
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
When the chalcone moiety and methylene spacers are bonded to polymer backbone, they are expected to incorporate photosensitive and liquid crystalline properties, respectively. The thermotropic side-chain liquid crystalline photosensitive polymers were synthesized using 4-phenyl styryl carbonyl (phenyl-4-oxy-4-hexyl) methacrylate (PSCPOHMA) and 4-phenyl styryl carbonyl (phenyl-4-oxy-4-decyl) methacrylate (PSCPODMA) monomers by free-radical polymerization using benzoyl peroxide (BPO) as an initiator. The structures of the synthesized monomers and polymers were confirmed by FT-IR, 1H NMR and 13C NMR techniques. The thermal stability and glass transition temperature of the poly (PSCPOHMA) were higher than that of the poly (PSCPODMA). The photocrosslinking of the polymers on irradiation with UV light established that the polymers followed 2π + 2π cyclo addition. The rate of crosslinking increased with increase in the length of the methylene chain. The mesophase of the polymers was also characterized using x-ray diffraction analysis. The liquid crystalline behavior of the polymers was confirmed by DSC and Hot stage optical polarized microscopy (HOPM).The polymers exhibited nematic phase in HOPM. So the polymers have high potential for application in photo resists and electro optic technology.
1. Introduction
Side-chain liquid crystalline polymers have attracted the attention of many researchers for both fundamental and applied reasons. On one hand, as they present an original architecture, they can potentially display interesting new structures, while on the other they are expected to exhibit properties that can lead to new applications such as long-term memory devices or non -linear optics [Citation1]. The non-linear optical organic single crystals take part in a vital role in optical research, such as fluorescence, optical signal processing, optical sensor, optical logic gates, laser radiation protection, etc. [Citation2–4].These materials are made by types of active building units consisting of typical organic and inorganic materials [Citation5,Citation6]. Such materials attracted considerable attention as key materials to produce coherent light via frequency conversion in solid state lasers [Citation7–9]. Chalcone moiety in the main- and side-chain polymers cross-link by 2π + 2π cycloaddition reaction on illumination with UV light appear to possess promising photo alignment characteristics [Citation10,Citation11].The photo reaction of methacrylate polymer films comprising 2-cinnamoyloxyethoxybiphenyl mesogenic side-groups by linearly polarized (LP) UV light and an alignment behavior of liquid crystal of the resultant film was studied by Nobuhiro et al [Citation12]. The objective of the present work is that, the synthesis, characterization and properties of novel thermotropic liquid crystalline photo sensitive polymers which show liquid crystalline properties as well as photo sensitive properties, are reported. The sophisticated characterization involves as FT-IR, 1H NMR, and 13C NMR. The differential scanning calorimeter (DSC) analysis was used to verify the existence of liquid crystalline phase and to confirm the nature of the phase observed by the hot stage optical polarized microscopy (HOPM). Because of their methylene spacers, they showed excellent liquid crystalline property and with the pendant chalcone moiety they exhibit excellent photosensitive properties, the obtained results are explained in a lucid manner.
2. Experimental
2.1. Materials
6-bromohexanol, 10-bromodecanol, Biphenyl-4-carboxaldehyde, and 4-hydroxy acetophenone (Aldrich) were used as received. Meth acryloyl chloride, Triethyl amine, N, N-dicyclohexyl carbodimide and 4-N, N-dimethyl amino pyridine were purchased from Spectrochem chemicals. Anhydrous potassium carbonate (Merk) and benzoyl peroxide (BPO) were freshly recrystallized from a chloroform/methanol (1:1) mixture. All other chemicals and solvents were of analytical grade, and they were used as received.
2.2. Measurements
The Furier Transformation Infrared (FT-IR) spectrum of polymer was recorded on Perkin Elmer FT-IR Spectrometer RXI. The specimen was prepared in the pellet form using KBr. High-resolution 1H NMR and 13C NMR spectra were recorded using a Brucker spectrometer of 300 and 75.4 MHz, respectively. CDCl3 and TMS were used as solvent and internal standard, respectively. The number average molecular weight (Mn) and weight average molecular weight (Mw) of the polymer were determined by PL-GPC 650.TGA was performed using a Mettler TA 3000 thermal analyzer in nitrogen atmosphere at a heating rate of 10 °C min−1 with a sample weight of 3–5 mg. DSC measurements were performed using a Mettler Toledo STAR e system for all the polymers in sealed aluminum pans in a dry nitrogen atmosphere with an empty aluminum pan as reference. The scans were carried out at a heating rate of 10 °C min−1. The photocrosslinking ability of all the polymers was monitored by irradiating under UV light at 420 nm at a distance of 10 cm from the light source for different time intervals. XRD measurements were performed with a nickel-fifiltered Cu-Kα radiation with a Rigaku (Rigaku, Japan) powder diffractometer. The textures of all the polymers were observed using a Euromex polarizing microscope equipped with a Linkem HFS91 heating stage and a TP-93 temperature programmer. All the polymer samples were placed between two thin microscopic slides. The textures were investigated during heating at a rate of 5 °C min−1. The samples were sliced after melting started. Images were obtained with a Nikon FM 10 camera and exposed on Konica film.
2.3. Synthesis of precursors and monomers
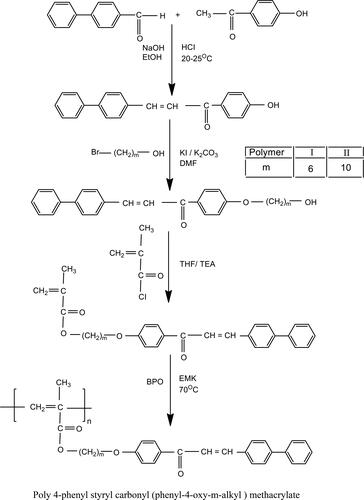
2.3.1. Synthesis of 4-phenyl styryl 4’-hydroxy phenyl ketone (PSHPK)
4-Hydroxyacetophenone (0.05 mol) in ethanol (100 ml) and sodium hydroxide (0.15 mol) was stirred at 40 °C. Biphenyl-4-carboxaldehyde (0.05 mol) in 25 ml of ethanol was added to it drop wise and the stirring continued for 24 h. The reaction mixture was neutralized with dil. hydrochloric acid, and the resulting yellow solid was filtered and washed with distilled water. The solid was recrystallized in methanol and its structure was identified by the spectroscopic techniques (IR, 1H-NMR and 13C NMR).
IR (KBr, cm−1): 3250 (phenolic-OH), 1627 (olefinic – CH = CH-), 1718 (pheny C = O), 1497 (aromatic ring)
1H-NMR (CDCl3, δ in ppm): 7–8 (m, 5H, aromatic phenyl), 6.7 (m, 4H, aromatic phenyl), 8.22 (s, 4H, aromatic carbon -CO- attached), 5.8 (d, 2H, -CH = CH-).
13C-NMR (CDC13, δ in ppm): 170 (keto carbon), 120–130 (aromatic carbon), 112 and 137 (olefinic carbon).
2.3.2. Synthesis of 4-phenyl styryl (m-hydroxy alkyloxy) phenyl ketone (m = 6, 10)
The typical procedure for the synthesis of 4-phenyl styryl (4- hydroxy hexyloxy) phenyl ketone (PSHHOPK) was as follows: a mixture of 4-phenyl styryl-4′-hydroxy phenyl ketone (0.055 mol) and anhydrous potassium carbonate (K2CO3) (0.025 mol) was dissolved in dry dimethylformamide (50 ml) and a pinch of KI was added to it. The mixture was heated to 90 °C with constant stirring. 6-Bromo-1-hexanol (0.15 mol) was added to it dropwise, refluxed overnight, cooled to room temperature, poured into ice-cold water and neutralized with 10% hydrochloric acid. The precipitated product was filtered, washed and dried in vacuum (Yield 73%, mp 83 − 85 °C). The titled precursors were synthesized according to the Williamson aryl ether synthesis method [Citation13]. By adopting a similar procedure 4-phenyl styryl (10-hydroxy decyl oxy) phenyl ketone (PSHDOPK) was synthesized (Yield 65%), mp 72–75 °C).The structures of the compounds, PSHBOPK and PSHDOPK, were identified by the spectroscopic techniques (IR, 1H-NMR, and 13C NMR).
IR (KBr, cm−1): 3310 (OH), 2230 (C-O stretching), 1606 (olefinic -CH = CH-), 1497 (aromatic ring), 2924 (aliphatic –CH2), 1715 (ester C = O), 1118 and 1050 (O-CH2).
1H- NMR (CDCl3, δ in ppm): 8.9 (O-H) ,7.45 (m, 4H, aromatic pendent phenyl), 7.05 (m,4H aromatic carbon, -CO- attached), 6.5 (d, 2H; -CH = CH-), 6.9 (m, 5H phenyl group), 3.7 (m, O-CH2), 1.7 (methylene group)
13C-NMR (CDCl3 δ in ppm): 175 (carbonyl) 120–129 (aromatic carbon), 115 and 133 (olefinic carbon) 10 − 50 (aliphatic carbon).
2.3.3. Synthesis of 4-phenyl styryl carbonyl (phenyl-4-oxy-m-alkyl) meth acrylate
In 250-mL two-neck flask, triethyl amine and 4-phenyl styryl (4- hydroxy hexyloxy) phenyl ketone (PSHHOPK) (0.015 mol), was dissolved in tetrahydrofuran (THF) and was allowed to cool between −5 °C and 0 °C. Methacryloylchloride (0.015 mol) was added drop wise at 0 °C.The above reaction mixture was allowed to stir for overnight and quaternary ammonium salt was filtered. THF was removed by rotary evaporation under reduced pressure by keeping the bath temperature below 40 °C.The residue was treated with petroleum ether to remove unreacted methacryloyl chloride. The precipitate was recrystallized with ethanol (Yield 65%, mp 72–75 °C). Another monomer 4-phenyl styryl carbonyl (phenyl-4-oxy-4-decyl) methacrylate (PSCPODMA) was also synthesized by the same procedure. The structure of the monomers, PSCPOHMA and PSCPODMA, was confirmed by IR, 1H-NMR, and 13C NMR spectra.
IR (KBr, cm−1): 2855 (aliphatic –CH2), 1723 (ester C = O), 1610 (olefinic CH2=CH-), 1590 (aromatic and C = C), 1230 (C-O stretching), 1236 (Ar-O)
1H- NMR (CDCl3, δ in ppm): 8.15 (m, 4H, aromatic pendent phenyl), 7.95 (m,4H aromatic carbon, -CO- attached) 6.5 (m, 2H; -CH = CH-). 7.30 (m, 5H phenyl group), 3.5 (m, O-CH2),1.6 (methylene group).
13C-NMR (CDCl3 δ in ppm): 179 (carbonyl) 120–140 (aromatic carbon), 116–119 (olefinic carbon) 10 − 55 (aliphatic carbon).
2.3.4. Polymerization
The typical procedure for the synthesis of poly 4-phenyl styryl carbonyl (phenyl-4-oxy-4-hexyl) methacrylate (PSCPOHMA) was as follows: PSCPOHMA (0.01 mol) and BPO (1 wt %) and the solvent dry THF were placed in a polymerization tube and gentle stream of nitrogen was purged into it. Then the tube was placed in a thermostat at 70 °C for 48 h. Similarly poly (PSCPODMA) was prepared with the same procedure by taking 4-phenyl styryl carbonyl (phenyl-4-oxy-4-decyl) methacrylate. The polymers were purified by dissolving in chloroform and re-precipitating with methanol. The polymers were dried under vacuum at 50 °C (Yield 50%, mp 78–81 °C) (Scheme 1).
Scheme 1. Synthesis of polymers PSCPOHMA PSCPODMA.
Poly 4-phenyl styryl carbonyl (phenyl-4-oxy-m-alkyl) methacrylate
3. Result and discussion
3.1. Solubility test
All the polymers were soluble in chlorinated solvents such as chloroform, methylene dichloride and in polar aprotic solvents like dimethylformamide and tetrahydrofuran.They were insoluble in hydrocarbons such as benzene, toluene and xylene.
3.2. Molecular weight data
shows that the number and weight average molecular weight. The (Mw)/(Mn) values of the polymers 1.98 and 1.93 which suggests the strong tendency of chain termination by radical combination rather than disproportionation .The theoretical values of PDI for polymer produced by radical combination and disproportionate were 1.5 and 2.0, respectively [Citation14].
Table 1. TGA and molecular weight data of Poly (PSCPOHMA) & Poly (PSCPODMA).
3.3. Fourier transform infrared spectroscopy
The aromatic ring and CH stretching vibrations of olefinic group gave peaks (very weak) just below 3010 cm−1 is shown in . The C = O stretching vibration of the ester group CO occur close to 1718 cm−1 and that of keto group close to 1606 cm−1 where assigned to aromatic ring and C = C stretching vibrations. The peaks at 1375 cm−1 were due to aromatic ring skeletal vibrations. The CH2 bending vibrations occurred close to 1285 cm−1. The ester COO vibration phenoxide C-O and ester C-O vibration gave a group of peaks at 1120 and between 990 and 1300 cm−1. The CH bending vibrations of mono substituted aromatic ring gave peaks close to 875 and 801 cm−1 and that of P-substituted aromatic ring at about 750 cm−1.
Figure 1 (a). FT-IR Spectrum of polymer PSCPOHMA. (b). FT-IR Spectrum of polymer PSCPODMA.
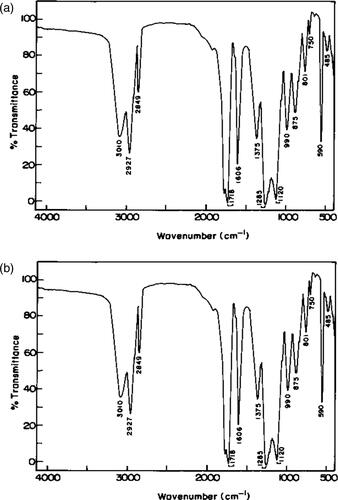
The aromatic and olefinic C-H stretching vibrations yielded peaks at 3032 cm−1 is shown in .The symmetric and asymmetric CH2 stretching vibrations occurred at 2927 and 2850 cm−1. The keto and ester C = O stretching vibrations showed peaks close to 1733 cm−1. The C = C stretching vibration, aromatic skeletal vibrations showed a broad band at 1617 cm−1. The peaks 1487, 1457 cm−1 were due to aromatic ring vibrations. The CH2 bending vibrations occurred at 1376 and 1457 cm−1. The ester –COO- vibration and phenoxide C-O vibration yielded a group of peaks between 1004 and 1345 cm−1.The C-H bending vibrations of 1, four disubstituted aromatic rings showed peaks at 692 and 762 cm−1. The monosubstituted aromatic ring vibrations occurred at 809 and 849 cm−1.
3.4. Nuclear magnetic resonance spectroscopy
The 1H NMR spectrum of the poly (PSCPOHMA) is shown in .The aromatic protons showed a group of multiplets between 7.9 and 8.8 ppm. The olefinic protons yielded their signals at 6.7 ppm. The backbone of methylene protons showed their characteristic signal between 1.8 and 1.6 ppm. The resonance signal at 3.6 ppm was due to methoxy protons.
Figure 2. (a). Proton NMR Spectrum of polymer PSCPOHMA. (b). Proton NMR Spectrum of polymer PSCPODMA.
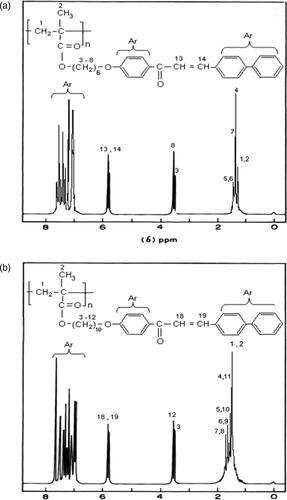
The 1H NMR spectrum of the poly (PSCPODMA) is shown in . The multiplet resonance signal between 7.9 and 8.9 ppm was due to the aromatic protons. The resonance signals at 6.9 ppm was due to olefinic protons. The signals at 1.8 and 1.5 were due to methylene protons .The methoxy protons yielded their signals at 3.7 ppm. Hence, the 1H NMR spectra of the polymers confirmed their structures.
3.5. 13C NMR spectra
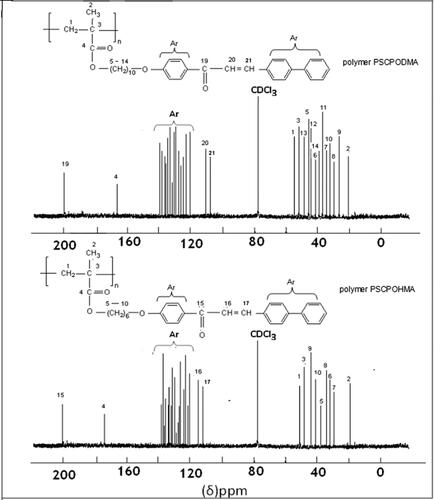
The proton decoupled 13C NMR spectra of poly (PSCPOHMA) and poly (PSCPODMA) are shown in . The ester carbonyl carbons showed their characteristic signals at 162 and 165 ppm. The resonance signals appeared at 189 and 191 ppm were to ketone carbonyl carbon. The olefinic carbon signals occurred at 115 and 116 ppm. The aromatic carbon signals appeared between 120 and 140 ppm. The resonance signals between 60 and 10 ppm were due to aliphatic carbons. Hence, 13C NMR spectra verified the existence of multiple environments in the polymeric backbone.
Figure 3. C13NMR Spectrum of polymer PSCPOHMA and polymer PSCPODMA.
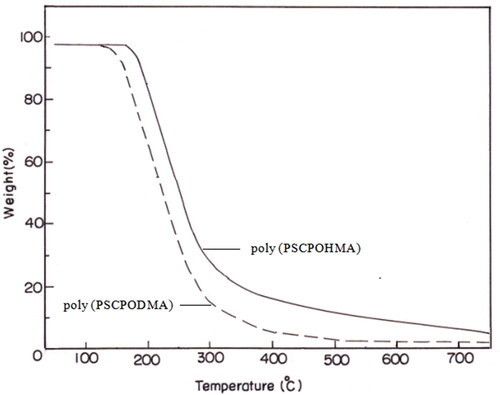
3.6. Thermogravimetric analysis
The results of the thermogravimetric analysis are illustrated in . The thermal stability was calculated for 5 to 80% weight loss. The polymers were stable up to 200 °C. The thermal stability of the poly (PSCPOHMA) was higher than that of poly (PSCPODMA) due to the shorter methylene chain of the polymer backbone.
Figure 4. TGA Thermogram of polymer PSCPOHMA and PSCPODMA.
3.7. Photocrosslinking studies
The polymer containing the mesogenic pendant chalcone unit undergoes photocrosslinking by UV irradiation and the structural changes were monitored by UV spectroscopy. The unique properties of these polymers are their ability to undergo rapid photocrosslinking without the addition of a photo initiator [Citation15]. The changes in the UV spectral pattern of the polymers due to irradiation at room temperature in the absence of sensitizer are illustrated in . The pendant chalcone moiety (>C = C<) of the polymers showed an UV absorption band at 190 nm due to the π- π* transition. Disappearance of the absorption band completed within 20 min of irradiation. The 2 π + 2π cycloaddition of the (>C = C<) group of the pendant chalcone unit is illustrated in Scheme 2. As the 2π + 2π cycloaddition destroyed conjugation in the entire π-electron system, the UV absorption intensity decreased with irradiation time [Citation16]. The photosensitivity of the polymers, poly (PSCPOHMA) and poly (PSCPODMA), containing an α, β- unsaturated carbonyl carbon was measured in terms of the rate of disappearance of (> C = C<) with irradiation time and the results are illustrated in , respectively. The photocrosslinking rate followed the order
Scheme 2. Photocrosslinking reactions (2π + 2π cycloaddition) of polymers.
Figure 5. (a). Change in UV spectral characteristics, during the photolysis of PSCPOHMA. (b). Change in UV spectral characteristics, during the photolysis of PSCPODA.
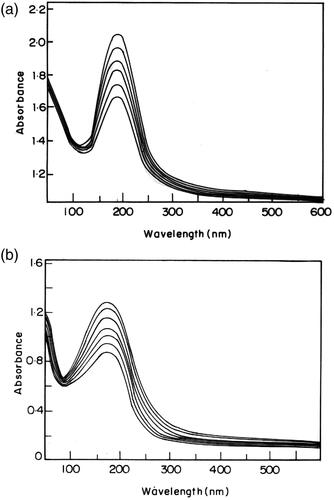
Figure 6. Dependence of the photocrosslinking rate on irradiation time of polymer PSCPOHA.
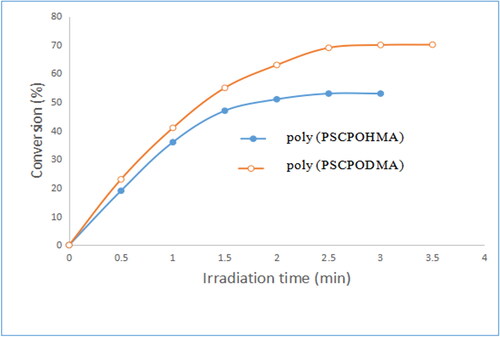
Hexamethylene < decamethylene.
The polymer containing hexamethylene spacer absorbed at shorter wavelength than the polymer containing decamethylene spacer. The photocrosslinking rate of the polymer containing decamethylene spacer was higher than hexamethylene spacer in accordance with the reported literature [Citation17]. The spacer unit between the photosensitive group and the polymer backbone provide more flexibility and free movement for the side-chain and accelerated rate of photocrosslinking.
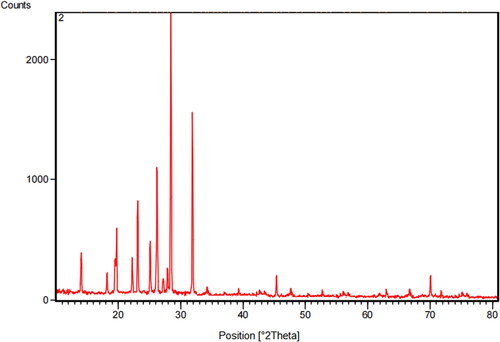
3.8. XRD analysis
Powder X-ray diffraction was used for the identification of the phase observed by the DSC and HOPM. There were a group of peaks between 10 and 32° (2θ) and the peaks were good resolved. The X- ray diffraction pattern of poly (PSCPOHMA) is shown in . The location and relative intensities were confirmed by the presence of a nematic phase [Citation18]. The well defined Braggs peaks at specific 2θ angles have the synthesized poly (PSCPOHMA) and poly (PSCPODMA) shown high liquid crystalline nature with nematic texture.
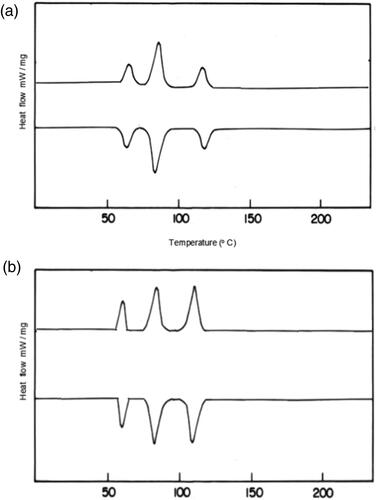
Figure 7. DSC Trace of polymer PSCPOHA and PSCPODA.
3.9. Polarized optical microscopic analysis
The DSC trace of the polymers showed three endothermic peaks one due to glass transition temperature (Tg) and other two due to melting (crystal –liquid Tm) and mesophase-isotropic phase transition (Ti) as shown in .
Figure 8. (a). HOPM photograph of polymer PSCPOHA. (b). HOPM photograph of polymer PSCPODA.
Tg curves are shown as first order transitions because it is a glass transition temperature.
The temperature at which solid changes into liquid is known as melting point. Below 130 °C it becomes soild. Above 132 °C it becomes liquid. Liquid crystals are partially an-isotropic fluid that are located in between solid and liquid state
In DSC Mesophase-isotropic phase transition temperature occurs at 130° C we are getting nematic phase by using Hot stage optical polarized microscope.Nematic phase will be show at 130 − 131 °C ().
Table 2. DSC data of poly (PSCPOHMA) & poly (PSCPODMA) both heating and cooling curve.
The glass transitions result revealed that the mesogenic transition temperature value decrease with an increase in methylene group from hexamethylene to decamethylene. Shorter Chain length exhibit nematic phase flexibility should be arrested [Citation19].
Poly (PSCPOHMA) showed higher mesogenic transition temperature than poly (PSCPODMA). This is due to the effect of shorter methylene chain on the thermal properties of the polymers. Both the polymers exhibited liquid crystalline nature with nematic textures. The polymers were heated to 130 − 131°C and 112 − 113°C as shown in . On the cooling of the polymer from isotropic melt (Ti), the reproducibility of the phase was satisfactory.
Figure 9. (a). HOPM photograph of polymer PSCPOHMA with nematic texture at 130 °C and 131 °C. (b). HOPM photograph of polymer (PSCPODMA) with nematic texture at 112 °C and 113 °C.


In general the melting temperature decreased with increase in the length of the methylene spacers. The liquid crystalline phases were observed between the Tm and Ti. The two polymers exhibited enantiotropic birefringence behavior and their mesophase was identified on the basis of the optical texture observed through a polarizing microscope. Both the polymers exhibited liquid crystalline nature with nematic textures.
4. Conclusions
Two series of liquid-crystalline-cum-photocrosslinkable polymers that differ in their spacer length attached to the cinnamoyl moiety were synthesized by free-radical solution polymerization from the corresponding monomers using benzoyl peroxide as an initiator in THF at 70 °C with good yield. These polymers were then characterized by the IR, 1 H NMR, 13C NMR spectral techniques. Thermal stability of the polymer PSCPOHMA was higher than the polymer PSCPODMA due to decreases with an increase in the methylene spacer. The photocrosslinkable liquid crystalline polymers containing cinnamoyl moiety with various methylene spacer and biphenyl groups in the side chains, which act as mesogen, were synthesized and characterized spectroscopically. The Tm and Ti of the polymers decrease with an increase in even number of methylene spacer. Para position phenoxy group was connected with polymer backbone through various even numbers of methylene spacer. Increasing the methylene chain always adds flexibility to the polymer backbone and increasing the collision probability there by crosslinking reaction is accelerated .So we are selected m = 6 and m = 10.
4-phenyl styryl carbonyl (phenyl-4-oxy-4-hexyl) methacrylate (PSCPOHMA) and 4-phenyl styryl carbonyl (phenyl-4-oxy-4-decyl) methacrylate (PSCPODMA). (PSCPOHMA) contains shorter methylene chain and (PSCPODMA)contains longer methylene chain
The photocrosslinking properties of two polymers were examined by UV spectroscopy, and the photocrosslinking polymers proceed through (2π + 2π) cycloaddition reaction of the olefinic group present in the mesogen of the polymer. The two polymers showed photocrosslinking behavior under UV radiation and the photolysis reaction completed within 5–7 min. The photolysis studies of various methylene-spacer containing polymers reveal that the rate of photocrosslinking depends on the length of methylene chain. The HOPM (Hot stage polarized microscopy) study indicates that the two prepared polymers exhibit LC properties with nematic texture. Finally, it may be concluded that these polymers can be suitably exploited for developing photo resists, photo fabrication, surface coating, energy exchange materials, printing plates, integrated circuit technology, etc.
Acknowledgments
The authors are thankful to sophisticated Analytical Instrument Facility (SAIF), Indian Institute of Technology, Chennai.
Data availability
The data are used to support the finding of this study are included within the article.
Conflict of interest
The authors declare that there is no conflict of interest regarding the publication of this article.
References
- G. D. Crevoisier et al., Macromolecules 35 (10), 3880 (2002). doi:10.1021/ma010459t
- S. Adhikari, T. Kar, and S. K. Seth, RSC Adv. 6 (101), 99139 (2016). doi:10.1039/C6RA21466E
- S. Adhikari, S. K. Seth, and T. Kar, Cryst. Eng.Comm. 15 (36), 7372 (2013). doi:10.1039/c3ce41079j
- S. Moitra, S. K. Seth, and T. Kar, J. Cryst. Growth 312 (12–13), 1977 (2010). doi:10.1016/j.jcrysgro.2010.03.016
- B. Zhang et al., Angew Chem. Int. Ed. Engl. 56 (14), 3916 (2017). doi:10.1002/anie.201700540
- Z. Yang et al., J. Mater. Chem. C 6 (10), 2435 (2018). doi:10.1039/C7TC04857B
- P. Becker, Adv. Mater. 10 (13), 979 (1998). doi:10.1002/(SICI)1521-4095(199809)10:13<979::AID-ADMA979>3.0.CO;2-N
- I. Chung, and M. G. Kanatzidis, Chem. Mater. 26 (1), 849 (2014). doi:10.1021/cm401737s
- W. J. Yao et al., Adv. Opt. Mater. 2 (5), 411 (2014). doi:10.1002/adom.201300535
- D. H. Choi, S. Y. Ban, and J. H. Kim, Bull. Kor. Chem. Soc. 24, 441 (2003).
- K. H. Jung et al., Opt. Mater. 21 (1–3), 663 (2003). doi:10.1016/S0925-3467(02)00219-7
- K. Nobuhiro et al., Chem Mater. 12, 1549 (2000).
- P. Sakthivel, and P. Kannan, J. Polym. Sci. A Polym. Chem. 42 (20), 5215 (2004). doi:10.1002/pola.20350
- S. Teramacht et al., Macromolecules 11 (6), 1206 (1978).
- P. Gupta et al., Macromolecules 37 (24), 9211 (2004). doi:10.1021/ma048844g
- D. Madheswari, K. Subramanian, and A. Reddy, Eur. Polym. J. 32 (4), 417 (1996). doi:10.1016/0014-3057(95)00154-9
- S. Kumaresan, and P. Kannan, J. Polym. Sci. A Polym. Chem. 41 (20), 3188 (2003). doi:10.1002/pola.10910
- H. Changhai et al., Cryst. 35, 1321 (2008).
- U. Shukla, K. V. Rao, and A. K. Rakshit, J. Appl. Polym. Sci. 88 (1), 153 (2003). doi:10.1002/app.11618