
Abstract
Mitogen-activated protein kinase p38 is a key regulator in the inflammation pathway and is activated in the lungs of chronic obstructive pulmonary disease (COPD) patients. Acumapimod is a potent, selective, oral, p38 inhibitor under investigation for treatment of acute exacerbations of COPD (AECOPD). In this Phase II, double-blind, randomized, placebo-controlled dose-exploration study of acumapimod in patients with moderate or severe AECOPD (NCT01332097), patients presenting with AECOPD were randomized to receive single-dose acumapimod (20 mg or 75 mg) on Day 1, repeated single-dose acumapimod (20 mg or 75 mg) on Days 1 and 6, oral prednisone 40 mg (10 days), or placebo. Primary outcome: improvement in forced expiratory volume in 1 s (FEV1) versus placebo at Day 5 (single doses) and Day 10 (repeated doses). N = 183 patients were randomized; 169 (92%) patients completed the study. Although the primary endpoint (FEV1 at Day 10) was not met (p = 0.082), there was a significant improvement in FEV1 with acumapimod repeat-dose 75 mg versus placebo at Day 8 (p = 0.022) which, though not a prespecified endpoint, was part of an overall trend. Differences at lower doses did not achieve significance. Mean change in FEV1 AUC from baseline to Day 14 in the 75 mg repeat-dose group was significantly higher versus placebo (p = 0.02), prednisone (p = 0.01), and 20 mg single-dose groups (p = 0.015) (post-hoc analysis). EXACT-PRO showed numerical differences versus placebo that did not reach significance. Acumapimod was well tolerated. In conclusion, repeated single-dose acumapimod showed a clinically relevant improvement in FEV1 over placebo at Day 8, along with consistent numerical differences in EXACT-PRO. These data can be used to determine dose regimens for a proof-of-clinical-concept trial.
Introduction
Chronic obstructive pulmonary disease (COPD) is currently the third leading cause of death worldwide, killing over three million people globally in 2016, and this is expected to rise [Citation1,Citation2]. The economic burden of COPD is high [Citation1], driven by substantial hospital costs [Citation3] with acute exacerbations of COPD (AECOPD) accounting for the greatest proportion of the COPD burden on healthcare systems [Citation1].
AECOPD are acute events characterized by deteriorating respiratory symptoms beyond the level of daily changes [Citation1], associated with progressive decline in lung function, increased mortality risk [Citation4] and poor quality of life [Citation5]. Exacerbation frequency rises with increasing COPD severity, where approximately 30% and 50% of patients with Global Initiative for Chronic Obstructive Lung Disease (GOLD) Stages 3 and 4, respectively, have ≥2 exacerbations annually [Citation6]. AECOPD are often triggered by viral or bacterial respiratory infections, or environmental pollutants [Citation1] leading to increased airway inflammation [Citation1].
AECOPD management, which aims to minimize the impact of the current exacerbation, includes antibiotics, bronchodilators and systemic corticosteroids [Citation7]. Although corticosteroid resistance is common [Citation8] and associated with poor outcomes, some current treatments can produce symptomatic improvement and may reduce duration of hospital stays [Citation1]. Nevertheless, there remains a need to improve recovery times, reduce treatment failures and mitigate the permanent worsening of lung function.
Mediators released during AECOPD activate several intracellular pathways, including the mitogen-activated protein kinase (MAPK) pathway whereby leukocytes are recruited to the site of inflammation. Here, they are stimulated by pro-inflammatory cytokines which subsequently activate the MAPK isoform, p38 [Citation9]. The p38 MAPK signaling pathway promotes inflammation by enhancing inflammatory gene transcription, stabilizing mRNAs and increasing protein translation [Citation10]. As increased lung p38 MAPK phosphorylation has been observed in COPD patients [Citation11,Citation12], it is an interesting target for treating AECOPD. Synthetic p38 MAPK kinase inhibitors have inhibited the inflammatory reaction in animal models of COPD [Citation13,Citation14]. Further p38 MAPK inhibition can attenuate pro-inflammatory cytokine production, such as tumor necrosis factor alpha (TNFα) [Citation15], in different lung cells from patients with COPD including alveolar macrophages [Citation16], lymphocytes and bronchial epithelial cells [Citation11]. Inhibition of p38 may reverse steroid insensitivity in vitro [Citation17]. Laboratory studies implicate p38 MAPK activation in COPD inflammation. This pathway is not covered by corticosteroids, thereby indicating a potential benefit of MAPK inhibitors in COPD treatment [Citation11]. Also, studies in patients with severe asthma and poor corticosteroid response have shown that corticosteroid insensitivity can be overcome or “reversed” by the use of p38 MAPK inhibitors [Citation18]. Such observations support the contribution of p38 MAPK activation to COPD inflammation and have encouraged p38 MAPK inhibitor development.
Acumapimod is an orally active, highly selective alpha/beta p38 MAPK inhibitor under investigation for the treatment of AECOPD [Citation19]. In animal COPD models, including those with steroid resistance, acumapimod potently decreases inflammatory responses to intratracheal lipopolysaccharides [Citation20]. An in vivo endotoxin challenge in healthy volunteers induced nearly complete suppression of circulating TNFα [Citation21]. We report the results of the first trial of acumapimod in patients with AECOPD. The aim was to explore the efficacy and safety through use of single doses, and repeated single doses, to support decision-making and define dose regimens for future clinical efficacy studies.
Materials and methods
Study design and patients
This was an exploratory, double-blind, randomized, placebo-controlled, multicenter, adaptive parallel-group trial carried out between March 2011 and May 2013 in eight centers across Russia, Bulgaria and Romania (ClinicalTrials.gov: NCT01332097) (Supplementary Table S1). The study was conducted in accordance with the Declaration of Helsinki [Citation22] and was approved by the Independent Ethics Committee or Institutional Review Board for each study center. Patients provided written informed consent before randomization. All mandatory laboratory health and safety procedures have been complied with in the course of conducting experimental work for this study.
Adult patients (aged 40–80 years, with a smoking history of ≥10 pack-years) were enrolled if they had a diagnosis of COPD (Stage II to IV according to GOLD [Citation1]) and presented with an exacerbation within the preceding 24 h.
Patients were excluded if they had: used any oral or intravenous corticosteroids (i.e. those given for an exacerbation and not conventional inhaled corticosteroids) within ≤30 days of randomization (re-exacerbation ≤30 days is usually considered to indicate treatment failure [Citation23] rather than recurrent exacerbation, however, the aim of the study was not to investigate treatment failures); arterial blood pH <7.26 at randomization; a history or presence of clinically significant uncontrolled left ventricular failure; clinical or radiologic evidence of pneumonia; elevated liver enzymes (1.5× the upper limit of normal) at study entry; history of liver toxicity with other drugs. Macrolide antibiotics were excluded due to potential drug interaction with acumapimod. Antibiotics within 24 h of randomization were not preferred but were not a formal exclusion.
Treatment
Treatment overview and rationale
This was an exploratory study with an adaptive design utilizing interim analysis to guide progression. The first cohort of the study used the maximum acumapimod single dose, 75 mg (as supported by pre-clinical toxicity testing, clinical safety and pharmacokinetic/pharmacodynamic data) to compare with a placebo and prednisolone benchmark to determine whether acumapimod showed sign of an effect, and with planned interim analyses to guide study progression decisions, as well as safety and tolerability in the target population. Two interim analyses were performed on this first cohort. After the second interim analysis, when the effect on FEV1 at Day 3 was established, Cohort 2 was initiated to determine if the lower acumapimod single dose (20 mg) showed an effect. Cohorts 3 and 4 were added when interim analysis three showed an offset of effect on patient FEV1 and the possibility that the duration of the exacerbation was not covered by the single dose. As re-dosing at the drug levels used in Cohorts 1 and 2 had not previously been performed, Cohort 3, with 20 mg acumapimod repeated at Day 6, was initially performed for safety evaluation; this was followed by Cohort 4 with 75 mg acumapimod repeated at Day 6.
Treatment cohorts
As mentioned above, the study recruited four sequential cohorts in which patients were randomly assigned to acumapimod treatment, with a placebo group included for each cohort. A placebo group for prednisone was also included in Cohort 1 where prednisone was a comparator. Single-dose cohorts were completed before repeated single-dose cohorts for safety monitoring. Patients in Cohort 1 were assigned 1:1:1 to acumapimod 75 mg single-dose (Day 1), oral prednisone 40 mg (10 days) or placebo. Patients in Cohort 2 were assigned 5:1 to acumapimod 20 mg single-dose (Day 1) or placebo. Cohorts 3 and 4 patients were randomly assigned (5:1) to acumapimod 20 mg and 75 mg repeated dose, respectively, on Days 1 and 6, or placebo (, Supplementary Figure S1). Placebo was provided in visually identical capsules to the active therapies.
Figure 1. Patient disposition and flow.
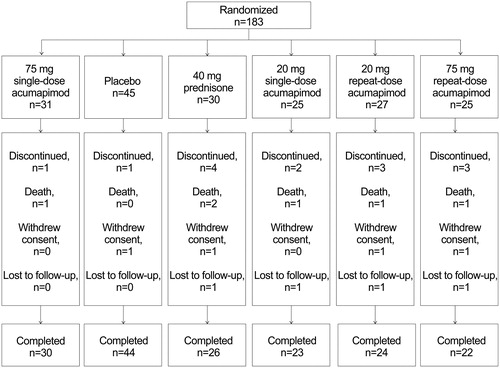
All patients received doxycycline or a non-macrolide antibiotic (according to local guidelines) for ≤10 days (Days 1–10 inclusive) and supportive therapy as required, including nebulized bronchodilators, supplementary oxygen, inhaled long-acting bronchodilators and inhaled steroids (excluding nebulized budesonide). The use of oral corticosteroid and nebulized budesonide (which may have similar efficacy to oral corticosteroids) was excluded, apart from the cohort randomized to prednisone as a benchmark arm [Citation24,Citation25]. Any requirement for systemic corticosteroid was documented as treatment failure. At Day 1, all eligible patients were randomized via the Interactive Response Technology provider to one of the treatment arms. Patients, investigator staff and all personnel performing the assessments remained blinded to the identity of the treatment from the time of randomization until database lock.
Endpoints and assessments
The primary endpoint was improvement in FEV1 for acumapimod versus pooled placebo data at Day 5 (single dose) or Day 10 (repeated doses). Secondary endpoints included: patient-reported outcomes; patients responding at Day 30 (defined as no need to treat with oral steroids, no re-admission to hospital for COPD-related symptoms [study admissions were by treating physician’s decision and not protocolized], no change in antibiotic therapy relating to COPD, nor any need for treatment for a further exacerbation in the attending physician’s opinion); length of hospital stay; and safety and tolerability (6-month follow-up). Biomarkers of inflammation, including high sensitivity C-reactive protein (hsCRP) and fibrinogen levels, were examined in an exploratory analysis (Supplementary Table S3).
Following a baseline assessment, all patients were assessed throughout their AECOPD to Day 14, with long-term follow-up to Day 30, Day 90, and 6 months. Clinical efficacy assessments were made on Day 1 (pre-dose), every day through to Day 10, then on Days 14, 30, 90, and at end of study (6 months) in all treatment groups. Adverse events (AEs) were assessed across the study. Baseline spirometry was performed in all patients before dosing, and subsequent spirometries were performed at Days 5, 10, 30 and 90, and 6 months after dosing. All measurements complied with the 2005 American Thoracic Society/European Respiratory Standards (ATS/ERS) Task Force recommendations [Citation26].
Patient-reported outcomes were assessed using the Exacerbations of Chronic Pulmonary Disease Tool (EXACT-PRO), a 14-point questionnaire designed to capture the frequency, severity, and duration of acute exacerbation symptoms in patients with COPD [Citation27]. Patients completed the e-Diary at the end of every day.
Statistical analyses
The primary analysis compared FEV1 change from baseline against placebo (pooled placebo group data). FEV1 values were analyzed in a model that included effects for baseline (Day 1, pre-dose FEV1 value), treatment (placebo, prednisone, acumapimod 75 mg and 20 mg [single dose], and acumapimod 20 mg and 75 mg [repeated dose]), time (class variable: Days 3, 5, 8, 10, 14 and 30), treatment-by-time interaction and baseline-by-time interaction. An unstructured covariance matrix was used to model the correlations of FEV1 values measured on the same patient.
An initial interim analysis was planned after 45 patients had completed 5 days in the study, with additional patients recruited if there was no statistically significant difference in FEV1 between treatment and placebo. A second interim analysis was planned after 91 patients had completed 5 days in the study with positive results leading to further patient recruitment.
As an exploratory study, only the first cohort (1:1:1 of acumapimod, placebo and prednisone) was formally powered and statistically analyzed. It was initially assumed that the FEV1 treatment effect for acumapimod versus placebo was 125 mL with a SD of 300 mL [Citation28]. Based on results from the first interim analysis, these values were updated to an 89% power to detect a treatment difference of 100 mL in FEV1 (SD 230 mL) between acumapimod and placebo. Subsequent cohorts were not formally powered, as the purpose of these was to assess effect size for future study planning. Each cohort was run with a placebo, and the placebo results from each part were combined.
A post-hoc analysis evaluated mean change in FEV1 area under the curve (AUC) from baseline to Day 14. The AUC calculation was based on using the scheduled day values for each patient. The least-squares means were derived from a mixed-effects model that included all treatment groups and which pooled placebo patients. The model included effects for all treatment groups.
For the EXACT-PRO score, the standardized AUC for Days 1–14 was derived and analyzed using analysis of covariance with effects for treatment and baseline (covariate). In addition, the EXACT-PRO 3-day rolling average was computed for Days 1–29, with improvement from Day 1 analyzed using the same model as described above for FEV1. The 3-day EXACT PRO rolling average standardized AUCs were also computed for Days 1–10, 1–14 and 1–29. These were analyzed in an analysis of covariance model with baseline as a continuous covariate and treatment group as a factor. Length of hospital stay following initial admission was assessed using Kaplan–Meier plots. Safety data were analyzed for all patients who received at least one dose of study drug. No corrections have been made for statistical multiplicity.
Results
Patient characteristics
Forty-five patients were initially recruited, with a further 45 patients recruited following failure to show a statistically significant difference in FEV1 between treatment and placebo after completion of 5 days of treatment. Positive results after the second interim analysis resulted in the additional recruitment of 92 patients. In total, 183 patients were randomized to treatment and 169 patients (92%) completed the study; four withdrew consent, four were lost to follow-up and six died (unrelated to study drug) (). While there were protocol deviations in the study, none were major deviations that led to a subject’s data being excluded from the analysis. As such, all 183 patients were included in the efficacy and safety analyses. Baseline characteristics were similar among treatment groups (). Patients had a mean age of 62 years (range: 41–77 years) and mean body mass index of 26.5 kg/m2 (range: 15.9–46.9 kg/m2). All were Caucasian and 80% were male.
Table 1. Patient demographics and baseline characteristics (all patients).
Table 2. Treatment effects on FEV1 (all patients).
Lung function
Numerical differences from baseline were observed for single-dose acumapimod, 20 mg and 75 mg on Day 5, and for the acumapimod 20 mg repeated dose on Day 10, but these did not reach statistical significance versus placebo (primary endpoint). The difference between acumapimod 75 mg repeated dose and placebo did not reach significance at Day 10 and thus the primary endpoint was not met (Day 10: 124 mL; 95% confidence interval (CI): −16, 263; p = 0.082) (). However, change from baseline in FEV1 did appear to show a dose effect of acumapimod, with a clinically relevant and statistically significant improvement in FEV1 over baseline was observed for acumapimod 75 mg repeat-dosing versus placebo at Day 8, although this was not a primary endpoint (difference vs. placebo: 152 mL; 95% CI: 21, 283; p = 0.022; ). The prednisone group showed no improvement in FEV1 from baseline on any of the study days.
Figure 2. Mean (95% CI) change from baseline in forced expiratory volume in 1 s (FEV1) with acumapimod versus standard of care and placebo. *p < 0.001 versus placebo.
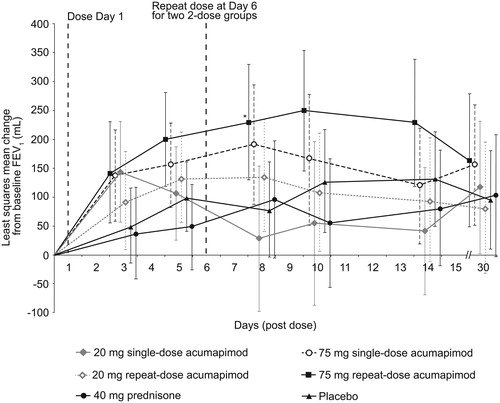
In a post-hoc analysis, the mean change in FEV1 AUC from baseline to Day 14 in the acumapimod 75 mg repeat-dose group was consistent with a dose-dependent effect over placebo (1.46 L, p = 0.02), prednisone (1.75 L, p = 0.01) and 20 mg single-dose acumapimod (1.72 L, p = 0.015) groups.
Patient-reported outcomes
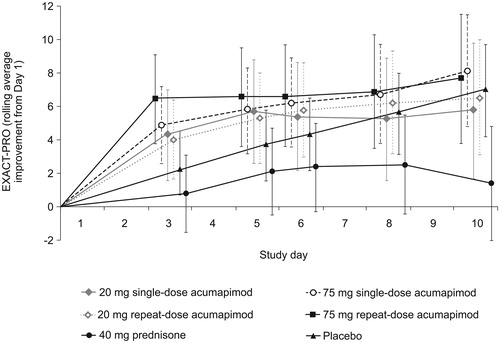
Patients receiving acumapimod experienced rapid improvements on the rolling average improvement score (RAIS) on EXACT-PRO (). In the early part of an exacerbation and on Day 3, the RAIS was higher for the acumapimod 75 mg single- and repeat-dose groups than for placebo (4.90 and 6.46 vs. 2.23, respectively), showing a similar pattern of response to the FEV1. The lowest improvement score was observed for prednisone on all days. However, overall, there was no statistically significant difference between the acumapimod and placebo groups in the RAIS on EXACT-PRO (). Moreover, there were no differences between treatment groups for rolling average for Days 1–10, Days 1–14 or Days 1–29.
Figure 3. Mean (95% CI) change in patient-reported outcomes (EXACT-PRO) from Day 1 to Day 10 (rolling average, increase is improvement).
Length of hospital stay
Across treatment groups, 28%–48% of patients were hospitalized for their initial exacerbation. Length of stay was variable and there were no statistically significant differences between the treatment groups (Supplementary Table S2).
Patient response to treatment at Day 30
There was no statistical difference in the exploratory endpoint of treatment response rates in acumapimod versus placebo or prednisone (87.5%–100% across all groups at Day 30).
Biomarkers
Reductions in hsCRP were numerically greater in acumapimod versus placebo groups from baseline to Day 5 (placebo [28%]; 20 mg acumapimod single-dose [51%]; 20 mg acumapimod repeat-dose [59%]; 75 mg acumapimod single-dose [61%]; 75 mg acumapimod repeat-dose [72%]; prednisone [74%]). No consistent trends in fibrinogen levels were observed for any of the treatment groups (Supplementary Table S3).
Safety and tolerability
Overall, 54% of patients experienced at least one AE during the study, most of which were mild or moderate in severity (). The most common AE was COPD exacerbation, with the highest incidence reported in the placebo group (27%) followed by the 75 mg repeat-dose acumapimod group (24%). Fecal occult blood and nasopharyngitis were also commonly reported; with no dose relationship to acumapimod.
Table 3. Summary of adverse events (all patients).
There were six deaths during the study (): one caused by COPD, myocardial infarction, cardiopulmonary failure, decompensated chronic respiratory failure and two caused by cardiac death/failure, No deaths were considered by the investigators to be related to the study drug. Thirteen patients experienced 15 serious AEs (SAEs), excluding deaths, none of which were suspected to be related to acumapimod. An SAE of COPD exacerbation was reported in 10 patients; three patients (one receiving prednisone, 20 mg or 75 mg repeat-dose acumapimod) were hospitalized twice for COPD exacerbations. All SAEs were considered to be moderate in severity except for one SAE that was severe (bladder cancer experienced by one patient).
No patients withdrew because of AEs. In the 75 mg repeat-dose group, two cases of pruritic rash were reported. There were no cases of the acneiform skin rash that has been associated with p38 inhibitors [Citation29] at any dose and no changes in renal function were observed. Two cases of mild (<2× upper limit of normal) and transient transaminase elevations were reported as AEs, one in the 20 mg and 75 mg repeat-dose groups.
Discussion
MAPK p38 is a key regulator in the inflammation pathway and is activated in COPD. This was an exploratory study to assess the clinical efficacy of acumapimod, an oral p38 inhibitor, in AECOPD treatment. Although the study did not reach statistical significance for its primary endpoint at Day 10, two single doses (Day 1 and Day 6) of the highest dose (75 mg) of the p38 MAPK inhibitor, acumapimod, showed clinically relevant and statistically significant 152 mL increase in FEV1 versus placebo in AECOPD at Day 8 (75 mg single-dose). This may be explained by acumapimod’s half-life of 30–35 h, suggesting that the second dose at Day 6 provided anti-inflammatory effects to Day 8, but was lost by Day 10. Post-hoc analyses were supportive of the physiological data for the 75 mg repeat-dose, demonstrating improvements in FEV1 AUC over placebo, prednisone and 20 mg single-dose acumapimod over 14 days. Although not statistically significant, the changes in both patient symptoms (as measured by EXACT-PRO) and anti-inflammatory biomarkers were consistent in supporting the effect of acumapimod observed on FEV1. Acumapimod was well tolerated with a manageable safety profile.
The failure to reach statistical significance compared with placebo and the apparent tailing-off of the effect of the 75 mg single-dose may partly be explained by the dosing regimen used. With a half-life of 30–34 h for acumapimod, doses given at Day 1 or at Day 1 and Day 6 would not have maintained adequate systemic exposure at trough for the half-maximal inhibitory concentration (IC50) and anti-inflammatory effect throughout the period of the exacerbation [Citation21]. Future trials will use alternate-day repeat-dose regimens in order to maintain acumapimod levels above IC50 for p38 inhibition throughout the expected duration of an AECOPD (e.g. ClinicalTrials.gov: NCT02700919).
Of note, outcomes in the prednisone-treated control group were consistently lower than those seen in all other treatment arms for most endpoints. There is controversy over the usefulness of systemic steroids in acute exacerbations [Citation8]. Several clinical trials have shown superiority of systemic steroids over placebo, with no significant efficacy difference between short and long treatment regimens [Citation30–33]. However, steroid resistance is of increasing concern in COPD [Citation34]. The observed limited effect of oral prednisone may have been due to the prohibition of the requirement for systemic corticosteroids in the study. It is possible that investigators enrolled patients who they believed did not require corticosteroids, and this may have biased the population. In addition, approximately, 50% of the enrolled population were of the low eosinophil phenotype. This sub-population is suggested to respond poorly to corticosteroids during exacerbation [Citation35], and some studies have shown systemic corticosteroids adversely affect the outcome of AECOPD [Citation36].
The use of oral corticosteroids and nebulized budesonide were excluded in this study, as demonstrating acumapimod’s anti-inflammatory superiority on top of oral corticosteroids would have required a larger study which, given the early stage of acumapimod’s development at that time, would not have been appropriate. Also, the exclusion of oral corticosteroids (and nebulized budesonide) allowed assessment of the potential of acumapimod as a steroid-sparing agent.
Previous studies with p38 MAPK inhibitors have been disappointing with no clear evidence of clinical benefit in chronic treatment of COPD or rheumatoid arthritis [Citation37–39]. This is possibly a result of “inflammatory escape” during chronic dosing [Citation40], as evidenced by positive effects on inflammatory markers declining over the course of a few weeks despite continued dosing [Citation21,Citation41]. Inhibition of p38 may therefore be best positioned in the acute short-term treatment of inflammation-driven exacerbations of disease, such as AECOPD.
Patients with COPD suffer both a lowered quality of life and an increased burden of disease associated with emergency department visits, hospitalizations, and death resulting from AECOPD [Citation3]. Improved treatment of acute exacerbations could both reduce healthcare burden and improve quality of life.
There was no evidence of hepatotoxicity for either of the study arms treated with acumapimod during the course of this study. In contrast, hepatotoxicity has been reported with p38 MAPK inhibitors in clinical trials, with elevations in liver transaminases among common AEs [Citation34]. There were two cases of pruritic rash in the 75 mg repeat-dose group. No incidence of acneiform skin rash was associated with acumapimod treatment in this study, whereas transient acneiform rashes have occurred in several Phase II trials with p38 inhibitors in patients with underlying chronic inflammatory conditions [Citation39,Citation42,Citation43]. SAEs or deaths during the study were unrelated to acumapimod and were predictable in this patient population. The short-term dosing of acumapimod in this study may explain the reduced class-specific AEs compared with other studies with p38 inhibitors [Citation40].
As well as being a Phase II study with the complexity of multiple therapy doses and placebos studied within small patient populations, the current trial had some limitations, which reflect the challenges generally experienced when designing and recruiting for, an exploratory study in AECOPD [Citation44]. There is inherent clinical and pathophysiological heterogeneity in COPD exacerbations [Citation45] (e.g. patients were recruited at different times during the exacerbation season with varying disease severity, and some exacerbations were eosinophilic while others were viral), as well as additional differences in medical care; such heterogeneity was evident in this study. Although this was generally a population with severe COPD with respect to baseline FEV1, patients did have varying disease severity; approximately half the patients were hospitalized because of the severity of the exacerbation and half were managed in the community. Furthermore, detailed information on patients’ disease severity (frequency of exacerbations) and smoking status were in collated form and therefore not available to present as separate data. Time from diagnosis and exacerbation history (disease severity) and specific details on current versus ex-smokers were not collected. We acknowledge also that acumapimod was administered as a single-dose regimen, despite knowing the exact half-life of the drug, as this was the highest acceptable dose, based on results of preclinical studies and clinical safety and pharmacokinetic/pharmacodynamic data. Using the EXACT-PRO tool to assess quality of life also had some limitations, as it requires a run-in training period during a stable disease state; thus using it as a symptom assessment tool in acute exacerbations only after an exacerbation had become clinically apparent may have affected the results [Citation27,Citation46]. Additionally, all the patients in this trial were Caucasian; thus, ethnicity-related differences in response were not captured. Of note, for exacerbation phenotypes, data were not collected on bacterial versus viral infection in this study. However, post-hoc analysis by eosinophil subtype (<2% vs. >2%) appeared to show that both phenotypes had similar FEV1 response (data not presented). This is aligned with data from another p38 inhibitor, which supported the effect of low eosinophil phenotypes [Citation47]. Finally, the variation of spirometry results in the acutely sick may have impacted the accuracy of FEV1 readings, the primary efficacy measure in this study. It should be noted, however, the FEV1 data were supported by the EXACT-PRO questionnaire which was developed to measure the symptoms associated with AECOPD [Citation27].
Patients were treated within 24 h after presenting with an acute exacerbation, enabling evaluation of the effects of acumapimod early in the evolution of the exacerbation. Other strengths include the use of placebo and prednisone arms to assess the effects of acumapimod without other anti-inflammatory agents and to compare acumapimod with the current standard of care for moderate and severe exacerbations. The study also benefited from the planned interim analysis, which allowed the treatment arms to be resized based on emergent FEV1 data, thereby ensuring sufficient statistical power to assess outcomes. However, while numerical differences were observed for many of the endpoints, large variations were observed and statistical significance was only reached for a single measurement. Also, consistency was noted between the physiologic, symptomatic and inflammatory biomarkers, and the timing of effects in relation to the known pharmacokinetics of acumapimod.
Overall, the data from this study suggest that acumapimod has the potential to provide meaningful clinical benefits in the treatment of acute exacerbations and will be well tolerated. Acute dosing with acumapimod allows the benefit of anti-inflammatory effects in AECOPD without the toxicities that have been observed with chronic p38 inhibitor dosing.
Treatment of acute exacerbations could lead to improved quality of life for these patients and reduced healthcare burden. Improved control and resolution of acute exacerbations may lead to better outcomes, such as reducing treatment failures, escalation of treatment and further decline in lung function. More effective inflammatory control has also been associated with increased time between exacerbations, in the long term [Citation48]. Data from the current trial have been used to determine the acumapimod doses in the ongoing dose-ranging, Phase II trial in patients with AECOPD (NCT02700919).
In conclusion, this exploratory, proof-of-concept study was designed to detect the activity of acumapimod when given along with antibiotics as an acute treatment for AECOPD. Short-term treatment with acumapimod showed a clinically relevant and statistically significant improvement in FEV1 over placebo in AECOPD in the highest dose tested (75 mg given on Day 1 and Day 6) at Day 8 of the exacerbation and AUC over 14 days of the exacerbation over both placebo and prednisone. The physiological effects were supported by non-significant trends in biochemical markers of inflammation and benefit in patient-reported outcomes.
Author contributions
I. R. S. contributed to acquisition of data for the work and drafting the manuscript. J. M. P. and Z. D. K. contributed to the drafting of the manuscript and critically reviewed for intellectual content. A. M. contributed to the interpretation of data, drafting of the manuscript and critically reviewed for intellectual content. B. P. M. contributed to the conception and design of the work, the acquisition of data, and the analysis and interpretation of data. All authors approved the final version submitted for publication.
Declaration of interest
I. R. S. reports personal and other fees received from Arensia Exploratory Medicine while the study was being conducted. J. M. P. and A. M. are employees of Mereo BioPharma Group plc. B. P. M. is an employee of Novartis. Z. D. K. reports personal and other fees from Ascent Clinical Research Solutions LLC while the study was being conducted. This study was originally sponsored by Novartis Institutes for BioMedical Research; this investigational medicinal product is now under clinical development sponsored by Mereo BioPharma 1 Limited.
Supplemental Material
Download PDF (357.7 KB)Acknowledgments
The authors would like to thank all patients and trial investigators for their contribution to the study (Arensia Bucharest team: Diana Ionită, Ileana Stoicescu, Camelia Nită, Daniela Dospinoiu, Felicia Coiocaru, Mădălina Burecu, Alina Croitoru). The authors would also like to thank Kunbi Ayo-Okanlawon, Cello Health MedErgy (Europe) for editorial assistance throughout the development of the manuscript.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for the diagnosis, management and prevention of COPD; 2017 [cited 2018 Aug 9]. Available from: https://goldcopd.org/gold-2017-global-strategy-diagnosis-management-prevention-copd/
- World Health Organisation. The top 10 causes of death. Global Health Observatory data; 2017 [cited 2018 Aug 9]. Available from: http://www.who.int/gho/mortality_burden_disease/causes_death/top_10/en/
- Guarascio AJ, Ray SM, Finch CK, et al. The clinical and economic burden of chronic obstructive pulmonary disease in the USA. Clinicoecon Outcomes Res. 2013;5:235–245. doi:10.2147/CEOR.S34321.
- Criner GJ, Bourbeau J, Diekemper RL, et al. Executive summary: prevention of acute exacerbation of COPD: American College of Chest Physicians and Canadian Thoracic Society Guideline. Chest. 2015;147(4):883–893. doi:10.1378/chest.14-1676.
- Suzuki M, Makita H, Ito YM, et al. Clinical features and determinants of COPD exacerbation in the Hokkaido COPD cohort study. Eur Respir J. 2014;43(5):1289–1297. doi:10.1183/09031936.00110213.
- Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363(12):1128–1138. doi:10.1056/NEJMoa0909883.
- Ko FW, Chan KP, Hui DS, et al. Acute exacerbation of COPD. Respirology. 2016;21(7):1152–1165. doi:10.1111/resp.12780.
- Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013;131(3):636–645. doi:10.1016/j.jaci.2012.12.1564.
- Herlaar E, Brown Z. p38 MAPK signalling cascades in inflammatory disease. Mol Med Today. 1999;5(10):439–447. doi:10.1016/S1357-4310(99)01544-0.
- Saklatvala J. The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr Opin Pharmacol. 2004;4(4):372–377. doi:10.1016/j.coph.2004.03.009.
- Gaffey K, Reynolds S, Plumb J, et al. Increased phosphorylated p38 mitogen-activated protein kinase in COPD lungs. Eur Respir J. 2013;42(1):28–41. doi:10.1183/09031936.00170711.
- Renda T, Baraldo S, Pelaia G, et al. Increased activation of p38 MAPK in COPD. Eur Respir J. 2008;31(1):62–69. doi:10.1183/09031936.00036707.
- Escott KJ, Belvisi MG, Birrell MA, et al. Effect of the p38 kinase inhibitor, SB 203580, on allergic airway inflammation in the rat. Br J Pharmacol. 2000;131(2):173–176. doi:10.1038/sj.bjp.0703605.
- Duan W, Chan JHP, McKay K, et al. Inhaled p38α mitogen-activated protein kinase antisense oligonucleotide attenuates asthma in mice. Am J Respir Crit Care Med. 2005;171(6):571–578. doi:10.1164/rccm.200408-1006OC.
- Westra J, Doornbos-van der Meer B, de Boer P, et al. Strong inhibition of TNF-α production and inhibition of IL-8 and COX-2 mRNA expression in monocyte-derived macrophages by RWJ 67657, a p38 mitogen-activated protein kinase (MAPK) inhibitor. Arthritis Res Ther. 2004;6(4):R384. doi:10.1186/ar1204.
- Armstrong J, Harbron C, Lea S, et al. Synergistic effects of p38 mitogen-activated protein kinase inhibition with a corticosteroid in alveolar macrophages from patients with chronic obstructive pulmonary disease. J Pharmacol Exp Ther. 2011;338(3):732–740. doi:10.1124/jpet.111.180737.
- Khorasani N, Baker J, Johnson M, et al. Reversal of corticosteroid insensitivity by p38 MAPK inhibition in peripheral blood mononuclear cells from COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:283–291. doi:10.2147/COPD.S72403.
- Bhavsar P, Khorasani N, Hew M, et al. Effect of p38 MAPK inhibition on corticosteroid suppression of cytokine release in severe asthma. Eur Respir J. 2010;35(4):750–756. doi:10.1183/09031936.00071309.
- Norman P. Investigational p38 inhibitors for the treatment of chronic obstructive pulmonary disease. Expert Opin Investig Drugs. 2015;24(3):383–392. doi:10.1517/13543784.2015.1006358.
- Parkin J, Hardaker EL. Effects of acumapimod (BCT197), an oral p38 inhibitor, on tobacco smoke and lipopolysaccharide-induced lung inflammation in a corticosteroid-resistant rat COPD model. Am J Respir Crit Care Med. 2017;195:A6305. doi:10.1164/ajrccm-conference.2017.195.1_MeetingAbstracts.A6305.
- De Buck S, Hueber W, Vitaliti A, et al. Population PK-PD model for tolerance evaluation to the p38 MAP kinase inhibitor BCT197. CPT Pharmacometrics Syst Pharmacol. 2015;4(12):691–700. doi:10.1002/psp4.12037.
- World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191–2194. doi:10.1001/jama.2013.281053.
- Sivapalan P, Lapperre TS, Janner J, et al. Eosinophil-guided corticosteroid therapy in patients admitted to hospital with COPD exacerbation (CORTICO-COP): a multicentre, randomised, controlled, open-label, non-inferiority trial. Lancet Respir Med. 2019;7(8):699–709. doi:10.1016/S2213-2600(19)30176-6.
- Gunen H, Hacievliyagil SS, Yetkin O, et al. The role of nebulised budesonide in the treatment of exacerbations of COPD. Eur Respir J. 2007;29(4):660–667. doi:10.1183/09031936.00073506.
- Maltais F, Ostinelli J, Bourbeau J, et al. Comparison of nebulized budesonide and oral prednisolone with placebo in the treatment of acute exacerbations of chronic obstructive pulmonary disease: a randomized controlled trial. Am J Respir Crit Care Med. 2002;165(5):698–703. doi:10.1164/ajrccm.165.5.2109093.
- Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338. doi:10.1183/09031936.05.00034805.
- Leidy NK, Wilcox TK, Jones PW, et al. Standardizing measurement of chronic obstructive pulmonary disease exacerbations. Reliability and validity of a patient-reported diary. Am J Respir Crit Care Med. 2011;183(3):323–329. doi:10.1164/rccm.201005-0762OC.
- Davies L, Angus RM, Calverley P. Oral corticosteroids in patients admitted to hospital with exacerbations of chronic obstructive pulmonary disease: a prospective randomised controlled trial. Lancet. 1999;354(9177):456–460. doi:10.1016/S0140-6736(98)11326-0.
- MacNee W, Allan RJ, Jones I, et al. Efficacy and safety of the oral p38 inhibitor PH-797804 in chronic obstructive pulmonary disease: a randomised clinical trial. Thorax. 2013;68(8):738–745. doi:10.1136/thoraxjnl-2012-202744.
- Abroug F, Ouanes I, Abroug S, et al. Systemic corticosteroids in acute exacerbation of COPD: a meta-analysis of controlled studies with emphasis on ICU patients. Ann Intensive Care. 2014;4:32. doi:10.1186/s13613-014-0032-x.
- Walters JA, Tan DJ, White CJ, et al. Different durations of corticosteroid therapy for exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2014;(12):CD006897. doi:10.1002/14651858.CD006897.pub4.
- Walters JA, Wang W, Morley C, et al. Different durations of corticosteroid therapy for exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2011;(10):CD006897. doi:10.1002/14651858.CD006897.pub2.
- Leuppi JD, Schuetz P, Bingisser R, et al. Short-term vs conventional glucocorticoid therapy in acute exacerbations of chronic obstructive pulmonary disease: the REDUCE randomized clinical trial. JAMA. 2013;309(21):2223–2231. doi:10.1001/jama.2013.5023.
- Crisafulli E, Barbeta E, Ielpo A, et al. Management of severe acute exacerbations of COPD: an updated narrative review. Multidiscip Respir Med. 2018;13(1):36. doi:10.1186/s40248-018-0149-0.
- Barnes PJ. Inflammatory endotypes in COPD. Allergy. 2019;74(7):1249–1256. doi:10.1111/all.13760.
- Camp J, Cane LJ, Bafadhel M. Shall we focus on the eosinophil to guide treatment with systemic corticosteroids during acute exacerbations of COPD?: PRO. Med Sci. 2018;6(3):pii: E74. doi:10.3390/medsci6030074.
- Watz H, Barnacle H, Hartley BF, et al. Efficacy and safety of the p38 MAPK inhibitor losmapimod for patients with chronic obstructive pulmonary disease: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2014;2(1):63–72. doi:10.1016/S2213-2600(13)70200-5.
- Damjanov N, Kauffman RS, Spencer-Green GT. Efficacy, pharmacodynamics, and safety of VX-702, a novel p38 MAPK inhibitor, in rheumatoid arthritis: results of two randomized, double-blind, placebo-controlled clinical studies. Arthritis Rheum. 2009;60(5):1232–1241. doi:10.1002/art.24485.
- Cohen SB, Cheng T-T, Chindalore V, et al. Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind, methotrexate-controlled study of patients with active rheumatoid arthritis. Arthritis Rheum. 2009;60(2):335–344. doi:10.1002/art.24266.
- Hammaker D, Firestein GS. “Go upstream, young man”: lessons learned from the p38 saga. Ann Rheum Dis. 2010;69(Suppl 1):i77–82. doi:10.1136/ard.2009.119479.
- Lomas DA, Lipson DA, Miller BE, et al. An oral inhibitor of p38 MAP kinase reduces plasma fibrinogen in patients with chronic obstructive pulmonary disease. J Clin Pharmacol. 2012;52(3):416–424. doi:10.1177/0091270010397050.
- Xu JJ, Hendriks BS, Zhao J, et al. Multiple effects of acetaminophen and p38 inhibitors: towards pathway toxicology. FEBS Lett. 2008;582(8):1276–1282. doi:10.1016/j.febslet.2008.01.063.
- Genovese MC, Cohen SB, Wofsy D, et al. A 24-week, randomized, double-blind, placebo-controlled, parallel group study of the efficacy of oral SCIO-469, a p38 mitogen-activated protein kinase inhibitor, in patients with active rheumatoid arthritis. J Rheumatol. 2011;38(5):846–854. doi:10.3899/jrheum.100602.
- Ferguson GT, Beck B, Clerisme-Beaty E, et al. Recruiting patients after hospital discharge for acute exacerbation of COPD: challenges and lessons learned. Chronic Obstr Pulm Dis. 2017;4(4):265–278. doi:10.15326/jcopdf.4.4.2016.0176.
- Parker CM, Voduc N, Aaron SD, et al. Physiological changes during symptom recovery from moderate exacerbations of COPD. Eur Respir J. 2005;26(3):420–428. doi:10.1183/09031936.05.00136304.
- Leidy NK, Murray LT. Patient-reported outcome (PRO) measures for clinical trials of COPD: the EXACT and E-RS. COPD. 2013;10(3):393–398. doi:10.3109/15412555.2013.795423.
- Marks-Konczalik J, Costa M, Robertson J, et al. A post-hoc subgroup analysis of data from a six month clinical trial comparing the efficacy and safety of losmapimod in moderate-severe COPD patients with ≤2% and >2% blood eosinophils. Respir Med. 2015;109(7):860–869. doi:10.1016/j.rmed.2015.05.003.
- Perera WR, Hurst JR, Wilkinson TMA, et al. Inflammatory changes, recovery and recurrence at COPD exacerbation. Eur Respir J. 2007;29(3):527–534. doi:10.1183/09031936.00092506.