is not needed (Citation71). Cellular cross-talk controls aspects of neutrophil activity. For example, during inflammatory events, activated platelets adhere to circulating blood neutrophils causing neutrophil-derived and arachidonic acid-containing extracellular vesicles to be taken up into the platelet in a Mac1-dependent fashion. Within the platelet, arachidonic acid is used to synthesise thromboxane A2 which increases ICAM-1 on endothelial cells, facilitating the movement of neutrophils from the blood stream into the tissues (Citation72). This co-operation between three cell types (platelet, neutrophil and endothelial cell) is thought to prevent unwarranted neutrophil migration into tissues and subsequent tissue damage. Degranulation is not an “all or nothing” response and neutrophils release different vesicles or granules depending on the environmental triggers encountered. Different granule sub-types are mobilised and released dependent on cytosolic-free calcium levels, which allows coordination of granule release to match cellular requirement, e.g. mobilisation of azurophilic granules occurs only once the neutrophil comes into contact with a significant inflammatory stimuli while mobilisation of secretory vesicles occurs during the initial stages of cell polarisation (Citation73).
Changing perceptions of the neutrophil
The classical view of neutrophils is that they are a uniform population of short lived cells and have a narrow range of responses, in contrast to the ever-expanding sub-populations of other cell types. However, this perception is being challenged and there is evidence that neutrophils may be more diverse than first thought. Neutrophils are more transcriptionally active than previously appreciated (Citation74) and their lifespan appears to change depending on activation status and environmental circumstances (Citation75). Furthermore, experimental models have described an increasing number of neutrophil “phenotypes” which have different functional characteristics and appear more distinct than simply being “young” (or freshly released from the bone marrow) or “old” (prior to returning to the marginated pools and apoptosis) cells. Not all neutrophils stay in tissues for clearance by efferocytosis, and a subset have been shown to “reverse transmigrate” back into the systemic circulation (Citation76). Studies have described anti-inflammatory neutrophils that are involved in tumour clearance (Citation77) and there are descriptions of neutrophils which appear to modulate T-cell function via Mac 1 signalling, leading to dampening of the immune response (Citation78), so called “anti-inflammatory” neutrophils. In murine models of ventilator-associated acute lung injury, there appear to be two distinct waves of neutrophil in the airways; the first is the classical neutrophil which has been associated with significant tissue damage (Citation79). The second appears to be a pro-angiogenic neutrophil (Citation80), characterised by increased MMP-9 release (Citation81, Citation82) with a distinctive pattern of surface markers and an association with improved clinical outcomes (Citation83). It remains unclear what the relevance of these cellular phenotypes are in clinical practice, including in COPD.
Neutrophils in COPD
Neutrophils are considered central to the pathogenesis of COPD. Airway neutrophilia is a feature of COPD regardless of the clinical phenotype, severity of disease, rapidity of decline or age of onset. Indeed, even in the subset of patients with eosinophilic COPD, neutrophils remain the predominant cell (Citation84). Their numbers and products in sputum and airway lavage fluid correlate with disease severity, as determined by the degree of airway obstruction, decline in FEV1 or severity of emphysema present (Citation85–88). Histological studies have not always reported the presence of neutrophils within the airway wall (Citation89, Citation90), however, neutrophils are not tissue resident but transitory visitors, migrating through to the airways.
The association between COPD and neutrophils is further evidenced by Alpha-1 Antitrypsin Deficiency (AATD). AATD is the most extensively documented genetic risk factor for COPD, where the reduction of AAT predisposes to increased proteinase-mediated degradation of extracellular matrix and development of emphysema (Citation91). Emphysema occurs even in the absence of smoking in some individuals with AATD and rates of decline in lung function are faster in those who smoke. Furthermore, there is evidence that emphysema progression is slowed by augmentation with replacement Alpha-1 Anti-trypsin (Citation92) although the effect of augmentation on FEV1 decline is less clear (Citation93).
Neutrophil products have been shown to cause all of the pathological features of COPD in animal and cell-based models. As described in and using NE as an exemplar, serine proteases not only degrade all components of the extracellular matrix but also activate matrix metalloproteinases (MMPs) from their inactive pro-forms and inhibit the inhibitors of MMPs, increasing the proteinase burden (Citation94) implicated in COPD (Citation95). Proteinases are potent stimulants of mucus secretion in the airways and reduce mucociliary clearance by impeding ciliary function leading to symptoms of chronic bronchitis (Citation96–98). They also cleave immunoglobulins and components of the complement cascade, potentially causing an opsono-receptor mismatch (Citation64). There is evidence to link neutrophil degranulation with bacterial colonisation. In murine models, neutrophil recruitment to the mucosa has been associated with a neutrophil-elastase-dependent change in the composition of the intestinal microbiota, facilitating colonisation (Citation99). In cystic fibrosis, neutrophil recruitment to the airways is associated with prolonged neutrophil survival, activation and altered functions consistent with a phenotypic change which permits the co-habitation of neutrophils and microbes (Citation100). In COPD, and sputum NETosis correlates with the dominance of Haemophilus species in the lung microbiome (Citation101). In theory, in COPD, this might result in a vicious circle, where inflamed or damaged tissue support bacterial colonisation and subsequent infection, which leads to more inflammation and more neutrophil activation and recruitment to the airways.
In support of this, there is consistent evidence of heightened neutrophil activity in COPD. AαVAL360 is a neutrophil elastase-specific fibrinogen degradation product, thus a footprint of elastase activity, and plasma levels have been shown to be raised in stable COPD. This suggests either systemic activity of neutrophils in COPD or potentially a significant overspill from the lungs into the circulation (Citation102). COPD sputum also contains neutrophil products including human neutrophil lipocalin (HNL) and myeloperoxidase (MPO) (Citation103).
Further evidence of the importance of neutrophils in COPD comes from imaging studies. 18-fluorodeoxyglucose positron emission tomography in COPD patients show enhanced uptake caused by active neutrophils in the emphysematous regions of the lungs and this correlates with measures of disease severity (Citation104). The release of neutrophil proteinases into these damaged lungs will contribute to the degradation of elastin and type III collagen, leading to the destruction of alveolar tissue and consequently centrilobular emphysema, apparent in these imaging studies and in many cases of COPD (Citation105, Citation106). Elegant pathological studies by Hogg et al. (Citation107) suggest small airways dysfunction and destruction may precede the development of emphysema and may reflect the earliest pathological changes in the lungs of patients with COPD. Neutrophils have also been implicated in small airways disease, showing a relationship with neutrophilic infiltration and tomographic measures of air trapping (Citation108).
Cellular dysfunction as a mediator of COPD
Neutrophil responses should be tightly controlled to prevent host damage and the high number of neutrophils present in airway secretions in COPD should mitigate against bacterial colonisation. Despite this, there is tissue damage and recurrent infections are common. There is evidence to suggest that neutrophils from patients with COPD are impaired in function which might explain these injurious effects. COPD neutrophils demonstrate migratory inaccuracy, able to migrate towards chemotactic signals with greater speed, but decreased accuracy than neutrophils from age-matched healthy controls (Citation109, Citation110). This defect was not present in smokers without COPD and has been described in mild to chronic disease, which suggests it is an early development in COPD and might theoretically precede the disease, although this has not been tested. In vitro modelling suggests the migratory defect results in a longer and more convoluted migratory paths, increasing the secretion of damaging enzymes and delaying bacterial killing (Citation109). There is limited data about the phagocytic functions of COPD neutrophils. In some studies, ingestion of opsonised species have been found to be reduced in COPD neutrophils compared to age-matched healthy controls (Citation111, Citation112). However, in other studies, there are no differences between the phagocytic abilities of COPD neutrophils and controls (Citation113, Citation114). This suggests any defect may be stimulant, environment (tissue or blood neutrophil) and assay dependent or only seen in a subset of patients. Flow cytometric studies demonstrate enhanced respiratory burst in neutrophils from patients with COPD as compared to healthy smokers (Citation115) and increased markers of oxidative activity both in COPD airways and systemically suggest increased ROS-producing capacity in stable disease (Citation116). Neutrophils from patients with COPD are also able to degranulate more readily to stimuli and increased quantities of NETs and NET-producing neutrophils are observed in the sputum of both stable and exacerbating COPD patients (Citation117, Citation118). All of these functions favour tissue damage.
It is unclear why neutrophil functions may be altered in COPD. Some other chronic inflammatory conditions report a profound change in neutrophil gene expression profile which might support a phenotypic shift in these cells (Citation83, Citation119) and there is some evidence to support a similar concept in COPD. A recent study identified different neutrophil phenotypes in COPD based on the expression of regulated proteins and hierarchical clustering without clear differences in clinical phenotype (Citation120) but this was cross-sectional and could not assess when neutrophil phenotype was altered. It might reflect differences in genetic traits between patients but also might reflect epigenetic changes within the neutrophil following long-term exposure to inflammation. DNA damage has been seen in COPD due to oxidative stress (Citation121) and in other conditions transcriptional changes have been described in neutrophils and are associated with poor neutrophil migratory accuracy (Citation122, Citation123). Epigenetic changes are long-lived, and the systemic inflammation present in COPD could theoretically affect neutrophil progenitor cells, leading to sustained aberrant functions and subsequent inflammation. In support of this concept, epigenetic changes have been described in bone marrow macrophage progenitor cells in type 2 diabetes (Citation124), which is also a pro-inflammatory disease.
Neutrophil functions during exacerbations of COPD
COPD exacerbations are heterogeneous in their cause (viral, bacterial, environmental, associated with co-morbidities), severity and duration (Citation125) and the majority of studies of inflammation during exacerbations have included small number of patients. However, they have confirmed the presence of neutrophils during these episodes (Citation126). Increased serum levels of damage-associated molecular patterns (DAMPs) have been described during COPD exacerbations and gene expression of DAMP receptors (including toll-like receptor (TLR2 and TLR4) in circulating neutrophils have been shown to be significantly increased during these events supporting neutrophil activation (Citation127). Furthermore, there is evidence of increased neutrophil elastase activity during COPD exacerbations compared with the stable state (Citation128) and spontaneous ROS production in sputum neutrophils is up to 45% higher during exacerbations of COPD compared with stable disease (Citation129). In summary, it appears that the potential for neutrophil-associated tissue damage seen in stable COPD is heightened in exacerbations, with more neutrophils, more activation and more proteinase and ROS release. It is unknown if the functional differences noted in stable patients with COPD are also present or even amplified during exacerbations.
Neutrophils and cellular cross talk in COPD
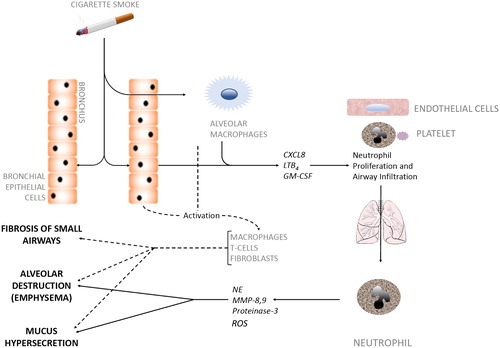
Neutrophils do not act in isolation, and many cells have altered functions in COPD, including macrophages (Citation130), T cells (Citation131), B cells (Citation131, Citation132), eosinophils (Citation133) and platelets (Citation134), which have all been implicated in tissue damage. The close cross talk between cells which allows an efficient response to infection but then limits host damage appears to be lost in COPD and it might be the accumulation of cell dysfunction which leads to such a damaging neutrophil response. For example, alveolar macrophages are less efficient at phagocytosing bacteria in COPD (Citation135). When activated, these cells release inflammatory mediators and macrophage-specific and scavenged proteinases, recruiting more neutrophils to the airways, which in turn promote monocyte recruitment. Activated CD8+ T cells in COPD demonstrate more aggressive, pro-inflammatory responses than seen in smokers without COPD in response to microbial TLR ligands (Citation136), leading to more neutrophil recruitment. Neutrophil clearance by macrophage efferocytosis is reduced in COPD (Citation137), increasing the likelihood that these cells will undergo necrosis, increasing the proteinase burden in lung tissue and thus leading to a vicious cycle of inflammatory damage. Also, fibroblasts in COPD have a reduced capability for tissue repair (Citation138) and endothelial and epithelial dysfunction have been widely described (Citation139). Perhaps a combination of diverse genetic mutations or transcriptional changes in patients, when associated with appropriate environmental inflammatory exposures (such as smoking) can cooperatively cause variations in immune, airway and endothelial cells which are sustained, lead to damaging neutrophil recruitment and activation and ultimately to COPD ().
Figure 2. Cellular interactions in COPD pathogenesis.
Inhaled irritants such as cigarette smoke stimulate bronchial epithelial cells and alveolar macrophages to secrete factors (CXCL8, LTB4, GM-CSF) that promote both neutrophil production in bone marrow and neutrophil migration towards inflamed airways, a process coordinated by activated platelets, endothelial cells and the neutrophil. Neutrophils infiltrate the airways in large numbers and produce serine proteases and elastolytic enzymes (e.g. NE, MMP-8,9, proteinase-3). These neutrophilic enzymes act alongside other activated immune cells (dotted line) to degrade alveolar tissue leading to emphysema, over-stimulate Goblet cells leading to hypersecretion of mucus, and cause fibrosis of the small airways, all of which are hallmark features of COPD. CXCL8, Chemokine (CXC motif) ligand 8; LTB4, Leukotriene B4; GM-CSF, Granulocyte-Macrophage Colony-Stimulating Factor; NE, Neutrophil Elastase; MMP, Matrix Metalloproteases.
Targeting the neutrophil in COPD
Despite a number of cells being implicated in COPD, arguably the cell with the most potential for damage is the neutrophil, due to cell numbers and the injurious potential of each cell. However, there is understandable caution in abrogating the function of this cell as this would place the host at risk of overwhelming infection. Instead, strategies have considered if numbers or functions (including pro-inflammatory responses) could be “normalised” in COPD.
Glucocorticosteroids are one of the most commonly prescribed anti-inflammatory medicines in obstructive lung diseases which mediate their effects in part through trans-repression of pro-inflammatory transcription factors such as nuclear factor-κB (NF-κB). There are a number of clinical and in vitro studies which describe reduced steroid responsiveness in COPD (including from COPD neutrophils due in part to reduced NF-κB activation) (Citation140) and large, late phase clinical trials of inhaled steroids do not suggest they prevent disease progression (Citation141). In light of this, steroids are not considered to be effective at targeting COPD-related neutrophilic inflammation.
Phoshodiesterase type-4 inhibitors (PDE4) such as Roflumilast have been shown to improve lung function, reduce exacerbation frequency and reduce inflammation in frequently exacerbating COPD (Citation142, Citation143). PDE4 is involved in the degradation of cyclic adenosine monophosphate (cAMP) and raised levels of cAMP promote airway smooth muscle relaxation and modulate inflammatory cell activity, reducing pro-inflammatory mediator release. In COPD, PDE4 inhibitors reduce neutrophil LTB4, protease and ROS release and reduce overall neutrophil recruitment to the airways (Citation144), suggesting potent and COPD-relevant anti-inflammatory actions.
Leukotriene B4 (LTB4) is a potent neutrophil chemoattractant, and it was hypothesised that blocking this chemokine might reduce neutrophil recruitment to the lungs. However, an open label study of an LTB4 antagonist in 17 COPD patients did not show a reduction in the chemotactic potential of sputum (Citation145), suggesting other mediators were able to compensate for the reduction in LTB4 activity. A six-month trial of a CXCR2 inhibitor (which is the receptor for a number of neutrophil chemoattractants including IL-8, Growth related oncogene-α and Epithelial-neutrophil activating peptide-78, all raised in COPD) reduced neutrophil counts, MPO and MMP-9 in sputum and improved FEV1 compared to placebo in over 600 patients with COPD but 18% of patients had to withdraw from the active arm due to neutropenia, suggesting perhaps this treatment was too potent in its effect (Citation146).
Although in its infancy, more recent studies have shifted paradigms in COPD treatment; focusing on correcting cell function rather than decreasing recruitment signals (Citation147). p38 mitogen-activated protein kinase activation is involved in neutrophil directional sensing (Citation148) and p38 signalling has been shown to be raised in COPD (Citation149) but Losmapimod (a p38 inhibitor) had no effect on endothelial function (Citation150), lung function, exercise tolerance (Citation151) or sputum neutrophils in COPD although inflammatory mediators were reduced (Citation152). Targeting matrix metalloproteinases in phase 2 trials has also provided no clear evidence of efficacy although a potential signal was seen in urinary desmosine (Citation153). Aberrant neutrophil functions have been linked to increased activity of the PI3K pathway within the neutrophil (Citation154) and more recent studies have focused on this as a potential target (Citation155), with on-going trials in acute exacerbations of COPD. Of note, the PI3K pathway includes a number of therapeutic targets, some of which can be modified using repurposed medications such as HMG Co-A reductase inhibitors (commonly called “statins”). In vitro, high concentration statins have been shown to reproduce the same positive effect on neutrophil migration seen using a PI3K inhibitor (Citation156) including in COPD (Citation157). Of interest, metformin, acting through AMP-activating kinase, may also impact on these cellular functions through the same signalling cascade (Citation158) and metformin has been associated with better clinical outcomes in some studies of COPD (Citation159).
The future
Current COPD treatment stratification using the most recent GOLD document suggests symptoms may help inform treatment decisions, with bronchodilation for breathlessness and inhaled steroids (with bronchodilators) for exacerbations, especially if there are eosinophils (Citation160), and recommends three main drug classes (beta2 agonists, muscurinic antagonists and corticosteroids). This approach to COPD management does not reflect the phenotype and endotype approach utilised in complex asthma or in stratified lung cancer trials, or the array of treatment options available for the treatment of cardiovascular disease.
It is likely that we will not modify the course of COPD if we do not treat the cellular causes of the pathology. Neutrophils and more recently neutrophil dysfunction has been implicated in many of the pathological features of COPD (from chronic bronchitis to emphysema). A potential analogy may be with type 2 diabetes. In this diverse condition there are likely to be many susceptibility factors, both genetic and environmental, but almost irrespective of the actual cause, controlling blood glucose to within normal levels can help prevent end organ damage. Neutrophil dysfunction may be a common cause or indeed a common outcome of the underlying defect in COPD which then contributes substantially to host damage. If these were true, maintaining the function of these cells to within normal parameters might control aspects of tissue damage for many patients with COPD. Neutrophil dysfunction may have many different causes in COPD (PI3K signalling in some, p38 in others) which could be targeted incrementally through a number of putative therapeutics. Alternatively, this defect might be associated with only a proportion of COPD patients. However, whatever the incidence of neutrophil dysfunction in COPD, clinical phenotypes are unlikely to identify the particular cellular cause of the disease in an individual (like breathlessness will not identify the cellular basis of lung cancer nor the exact genetic mutation present) and we will probably need a deeper approach to treatment stratification to advance our ability to halt disease progression.
Conclusions
Neutrophils are innate immune cells that have been widely implicated in the pathogenesis of COPD. They are a feature of all disease phenotypes and their ability to damage lung parenchyma as part of the inflammatory pathology of COPD has been well-described. There is mounting evidence of neutrophil dysfunction in COPD, which might explain the continued inflammatory response. All these data suggest the neutrophil may represent a unifying therapeutic target in COPD, but the necessary functions of these cells make this target a challenging one.
Normalising their activity whilst maintaining their ability to participate in host defence may be a crucial step in preventing COPD progression. However, it is unclear whether changing the function of one cell will have a positive or detrimental effect on other crucial cell functions; whether normalising neutrophils will have a positive effect on COPD clinically, and whether targeting a potential “end-cell” mechanism of COPD (such as aggressive and indiscriminate neutrophils) will help a wider range of patients than seen in studies of a particular inflammatory mediator. Understanding neutrophil signalling and how these cells interact with other cells in COPD may unlock new strategies for COPD treatment.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Calverley PMA, Rabe KF, Goehring U-M, Kristiansen S, Fabbri LM, Martinez FJ. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet. 2009;374(9691):685–94.
- Løkke A, Lange P, Scharling H, Fabricius P, Vestbo J. Developing COPD: a 25 year follow up study of the general population. Thorax. 2006;61(11):935.
- Jeffery PK. Comparison of the structural and inflammatory features of COPD and asthma. Giles F. Filley lecture. Chest. 2000;117(5 Suppl 1):251S–60S.
- Bignon J, Khoury F, Even P, Andre J, Brouet G. Morphometric study in chronic obstructive bronchopulmonary disease. Pathologic, clinical, and physiologic correlations. Am Rev Respir Dis. 1969;99(5):669–95.
- Wedzicha JA, Brill SE, Allinson JP, Donaldson GC. Mechanisms and impact of the frequent exacerbator phenotype in chronic obstructive pulmonary disease. BMC Med. 2013;11:181.
- Vestbo J, Edwards LD, Scanlon PD, Yates JC, Agusti A, Bakke P, et al. Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med. 2011;365(13):1184–92.
- Patel BD, Coxson HO, Pillai SG, Agustí AGN, Calverley PMA, Donner CF, et al. Airway wall thickening and emphysema show independent familial aggregation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178(5):500–5.
- Probert K, Miller S, Kheirallah AK, Hall IP. Developmental genetics of the COPD lung. COPD Res Pract. 2015;1(1):10.
- Wain LV, Shrine N, Artigas MS, Erzurumluoglu AM, Noyvert B, Bossini-Castillo L, et al. Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nature Genet. 2017;49:416.
- Ferkol T, Schraufnagel D. The global burden of respiratory disease. Ann Am Thoracic Soc. 2014;11(3):404–6.
- Barnes PJ, Chowdhury B, Kharitonov SA, Magnussen H, Page CP, Postma D, et al. Pulmonary biomarkers in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;174(1):6–14.
- Sapey E, Bayley D, Ahmad A, Stockley R. The validation of assays used to measure biomarkers in exhaled breath condensate. Eur Respir J. 2008;32(5):1408–9; author reply 9.
- Hoonhorst SJM, Timens W, Koenderman L, Lo Tam Loi AT, Lammers J-WJ, Boezen HM, et al. Increased activation of blood neutrophils after cigarette smoking in young individuals susceptible to COPD. Respir Res. 2014;15(1):121.
- Rutgers SR, Postma DS, H. tHN, Kauffman HF, W. vDMT, Koeter GH, et al. Ongoing airway inflammation in patients with COPD who Do not currently smoke. Chest. 2000;117(1931–3543 (Electronic)):262S.
- Churg A., Wang RD, Tai H, Wang X, Xie C, Wright JL. Tumour necrosis factor alpha drives 70% of cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med. 2004;170:492–8.
- Pauwels NS, Bracke KR, Dupont LL, Van Pottelberge GR, Provoost S, Vanden Berghe T, et al. Role of IL-1α and the Nlrp3/caspase-1/IL-1β axis in cigarette smoke-induced pulmonary inflammation and COPD. Eur Respir J. 2011;38(5):1019.
- Rennard SI, Fogarty C, Kelson S, Long W, Ramsdell J, Allison J, et al. The safety and efficacy of infliximab in moderate to severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175:926–34.
- Calverley PMA, Sethi S, Dawson M, Ward CK, Finch DK, Penney M, et al. A randomised, placebo-controlled trial of anti–interleukin-1 receptor 1 monoclonal antibody MEDI8968 in chronic obstructive pulmonary disease. Respir Res. 2017;18:153.
- Stone H, McNab G, Wood AM, Stockley RA, Sapey E. Variability of sputum inflammatory mediators in COPD and α1-antitrypsin deficiency. Eur Respir J. 2012;40(3):561–9.
- Sapey E, Wood AM, Ahmad A, Stockley RA. Tumor necrosis factor-{alpha} rs361525 polymorphism is associated with increased local production and downstream inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182(2):192–9.
- Xie Z-K, Huang Q-P, Huang J, Xie Z-F. Association between the IL1B, IL1RN polymorphisms and COPD risk: a meta-analysis. Sci Rep. 2014;4:6202.
- Sapey E, Bayley D, Ahmad A, Newbold P, Snell N, Stockley RA. Inter-relationships between inflammatory markers in patients with stable COPD with bronchitis: intra-patient and inter-patient variability. Thorax. 2008;63(6):493–9.
- Nish S, Medzhitov R. Host defense pathways: role of redundancy and compensation in infectious disease phenotypes. Immunity. 2011;34(5):629–36.
- Verstrepen L, Bekaert T, Chau TL, Tavernier J, Chariot A, Beyaert R. TLR-4, IL-1R and TNF-R signaling to NF-κB: variations on a common theme. Cell Mol Life Sci.. 2008;65(19):2964–78.
- Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol. 2010;31(8):318–24.
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol.. 2013;13:159.
- Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010;120(7):2423–31.
- Wagner C, Iking-Konert C, Denefleh B, Stegmaier S, Hug F, Hänsch GM. Granzyme B and perforin: constitutive expression in human polymorphonuclear neutrophils. Blood. 2004;103(3):1099.
- Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. 2012;30:459–89.
- Harada Y, Kato Y, Miyaji T, Omote H, Moriyama Y, Hiasa M. Vesicular nucleotide transporter mediates ATP release and migration in neutrophils. J Biol Chem. 2018;293(10):3770–3779. doi: 10.1074/jbc.M117.810168. Epub 2018 Jan 23
- Wagner JG, Roth RA. Neutrophil migration mechanisms, with an emphasis on the pulmonary vasculature. Pharmacol Rev. 2000;52:349–74.
- Kienle K, Lämmermann T. Neutrophil swarming: an essential process of the neutrophil tissue response. Immunol Rev. 2016;273(1):76–93.
- Reátegui E, Jalali F, Khankhel AH, Wong E, Cho H, Lee J, et al. Microscale arrays for the profiling of start and stop signals coordinating human-neutrophil swarming. Nat Biomed Eng. 2017;1:0094.
- Greenberg S, Grinstein S. Phagocytosis and innate immunity. Curr Opin Immunol. 2002;14(1):136–45.
- Segal AW, Dorling J, Coade S. Kinetics of fusion of the cytoplasmic granules with phagocytic vacuoles in human polymorphonuclear leukocytes. Biochemical and morphological studies. J Cell Biol. 1980;85(1):42–59.
- Henry RM, Hoppe AD, Joshi N, Swanson JA. The uniformity of phagosome maturation in macrophages. J Cell Biol. 2004;164(2):185.
- Lee WL, Harrison RE, Grinstein S. Phagocytosis by neutrophils. Microbes Infect.. 2003;5(14):1299–306.
- Silverstein SC, Greenberg S, Di Virgilio F, Steinberg TH. Phagocytosis. In: William PE, editor. Fundamental immunology. New York: Raven Press; 1989. p. 703.
- van Kessel KP, Bestebroer J, van Strijp JA. Neutrophil-mediated phagocytosis of Staphylococcus aureus. Front Immunol. 2014;5:467.
- Ehlers MR. CR3: a general purpose adhesion-recognition receptor essential for innate immunity. Microbes Infect. 2000;2(3):289–94.
- Jankowski A, Scott CC, Grinstein S. Determinants of the phagosomal pH in neutrophils. J Biol Chem. 2002;277(8):6059–66.
- Segal AW, Geisow M, Garcia R, Harper A, Miller R. The respiratory burst of phagocytic cells is associated with a rise in vacuolar pH. Nature. 1981;290(5805):406–9.
- Korkmaz B, Moreau T, Gauthier F. Neutrophil elastase, proteinase 3 and cathepsin G: physicochemical properties, activity and physiopathological functions. Biochimie. 2008;90(2):227–42.
- Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197–223.
- Winterbourn CC, Hampton MB, Livesey JH, Kettle AJ. Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: implications for microbial killing. J Biol Chem. 2006;281(52):39860–9.
- McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem. 1969;244(22):6049–55.
- Klebanoff SJ. Myeloperoxidase. Proc Assoc Am Physicians. 1999;111(5):383–9.
- Levine AP, Segal AW. The NADPH oxidase and microbial killing by neutrophils, with a particular emphasis on the proposed antimicrobial role of myeloperoxidase within the phagocytic vacuole. Microbiol Spect. 2016;4(4): doi:10.1128/microbiolspec.MCHD-0018-2015. DOA 3/2/2017.
- Klebanoff SJ. Iodination of bacteria: a bactericidal mechanism. J Exp Med. 1967;126(6):1063–78.
- Klebanoff SJ, Clark RA. Iodination by human polymorphonuclear leukocytes: a re-evaluation. J Lab Clin Med. 1977;89(3):675–86.
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5.
- Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, et al. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol. 2006;16(4):396–400.
- Urban CF, Reichard U, Brinkmann V, Zychlinsky A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol. 2006;8(4):668–76.
- Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol. 2007;5(8):577–82.
- Francke U, Hsieh CL, Foellmer BE, Lomax KJ, Malech HL, Leto TL. Genes for two autosomal recessive forms of chronic granulomatous disease assigned to 1q25 (NCF2) and 7q11.23 (NCF1). Am J Hum Genet. 1990;47(3):483–92.
- Ganz T, Metcalf JA, Gallin JI, Boxer LA, Lehrer RI. Microbicidal/cytotoxic proteins of neutrophils are deficient in two disorders: Chediak-Higashi syndrome and "specific" granule deficiency. J Clin Invest. 1988;82(2):552–6.
- Lomax KJ, Gallin JI, Rotrosen D, Raphael GD, Kaliner MA, Benz EJ, et al. Selective defect in myeloid cell lactoferrin gene expression in neutrophil specific granule deficiency. J Clin Invest. 1989;83(2):514–19.
- Leiding JW. Neutrophil evolution and their diseases in humans. Front Immunol. 2017;8:1009.
- Damiano VV, Kucich U, Murer E, Laudenslager N, Weinbaum G. Ultrastructural quantitation of peroxidase- and elastase-containing granules in human neutrophils. Am J Pathol. 1988;131(2):235–45.
- Liou TG, Campbell EJ. Quantum proteolysis resulting from release of single granules by human neutrophils: a novel, nonoxidative mechanism of extracellular proteolytic activity. J Immunol. 1996;157(6):2624–31.
- Kessenbrock K, Fröhlich L, Sixt M, Lämmermann T, Pfister H, Bateman A, et al. Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J Clin Invest. 2008;118(7):2438–47.
- McGuinness AJ, Sapey E. Oxidative stress in COPD: sources, markers, and potential mechanisms. J Clin Med. 2017;6(2):21. https://doi.org/10.3390/jcm6020021. DOA 2/2/2017
- Ricciardolo FLM, Caramori G, Ito K, Capelli A, Brun P, Abatangelo G, et al. Nitrosative stress in the bronchial mucosa of severe chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2005;116(5):1028–35.
- Tosi MF, Zakem H, Berger M. Neutrophil elastase cleaves C3bi on opsonised pseudomonas as well as CR1 on neutrophils to create a functionally important opsonin receptor mismatch. J Clin INvest. 1990;86:300–8.
- Smallman LA, Hill SL, Stockley RA. Reduction of ciliary beat frequency in vitro by sputum from patients with bronchiectasis: a serine proteinase effect. Thorax. 1984;39:663–7.
- Hall CHT, Campbell EL, Colgan SP. Neutrophils as components of mucosal homeostasis. Cell Mol Gastroenterol Hepatol.. 2017;4(3):329–37.
- Slater TW, Finkielsztein A, Mascarenhas LA, Mehl LC, Butin-Israeli V, Sumagin R. Neutrophil microparticles deliver active myeloperoxidase to injured mucosa to inhibit epithelial wound healing. J Immunol. 2017;198(7):2886–97.
- Parmar JS, Mahadeva R, Reed BJ, Farahi N, Cadwallader KA, Keogan MT, et al. Polymers of α1-antitrypsin are chemotactic for human neutrophils. Am J Respir Cell Mol Biol. 2002;26(6):723–30.
- Nakajoh M, Fukushima T, Suzuki K, Yamaya M, Nakayama K, Sekizawa K, et al. Retinoic acid inhibits elastase-induced injury in human lung epithelial cell lines. Am J Respir Cell Mol Biol. 2002;28(3):296–304.
- Hubbard RC, Fells G, Gadek J, Pacholok S, Humes J, Crystal RG. Neutrophil accumulation in the lung in alpha 1-antitrypsin deficiency. Spontaneous release of leukotriene B4 by alveolar macrophages. J Clin Invest. 1991;88(3):891–7.
- Sapey E, Stockley RA. Red, amber and green: the role of the lung in de-priming active systemic neutrophils. Thorax. 2014;69(7):606–8.
- Rossaint J, Kühne K, Skupski J, Van Aken H, Looney MR, Hidalgo A, et al. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat Commun. 2016;7:13464.
- Sengelov H, Kjeldsen L, Borregaard N. Control of exocytosis in early neutrophil activation. J Immunol. 1993;150(4):1535–43.
- Yost CC, Denis MM, Lindemann S, Rubner FJ, Marathe GK, Buerke M, et al. Activated polymorphonuclear leukocytes rapidly synthesize retinoic acid receptor-alpha: a mechanism for translational control of transcriptional events. J Exp Med. 2004;200(5):671.
- Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 2005;201(1):105–15.
- Buckley CD, Ross EA, McGettrick HM, Osborne CE, Haworth O, Schmutz C, et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J Leukoc Biol. 2006;79(2):303–11.
- Sagiv Jitka Y, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015;10(4):562–73.
- Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers JW, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest. 2012;122(1):327–36.
- Yang K-Y, Arcaroli JJ, Abraham E. Early alterations in neutrophil activation are associated with outcome in acute lung injury. Am J Respir Crit Care Med. 2003;167(11):1567–74.
- Bekes EM, Schweighofer B, Kupriyanova TA, Zajac E, Ardi VC, Quigley JP, et al. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am J Pathol. 2011;179(1525–2191 (Electronic)):1455–70.
- Christoffersson G, Vågesjö E, Vandooren J, Lidén M, Massena S, Reinert RB, et al. VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood. 2012;120(23):4653.
- Massena S, Christoffersson G, Vågesjö E, Seignez C, Gustafsson K, Binet F, et al. Identification and characterization of VEGF-A-responsive neutrophils expressing CD49d, VEGFR1, and CXCR4 in mice and humans. Blood. 2015;126(17):2016.
- Blázquez-Prieto J, López-Alonso I, Amado-Rodríguez L, et al. Impaired lung repair during neutropenia can be reverted by matrix metalloproteinase-9. Thorax. 2018;73:321–330.
- Brightling CE, Monteiro W, Ward R, Parker D, Morgan MDL, Wardlaw AJ, et al. Sputum eosinophilia and short-term response to prednisolone in chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 2000;356(9240):1480–5.
- Thompson AB, Daughton D, Robbins RA, Ghafouri MA, Oehlerking M, Rennard SI. Intraluminal airway inflammation in chronic bronchitis. Characterization and correlation with clinical parameters. Am Rev Respir Dis. 1989;140(6):1527–37.
- Pilette C, Colinet B, Kiss R, Andre S, Kaltner H, Gabius HJ, et al. Increased galectin-3 expression and intra-epithelial neutrophils in small airways in severe COPD. Eur Respir J. 2007;29(5):914–22.
- Donaldson GC, Seemungal TA, Patel IS, Bhowmik A, Wilkinson TM, Hurst JR, et al. Airway and systemic inflammation and decline in lung function in patients with COPD. Chest. 2005;128(4):1995–2004.
- Parr DG, White AJ, Bayley DL, Guest PJ, Stockley RA. Inflammation in sputum relates to progression of disease in subjects with COPD: a prospective descriptive study. Respir Res. 2006;7:136.
- Di Stefano A, Capelli A, Lusuardi M, Balbo P, Vecchio C, Maestrelli P, et al. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med. 1998;158(4):1277–85.
- O'Shaughnessy TC, Ansari TW, Barnes NC, Jeffery PK. Inflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+ T lymphocytes with FEV1. Am J Respir Crit Care Med. 1997;155(3):852–7.
- Stoller JK, Aboussouan LS. A review of α1-antitrypsin deficiency. Am J Respir Crit Care Med. 2012;185(3):246–59.
- Balbi B, Ferrarotti I, Miravitlles M. Efficacy of augmentation therapy for emphysema associated with alpha1-antitrypsin deficiency: enough is enough. Eur Respir J. 2016;47(1):35–8.
- Stockley RA, Miravitlles M, Vogelmeier C. Augmentation therapy for alpha-1 antitrypsin deficiency: towards a personalised approach. Orphanet J Rare Dis. 2013;8:149.
- Shamamian P, Schwartz Jess D, Pocock Ben JZ, Monea S, Whiting D, Marcus Stuart G, et al. Activation of progelatinase A (MMP‐2) by neutrophil elastase, cathepsin G, and proteinase‐3: A role for inflammatory cells in tumor invasion and angiogenesis.J Cell Physiol Biochem. 2001;189(2):197–206.
- Demedts IK, Morel-Montero A, Lebecque S, Pacheco Y, Cataldo D, Joos GF, et al. Elevated MMP-12 protein levels in induced sputum from patients with COPD. Thorax. 2006;61(3):196.
- Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. 2010;363(23):2233–47.
- Leopold PL, O'Mahony MJ, Lian XJ, Tilley AE, Harvey BG, Crystal RG. Smoking is associated with shortened airway cilia. PLoS one. 2009;4(12):e8157.
- Tamashiro E, Xiong G, Anselmo-Lima WT, Kreindler JL, Palmer JN, Cohen NA. Cigarette smoke exposure impairs respiratory epithelial ciliogenesis. Am J Rhinol Allergy. 2009;23(2):117–22.
- Gill N, Ferreira RBR, Antunes LCM, Willing BP, Sekirov I, Al-Zahrani F, et al. Neutrophil elastase alters the murine gut microbiota resulting in enhanced Salmonella colonization. PLoS ONE. 2012;7(11):e49646.
- Margaroli C, Tirouvanziam R. Neutrophil plasticity enables the development of pathological microenvironments: implications for cystic fibrosis airway disease.Mol Cell Pediatr. 2016;3:38.
- Dicker AJ, Crichton ML, Pumphrey EG, Cassidy AJ, Suarez-Cuartin G, Sibila O, et al. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol Pract. 2018;141(1):117–27.
- Carter RI, Ungurs MJ, Mumford RA, Stockley RA. Aα-Val360: a marker of neutrophil elastase and COPD disease activity. Eur Respir J. 2012;41(1):31.
- Keatings VM, Barnes PJ. Granulocyte activation markers in induced sputum: comparison between chronic obstructive pulmonary disease, asthma, and normal subjects. Am J Respir Crit Care Med. 1997;155(2):449–53.
- Subramanian DR, Jenkins L, Edgar R, Quraishi N, Stockley RA, Parr DG. Assessment of pulmonary neutrophilic inflammation in emphysema by quantitative positron emission tomography. Am J Respir Crit Care Med. 2012;186(11):1125–32.
- Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27.
- Abboud RT, Vimalanathan S. Pathogenesis of COPD. Part I. The role of protease-antiprotease imbalance in emphysema. Int J Tuberc Lung Dis. 2008;12(4):361–7.
- McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011;365(17):1567–75.
- Berger P, Laurent F, Begueret H, Perot V, Rouiller R, Raherison C, et al. Structure and function of small airways in smokers: relationship between air trapping at CT and airway inflammation. Radiology. 2003;228(1):85–94.
- Sapey E, Stockley JA, Greenwood H, Ahmad A, Bayley D, Lord JM, et al. Behavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(9):1176–86.
- Burnett D, Chamba A, Hill SL, Stockley RA. Neutrophils from subjects with chronic obstructive lung disease show enhanced chemotaxis and extracellular proteolysis. Lancet. 1987;2(8567):1043–6.
- Prieto A, Reyes E, Bernstein ED, Martinez B, Monserrat J, Izquierdo JL, et al. Defective natural killer and phagocytic activities in chronic obstructive pulmonary disease are restored by glycophosphopeptical (inmunoferón) Am J Respir Crit Care Med. 2001;163(7):1578–83.
- Shanmugam L, Ravinder SS, Johnson P, Padmavathi R, Rajagopalan B, Kindo AJ. Assessment of phagocytic activity of neutrophils in chronic obstructive pulmonary disease. Lung India. 2015;32(5):437–40.
- Venge P, Rak S, Steinholtz L, Hakansson L, Lindblad G. Neutrophil function in chronic bronchitis. Eur Respir J. 1991;4(5):536–43.
- Muns G, Rubinstein I, Bergmann KC. Phagocytosis and oxidative burst of blood phagocytes in chronic obstructive airway disease. Scand J Infect Dis. 1995;27(4):369–73.
- Noguera A, Batle S, Miralles C, Iglesias J, Busquets X, MacNee W, et al. Enhanced neutrophil response in chronic obstructive pulmonary disease. Thorax. 2001;56(6):432–7.
- Rahman I. The role of oxidative stress in the pathogenesis of COPD: implications for therapy.Treatment Respir Med. 2005;4(3):175–200.
- Grabcanovic-Musija F, Obermayer A, Stoiber W, Krautgartner WD, Steinbacher P, Winterberg N, et al. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir Res. 2015;16:59.
- Obermayer A, Stoiber W, Krautgartner WD, Klappacher M, Kofler B, Steinbacher P, et al. New aspects on the structure of neutrophil extracellular traps from chronic obstructive pulmonary disease and in vitro generation. PLoS one. 2014;9(5):e97784.
- Adib-Conquy M, Pedron T, Petit-Bertron A-F, Tabary O, Corvol H, Jacquot J, et al. Neutrophils in cystic fibrosis display a distinct gene expression pattern. Molec Med. 2008;14(1–2):36–44.
- Loi ALT, Hoonhorst S, van Aalst C, Langereis J, Kamp V, Sluis-Eising S, et al. Proteomic profiling of peripheral blood neutrophils identifies two inflammatory phenotypes in stable COPD patients. Respir Res. 2017;18(1):100.
- Neofytou E, Tzortzaki EG, Chatziantoniou A, Siafakas NM. DNA damage due to oxidative stress in chronic obstructive pulmonary disease (COPD). Int J Mol Sci. 2012;13(12):16853–64.
- Kato T, Suzuki K, Okada S, Kamiyama H, Maeda T, Saito M, et al. Aberrant methylation of PSD disturbs Rac1-mediated immune responses governing neutrophil chemotaxis and apoptosis in ulcerative colitis-associated carcinogenesis. Int J Oncol. 2012;40(4):942–50.
- Coit P, Yalavarthi S, Ognenovski M, Zhao W, Hasni S, Wren JD, et al. Epigenome profiling reveals significant DNA demethylation of interferon signature genes in lupus neutrophils.J Autoimmun. 2015;58:59–66.
- Gallagher KA, Joshi A, Carson WF, Schaller M, Allen R, Mukerjee S, et al. Epigenetic changes in bone marrow progenitor cells influence the inflammatory phenotype and alter wound healing in type 2 diabetes. Diabetes. 2015;64(4):1420–30.
- Hurst JR, Bafadhel M, Bolton CE, Quint JK, Sapey E, Wilkinson TMA. COPD exacerbations: transforming outcomes through research. Lancet Respir Med. 2018;6(3):172–4.
- Qiu Y, Zhu J, Bandi V, Atmar RL, Hattotuwa K, Guntupalli KK, et al. Biopsy neutrophilia, neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;168(8):968–75.
- Pouwels SD, van Geffen WH, Jonker MR, Kerstjens HA, Nawijn MC, Heijink IH. Increased neutrophil expression of pattern recognition receptors during COPD exacerbations. Respirology. 2017;22(2):401–4.
- Thulborn SJ, Akram N, Mistry V, Brightling CE, Moffitt K, Ribeiro D, et al. S45 Evaluating the sensitivity and specificity of active neutrophil elastase as a biomarker for bacterial infection in subjects with copd. Thorax. 2016;71(Suppl 3):A28.
- Vaitkus M, Lavinskiene S, Bieksiene K, Jeroch J, Sakalauskas R. Analysis of reactive oxygen species in sputum neutrophils during acute exacerbation of COPD. Eur Respir J. 2012;40(Suppl 56):P793.
- Barnes PJ. Alveolar macrophages as orchestrators of COPD. COPD. 2004;1(1):59–70.
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–53.
- Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, Shamji B, et al. Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;184(7):796–802.
- Brightling CE, Monteiro W, Ward R, Parker D, Morgan MD, Wardlaw AJ, et al. Sputum eosinophilia and short-term response to prednisolone in chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 2000;356(9240):1480–5.
- Maclay JD, McAllister DA, Johnston S, Raftis J, McGuinnes C, Deans A, et al. Increased platelet activation in patients with stable and acute exacerbation of COPD. Thorax. 2011;66(9):769.
- Taylor AE, Finney-Hayward TK, Quint JK, Thomas CMR, Tudhope SJ, Wedzicha JA, et al. Defective macrophage phagocytosis of bacteria in COPD. Eur Respir J. 2010;35(5):1039.
- Freeman CM, Martinez FJ, Han MK, Washko GR, McCubbrey AL, Chensue SW, et al. Lung CD8+ T cells in COPD have increased expression of bacterial TLRs. Respir Res. 2013;14(1):13.
- Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol. 2003;81(4):289–96.
- Togo S, Holz O, Liu X, Sugiura H, Kamio K, Wang X, et al. Lung fibroblast repair functions in patients with chronic obstructive pulmonary disease are altered by multiple mechanisms. Am J Respir Crit Care Med. 2008;178(3):248–60.
- Polverino F, Celli BR, Owen CA. COPD as an endothelial disorder: endothelial injury linking lesions in the lungs and other organs? (2017 Grover Conference Series). Pulm Circ. 2018;8(1):2045894018758528.
- Caramori G, Romagnoli M, Casolari P, Bellettato C, Casoni G, Boschetto P, et al. Nuclear localisation of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbations. Thorax. 2003;58(4):348–51.
- Calverley PA, Anderson JA, Celli BC, Ferguson GT, Jenkins C, Jones PW, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. NEJM. 2007;356:775–89.
- Rabe KF. Update on roflumilast, a phosphodiesterase 4 inhibitor for the treatment of chronic obstructive pulmonary disease. Br J Pharmacol. 2011;163(1):53–67.
- Martinez FJ, Rabe KF, Sethi S, Pizzichini E, McIvor A, Anzueto A, et al. Effect of roflumilast and inhaled corticosteroid/long-acting β2-agonist on chronic obstructive pulmonary disease exacerbations (RE2SPOND). A randomized clinical trial. Am J Respir Crit Care Med. 2016;194(5):559–67.
- Wedzicha JA, Calverley PMA, Rabe KF. Roflumilast: a review of its use in the treatment of COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:81–90.
- Gompertz S, Stockley RA. A randomised controlled trial of a Leukotriene synthesis inhibitor in patients with COPD. Chest. 2002;112:289–94.
- Rennard SI, Dale DC, Donohue JF, Kanniess F, Magnussen H, Sutherland ER, et al. CXCR2 antagonist MK-7123. A phase 2 proof-of-concept trial for chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;191(9):1001–11.
- Bewley MA, Belchamber KB, Chana KK, Budd RC, Donaldson G, Wedzicha JA, et al. Differential effects of p38, MAPK, PI3K or Rho kinase inhibitors on bacterial phagocytosis and efferocytosis by macrophages in COPD. PLoS One. 2016;11(9):e0163139.
- Kim D, Haynes CL. The role of p38 MAPK in neutrophil functions: single cell chemotaxis and surface marker expression. Analyst. Eur Respir J. 2008 Jan;31(1):62–9.
- Renda T, Baraldo S, Pelaia G, Bazzan E, Turato G, Papi A, et al. Increased activation of p38 MAPK in COPD. Eur Respir J. 2008;31(1):62.
- Fisk M, Cheriyan J, Mohan D, Forman J, Mäki-Petäjä KM, McEniery CM, et al. The p38 mitogen activated protein kinase inhibitor losmapimod in chronic obstructive pulmonary disease patients with systemic inflammation, stratified by fibrinogen: a randomised double-blind placebo-controlled trial. PLOS ONE. 2018;13(3):e0194197.
- Watz H, Barnacle H, Hartley BF, Chan R. Efficacy and safety of the p38 MAPK inhibitor losmapimod for patients with chronic obstructive pulmonary disease: a randomised, double-blind, placebo-controlled trial Watz, Henrik et al. The Lancet Respiratory Medicine, Volume 2, Issue 1, 63–72.
- Lomas David A, Lipson David A, Miller Bruce E, Willits L, Keene O, Barnacle H, et al. An oral inhibitor of p38 MAP kinase reduces plasma fibrinogen in patients with chronic obstructive pulmonary disease. J Clin Pharmacol Toxicol. 2013;52(3):416–24.
- Dahl R, Titlestad I, Lindqvist A, Wielders P, Wray H, Wang M, et al. Effects of an oral MMP-9 and -12 inhibitor, AZD1236, on biomarkers in moderate/severe COPD: A randomised controlled trial. Pulm. Pharmacol. Ther. 2012;25(2):169–77.
- Sapey E, Greenwood H, Walton G, Mann E, Love A, Aaronson N, et al. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood. 2014;123(2):239–48.
- Sriskantharajah S, Hamblin N, Worsley S, Calver Andrew R, Hessel Edith M, Amour A. Targeting phosphoinositide 3‐kinase δ for the treatment of respiratory diseases. Ann N Y Acad Sci. 2013;1280(1):35–9.
- Sapey E, Patel JM, Greenwood HL, Walton GM, Hazeldine J, Sadhra C, et al. Pulmonary infections in the elderly lead to impaired neutrophil targeting, which is improved by simvastatin. Am J Respir Crit Care Med. 2017;196(10):1325–36.
- Stockley JA, Walton GM, Lord JM, Sapey E. Aberrant neutrophil functions in stable chronic obstructive pulmonary disease: the neutrophil as an immunotherapeutic target. Int Immunopharmacol. 2013;17(4):1211–17.
- Park DW, Jiang S, Tadie J-M, Stigler WS, Gao Y, Deshane J, et al. Activation of AMPK enhances neutrophil chemotaxis and bacterial killing. Molec Med. 2013;19(1):387–98.
- Bishwakarma R, Zhang W, Lin Y-L, Kuo Y-F, Cardenas VJ, Sharma G. Metformin use and health care utilization in patients with coexisting chronic obstructive pulmonary disease and diabetes mellitus. Int J Chron Obstruct Pulmon Dis. 2018;13:793–800.
- GOLD. The global strategy for the diagnosis, management and prevention of COPD, global initiative for chronic obstructive lung disease (GOLD) 2017. Available from: http://goldcopdorg. 2017;DOA 11.11.2017.
- Nordenfelt P, Tapper H. Phagosome dynamics during phagocytosis by neutrophils. J Leukoc Biol. 2011;90(2):271–84.
- Uriarte SM, Powell DW, Luerman GC, Merchant ML, Cummins TD, Jog NR, et al. Comparison of proteins expressed on secretory vesicle membranes and plasma membranes of human neutrophils. J Immunol. 2008;180(8):5575.
- Borregaard N, Cowland JB. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood. 1997;89:350321.