
Abstract
Protein-coding and non-coding RNA transcripts perform a wide variety of cellular functions in diverse organisms. Several of their functional roles are expressed and modulated via RNA structure. A given transcript, however, can have more than a single functional RNA structure throughout its life, a fact which has been previously overlooked. Transient RNA structures, for example, are only present during specific time intervals and cellular conditions. We here introduce four RNA families with transient RNA structures that play distinct and diverse functional roles. Moreover, we show that these transient RNA structures are structurally well-defined and evolutionarily conserved. Since Rfam annotates one structure for each family, there is either no annotation for these transient structures or no such family. Thus, our alignments either significantly update and extend the existing Rfam families or introduce a new RNA family to Rfam. For each of the four RNA families, we compile a multiple-sequence alignment based on experimentally verified transient and dominant (dominant in terms of either the thermodynamic stability and/or attention received so far) RNA secondary structures using a combination of automated search via covariance model and manual curation. The first alignment is the Trp operon leader which regulates the operon transcription in response to tryptophan abundance through alternative structures. The second alignment is the HDV ribozyme which we extend to the 5′ flanking sequence. This flanking sequence is involved in the regulation of the transcript's self-cleavage activity. The third alignment is the 5′ UTR of the maturation protein from Levivirus which contains a transient structure that temporarily postpones the formation of the final inhibitory structure to allow translation of maturation protein. The fourth and last alignment is the SAM riboswitch which regulates the downstream gene expression by assuming alternative structures upon binding of SAM. All transient and dominant structures are mapped to our new alignments introduced here.
Introduction
Living organisms either have genomes that express RNA sequences as their primary products or genomes made of RNA. Understanding how RNA molecules convey a multitude of functional roles is thus key to understanding life. RNA molecules have the remarkable ability to form RNA structures which is one key mechanism for assigning a functional role to an RNA. These RNA structures and the functional roles they play, for example in regulating the transcription and translation of eukaryotic genes, have been the subject of intense study for several decades. Databases such as RfamCitation1 provide a catalogue of RNA families and the corresponding key RNA secondary-structure features across a range of evolutionarily related organisms. This has, for example, helped to automatically search newly sequenced genomes for members of known RNA families.
The need to go beyond the one-sequence-one-structure dogma
As the sequencing of entire transcriptomes continues using increasingly powerful high-throughput sequencing techniques, it transpires that the transcriptomes of many organisms, including the human, are much more complex than initially thought. Several studies have discovered RNA molecules with multiple structures which each plays a distinct functional role at different times of the molecule's life. One early example of sequences with more than one functional RNA structure is so-called riboswitchCitation2–4 which consists of two distinct, mutually exclusive RNA structures each with a distinct functional role. We thus need to start looking beyond the one-sequence-one-structure dogma to appreciate that one RNA sequence can have more than one functional structure throughout its cellular life and to understand the mechanisms underlying their regulation.
We know by now that RNA structure formation in vivo involves transient RNA structure elements which can not only help to define co-transcriptional folding pathways, but can also play distinct functional roles of their own.Citation5 These transient structures can, for example, aid the correct formation of long range interaction (LRI), as seen in Bacterial RNase P Type A RNA and Bacterial SRP 4.5S RNA where the transient structures are formed preceding a transcriptional pausing site to sequester the 5' portion of a LRI until the cognate 3' portion is synthesised.Citation6 Moreover, such transient structures can be employed to regulate gene expression either via translational control as exemplified in LevivirusCitation7,Citation8 or via a transcriptional mechanism as exhibited by Tryptophan operon leaderCitation9,Citation10 and SAM riboswitch.Citation11 Last but not the least, transient structures incorporating the 5' flanking sequence are involved in adjusting the self-cleavage activity of HDV ribozyme, CPEB3 ribozyme and group I intron.Citation12,Citation13
Recent statistical evidence suggests that some transient structures are evolutionarily conserved across homologous sequences thus confirming their potential functional importance.Citation14 It is thus possible to provide entries in Rfam with a more complete structural annotation which should in turn allow us to gain a better understanding of the underlying regulatory mechanisms.
Purpose
For each of the four alignments introduced in this study, evidence from previous studies (as cited in each individual section below) shows that the formation of the experimentally confirmed transient/alternative structures is critical to confer to the RNA molecule the ability to modulate gene expression or regulate ribozyme activity. Moreover, our previous researchCitation14 shows that the computationally predicted co-transcriptional folding pathways for homologous RNA sequences go through similar transient structural configurations, thus supporting our hypothesis that evolutionarily related RNA sequences co-transcriptionally fold in similar ways whose features have been partly conserved. Overall, the functional and structural annotation of any RNA family should thus naturally include any conserved transient and alternative strutures with functional roles. Right now, however, RfamCitation1 only specifies a single functional structure for each RNA family. The Rfam database of RNA familiesCitation1 features three of them – Trp operon leader (RF00513), HDV ribozyme (RF00094), SAM riboswitch (RF00162) – but lacks the annotation and alignment for the alternative structures. More specifically, Rfam features the terminator structure of Trp operon leader, but misses the anti-terminator structure; for HDV ribozyme, the active self-cleavage structure is included, but the repressive and permissive structures involving the 5' flanking sequence of the cleavage site are missing; for the SAM riboswitch, though most of the SAM-bound structure is included, the corresponding alignment and annotation for the terminator hairpin and the SAM-unbound structure are absent. The Levivirus family is completely new and not yet part of Rfam. In order to provide more complete structural annotations including conserved transient and alternative structures, we set up a pipeline involving Infernal programCitation92 to structurally align sequences for multiple structures. We here show that it is possible to go beyond the one-sequence-one-structure dogma by providing carefully curated alignment annotated by both transient/alternative and dominant structures for the four RNA alignments (section 1 of Supplementary Material contains the alignments, CM files and initial structures/sequence identified from literature).
Trp Operon Leader
Transcriptional control of the tryptophan operon
Layout of the functional domains in trp operon
The E.coli trp operon spans approximately 7000 nucleotides (nt) which consecutively encode the promoter containing an operator,Citation15,Citation16 the transcribed leader region, and structural genes essential for the biosynthesis of tryptophan(trp), i.e. E, D, C, B, A.Citation17 The leader transcript refers to the 162 nt long untranslated region (UTR) preceding the structural genes.Citation10 Along this leader transcript, the ribosomal binding site resides at the 5' end,Citation18 and an internal transcription termination signal is located distally before the first structural gene E.Citation10,Citation19 This termination signal, lying within the attenuator, can be recognised by RNA polymerase to produce a transcript of about 140 nt (137-141) length.Citation9.Citation20 This shorter transcript generates a leader peptide consisting of 14 residues whose distal end has a tandem of trp residues.Citation10 This leader peptide is involved in the attenuation regulatory strategy where the Trp operon leader is utilised for adaptation to the metabolic condition concerning the biosynthesis of trp.Citation9
Attenuation in Tryptophan operon leader
The trp-activated repressor protein(trpR) is stimulated upon trp binding to compete against RNA polymerase,Citation21 and consequently switches off the initiation of trp operon transcription.Citation22 An additional repression mechanism is postulated to operate directly on the progressing transcription along the starting segment of the operon.Citation23,Citation24 Further deletion mutagenesis studies narrow down the regulatory region onto the leader region of the trp operonCitation25,Citation26 wherein transcription stops at a distal transcription termination site.Citation19,Citation27 The transcription and translation of this short leader transcript have been demonstrated in vivo and in vitro.Citation9,Citation28,Citation29 The corresponding leader peptide is the byproduct of the attenuation mechanism which is essentially an internal transcription termination signal that is modulated in response to the abundance of metabolites relevant to the products of this operon.Citation25 Consequently, the ongoing transcription of the downstream operon genes could be regulated accordingly and promptly in an “economical” way.Citation9,Citation25
The attenuation mechanism requires the translation of the leader peptide as shown by the altered termination frequency observed in mutants with deficient components or targets of the translation machinery, e.g., tRNA, tryptophanyl-tRNA synthetase and start codon.Citation10,Citation18,Citation30,Citation31 Whether the ribosome stalls during translation and where this occurs is likely to influence this regulatory attenuation mechanism because it depends on the abundance of the corresponding loaded tRNA; given the trailing trp tandem observed in this trp operon, the trp codon is found to be the codon responsible for this.Citation10,Citation32 Where the ribosome stalls dictates which alternative RNA structure the leader transcript forms: either the transcription termination hairpin forms or it is disrupted to permit transcriptional read-through.Citation30 In essence, the choice between terminator and anti-terminator structures bridges the communication between the translation of the leader peptide and the transcription of operon genes in exchanging the message for the abundance of trp.Citation10,Citation33
Dual regulatory systems for the operon expression
When trp is abundant, the cell uses a repressive system to promote the synthesis of other amino acids in starvation; here, the trpR repressor operates on the transcription initiation whereas the attenuation operates on the progressing transcription.Citation10 The repression system targets the intracellular trp concentration which depends on the influx trp, the newly synthesised trp, and finally the consumption of trp for protein synthesis.Citation10 In contrast, the attenuation measures the concentration of charged tRNATrp, which is contributed by cellular capability of protein synthesis, trp concentration, tryptophanyl-tRNA synthetase, and tRNATrp.Citation10 The intracellular trp concentration does not always correlate with the concentration of charged tRNATrp.Citation9 This dual system thus acts in concert to tune the biosynthesis level of trp on a wide spectrum.Citation10,Citation34
Terminator Structure
The terminator structure consists of two hairpins
The 5' portion of the first hairpin encompasses the region encoding the trailing residues of the leader peptide: Trp, Trp, Arg, Thr, SerCitation10 (). Other than the trp tandem required for sensing trp deficiency, base pairs embedded in this region also impose constraints on the sequence composition; indeed, conservation is observed in these five successive codons and mutating the upstream codons does not alter the operon expression.Citation9,Citation10,Citation35–37 The second hairpin, enriched in G/C and immediately followed by several uracil residues, comprises the termination signal that attenuates the operon transcription.Citation10,Citation20
Figure 1. Schematic drawing for Tryptophan operon leader. The Trp codon (W) tandem is highlighted in green color in both the gene structure (the leader peptide sequence is shown) and the RNA secondary structures. The stem of the termination hairpin is colored in red in both terminator and anti-terminator structures. In the anti-terminator structure, the ribosome, stalling on the Trp tandem, impedes the formation of the first hairpin. The 5' portion (red) of the termination hairpin is then sequestered in an anti-terminator hairpin. The RNA secondary structures in are drawn via VARNA[Citation98].
![Figure 1. Schematic drawing for Tryptophan operon leader. The Trp codon (W) tandem is highlighted in green color in both the gene structure (the leader peptide sequence is shown) and the RNA secondary structures. The stem of the termination hairpin is colored in red in both terminator and anti-terminator structures. In the anti-terminator structure, the ribosome, stalling on the Trp tandem, impedes the formation of the first hairpin. The 5' portion (red) of the termination hairpin is then sequestered in an anti-terminator hairpin. The RNA secondary structures in Figures 1–4 are drawn via VARNA[Citation98].](/cms/asset/e57297a8-d7d7-4dd0-af4c-f901318e3453/krnb_a_1008373_f0001_oc.gif)
The terminator structure forms when no ribosome stalls in the vicinity of the Trp tandem (i.e., Trp or Arg codon); that is, either the leader peptide is not translated or the translation proceeds smoothly along the 5′ portion of the first hairpin with abundant charged tRNATrp.Citation9,Citation10 More specifically, the ribosome is proposed to sterically mask about 10 nts downstream, thus ribosome stalling in either the upstream Gly or further downstream Thr does not disrupt the formation of the termination hairpin.Citation9,Citation10 Thereafter, co-transcriptional folding only allows the two hairpins to form sequentially; the first hairpin forms right after its pairing portions are transcribed, rendering the 3′ portion of the first hairpin unavailable to pair with the newly synthesised 5′ half of the second termination hairpin.Citation10
Experimental evidence. In the past, the terminator structure responsible for producing the 140 nt-long attenuated leader transcript has been investigated via experimental approaches. Lee and Yanofsky (1977) concluded that the termination efficiency at the attenuator is correlated with the stability of an embedded secondary structure which is conserved between E. coli and Salmonnella typhimurium; the proposed structures agree with results of a partial RNase T1 digestion that exhibit digestion resistance in the distal portion of this transcript, i.e., where the structural features are located.Citation32 Thereafter, Oxender et al. (1979) conducted structural probing with RNase T1 partial digestion followed by isolation of the co-migrating pairing regions in a non-denaturing gel electrophoresis; the base-pairing regions were subsequently identified via denaturing gel electrophoresis and fingerprinting, based on which the two hairpins comprising the terminator structure were drawn.Citation33 Later on, the secondary structure of DNA template was ruled out as a contributor for this termination signal so the RNA structural features are the one causing the termination.Citation9,Citation38,Citation39 Indeed, the functional importance of the second hairpin for the transcriptional termination is illustrated by the reduced transcription termination frequency observed in experiments destabilising the central G+C pairing of this hairpin, such as by in vivo mutational analysis or in vitro substitution of G-C bond by I-C bond.Citation32,Citation40-42 Moreover, mutational analysis progressively disrupting the first hairpin still preserves the production of the attenuated transcript, suggesting that the second hairpin itself is sufficient for the termination.Citation9,Citation42
Anti-terminator structure
Anti-terminator structure disrupts the two terminator hairpins
The anti-terminator hairpin is formed by the pairing between the 3′ portion of the first hairpin and the 5′ portion of the second hairpin from the terminator structure.Citation10
This structure occurs when charged tRNATrp (or tRNAArg) is starving, and the ribosome is impeded around the tandem Trp codon where the 5′ portion of the first terminator hairpin resides.Citation10 This stalling ribosome spans approximately 10 nucleotides downstream and thereby prevents the formation of the first terminator hairpin.Citation10 Instead, it promotes the base-pairing between the 3′ half of the first terminator hairpin and the 5′ half of the second terminator hairpin once they are transcribed.Citation10 Co-transcriptionally, this removes the pairing option held by the 3′ of the second terminator hairpin, rendering it single-stranded.Citation10 Since the transcriptional termination hairpin is sequestered under this circumstance, the progressing RNA polymerase no longer dissociates at the attenuation site, and the mRNA encoding the trp operon poly-peptides gets fully transcribed.Citation9,Citation10 Nonetheless, the anti-terminator hairpin is speculated to still form occasionally after the ribosome dissociates from the leader transcript even in the absence of ribosome stalling.Citation9
Experimental evidence
Both the translation of the leader peptide and ribosomal stalling are necessary for inhibiting the transcription termination.Citation31 Moreover, mutational analysis destabilising or disrupting the base-pairing of the anti-terminator hairpin, e.g., trpL75 mutant, demonstrates increased termination of several folds; consistent with the attenuation model, this mutation prevents the relief of attenuation even with Trp starvation.Citation9,Citation31 In contrast, complementary oligonucleotides targeting the 5′ portion of the first terminator hairpin increase the operon expression, presumably promoting the anti-terminator formation.Citation9,Citation43 However, there is no direct experimental evidence confirming the base-pairing of the anti-terminator hairpin (i.e., structural probing) due to the co-transcriptional nature, i.e. the other two terminator hairpins render the anti-terminator formation infeasible.Citation9
Half-life of the structures
The formation of the alternative structures is determined by whether or not the translating ribosome is impeded, which must be concomitantly captured by the transcribing polymerase.Citation9 The time scale of the structural modulation must thus be comparable to that of the transcribing polymerase.Citation10 Evidence supporting this requirement is a transcriptional pausing site located at the end of the first terminator hairpin which allows time to put the ribosome in sync with the RNA polymerase along the same transcript.Citation10 The amount of trp abundance can thus be measured in a timely manner via the proper formation of the alternative structure. Subsequently, either attenuation or read-through occurs accordingly.Citation10
5′UTR of Leviviridae Levivirus
Translational control of maturation protein in Leviviridae Levivirus
Phylogeny and host specificity of family Leviviridae
The family Leviviridae, a prevalent family targeting gram-negative bacteria, comprises positive single-stranded (ss) RNA bacteriophages with one of the smallest genome sizes (around 3500 to 4200 nt).Citation44-47 Most of the family members exhibit infection specificity for E. coli bearing F pilus receptors; moreover, the family members exploit similar mechanisms and host factors for replication and translational regulation.Citation44 This family was proposed to be a monophyletic group with main constituent genera Levivirus and Allolevivirus based on maximum likelihood and Bayesian estimation using the coat and replicase genes of nine species.Citation44 Levivirus and Allolevivirus have difference in the genes encoded and the orientation of the open reading frame (ORF), albeit both having four genes.Citation44 Each of these two genera is further sub-divided into two groups according to their serological cross-reactivity and the characteristics of the virion.Citation44,Citation45,Citation48,Citation49 Among them, MS2 and GA are the typical species of Group I, II of the Levivirus, respectively; Qβ and SP are the representative ones of Group III and IV of the Allolevivirus, respectively.Citation44,Citation50
Structure of 5′ UTR of maturation gene solves the competition between translation and replication on ssRNA template
MS2, a model organism for Group I coli-phage of Levivirus, has a sense RNA genome encoding four proteins (starting from the 5′ end): maturation, coat, lysis, and replicase.Citation8,Citation51 Both maturation and coat proteins serve as the structural component of the icosahedral virion, presenting a per-virion ratio of 1:180.Citation8 The lysis protein lyses the host cell; the replicase and host factors comprise a holoenzyme responsible for the replication of strands in both polarities.Citation8 As a result of the RNA genome, transcriptional regulatory tools are no longer available and the viral gene expression is thus regulated translationally to achieve the desired quantity and timing pertaining to these proteins.Citation8
As Levivirus has a ss RNA genome serving as the template for both translation and replication, the ribosome and the replicase tend to compete in binding the template.Citation8 To solve this conflict, the three distal genes share a single ribosomal entry site; moreover, the ribosome and replicase share the same binding site around the start codon of the coat gene.Citation7,Citation8,Citation52,Citation53 The translation of the three distal genes is therefore coupled as the binding of replicase and of ribosome is mutually exclusive.Citation7 Nevertheless, ribosome bound to the ORF of the maturation gene could potentially dislodge the replicase travelling to the 5′ end.Citation7 To prevent this, transcript folding can exert a translational control by gate-keeping the ribosomal binding site(RBS) in a long-distance interaction (LDI) via an inhibitory upstream complementary sequence (UCS).Citation8 This structure prevents the binding of the ribosome and yields way to the replicase.Citation8,Citation54 The translation of maturation protein is thus usually suppressed. When the formation of the secondary structure is prevented, translation increases.Citation8,Citation55 Expression of the maturation protein is key for the viral infection process as it can proteolytically trigger the releasing of viral genome through contacting the F-pili of male; this only requires low copies of the maturation protein in Escherichia coli.Citation8,Citation56 The virus employs a co-transcriptional folding strategy to postpone the formation of this inhibitory LDI via sequestering the UCS in a metastable hairpin, enabling a transient translation of maturation protein.Citation7 Moreover, studying this translational control of the maturation protein brings deeper insight into the evolutionary divergence of Levivirus and Allolevivirus in the Family Leviviridae.Citation44 Gene expansion is proposed to have occurred in Allolevivirus, which postpones the formation of the inhibitory LDI and therefore up-regulates the maturation protein; subsequent mutations accumulate and restore the virus fitness, resulting in the difference between these two genera.Citation44,Citation57
Final inhibitory Structure
The final cloverleaf-like structure consists of four hairpins
The cloverleaf-like structure assumed by the 5′ UTR of equilibrated RNA from MS2 was first proposed by Groeneveld et al. (1995), consisting of a 5′ hairpin, and the downstream West (W), South (S) and East (E) armsCitation7,Citation8 (). An inhibitory UCS, immediately 3′ to the 5′ hairpin, pairs with the 7 nt Shine-Dalgarno(SD, AGGAGGU) sequence, forming a LDI.Citation8 This cloverleaf-like structure, even including the bulge in the bottom of the S arm, is evolutionarily conserved between MS2 and KU1 which is a Group II Levivirus that varies from MS2 in terms of primary sequence.Citation8
Figure 2. Schematic drawing for Levivirus (MS2). The UCS is highlighted in red color, the SD sequence is in yellow color, and the start codon is colored in green. In the left structure, the SD sequence is accessible to ribosome for translation. In the right structure, the SD pairs with the UCS, forming the LDI to impede ribosome binding.
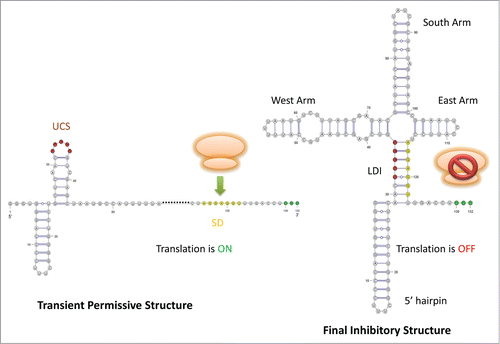
The cloverleaf-like and inactive structure forms once the first 123 nt of the plus-strand is synthesised.Citation7 Meanwhile, the start codon is not replicated yet, and the translation initiation complex requires the transcript up to the first 145 nt.Citation7 The maturation protein can therefore not be translated.Citation7 The RBS of the maturation protein spanning from nucleotide 110 to 145 can also pair with a stretch of downstream sequence to further sequester the RNA molecule in an inactive form.Citation8 This ensures that the RBS is not accessible to the ribosome once the inactive cloverleaf structure is formed, otherwise the ribosome could be there to dislodge the progressing replicase.Citation8
Experimental evidence
Phylogenetic analysis reveals the conservation of the cloverleaf structure in the 5′ UTR sequences of the maturation gene among Levivirus members: Groeneveld et al. (1995) assembled an alignment consisting of four group I phages (fr, M12, JP501, MS2), and two group II phages (KU1, GA).Citation8 They noticed that the structural features are generally preserved, albeit some variations in the W arm comparing the two groups and bulge shifting in the S arm comparing MS2 and M12.Citation8,Citation58,Citation59 They also reported covariation being observed in all arms and, most importantly, in the LDI; particularly, the amount of covariations present in Group I and II are similar.Citation8 In addition, they were able to employ the secondary-structure prediction program Mfold (GCG sofware, Genetics Computer Group, Madison, WI) to predict the cloverleaf structure.Citation8
Biochemical probing of the structure from a reference MS2 sequence is initialised as well using a combination of DMS, DEP, CMCT, and RNase T1/T2/VT.Citation8 The resultant probing pattern is consistent with the proposed structure in terms of sensitivity to modification or cleavage, including the bulge region.Citation8
Functional analyses have been conducted on the components of this cloverleaf structure through a series of mutants with deletions in variable portions of the arms.Citation8 Any changes in the synthesis of the maturation protein were thus attributed to the structural feature being mutated because the stability of these mutant RNAs was confirmed to be unchanged.Citation8 Only the base-pairing potential rather than the primary sequence composition of the W arm is required for maturation protein translation.Citation8 Moreover, deleting the entire UCS sequence or destabilising the LDI via mutations significantly enhances the maturation protein synthesis, which is further evidence for the negative regulatory role of the UCS strand.Citation8
Transient permissive Structure
The transient structure consists of a metastable hairpin
After Groeneveld et al. (1995) and Poot et al. (1997) proposed a kinetic model to explain the brief translation of maturation protein, a series of MS2 mutants were designed by Van Meerten et al. (2001) to progressively locate the kinetic trap that contributes to the slow folding of the cloverleaf-like structure.Citation7,Citation60 This temporary kinetic trap, essentially a transient structure, is located in the 5′ UTR.Citation7 The precise position was further explored by replacing the W, S and E arms of MS2 arm by arm by the cognate arm from KU1.Citation7 Finally, nt 37–45 of MS2, residing in the 5′ segment of the W arm, was identified as the functional sequence corresponding to the 3′ portion of a metastable hairpin which is conserved among MS2, KU1 and fr.Citation7
This transient hairpin encompasses 4 nts from the 3′ portion of the 5′ hairpin, the UCS, and 7 nts from the 5′ portion of the West arm originally in the cloverleaf-like structure.Citation7 Therefore, it disrupts the inhibitory LDI and thereafter frees the SD sequence, temporarily permitting the non-equilibrated RNA to be captured by the translation machinery for a brief translation of the maturation protein during the limited time window, i.e., after the synthesis of the RBS but before the LDI forms.Citation7 Moreover, this truncates the 5′ hairpin and exposes the G′s at the start of the 5′UTR as ss, which is stipulated by the viral replication; maturation protein complemented in trans is not enough to rescue a mutant with no metastable hairpin, which agrees with this additional role.Citation7
This structure forms only co-transcriptionally during the synthesis of the positive strand from an antisense template, and will be eventually replaced by the mutually exclusive LDI.Citation7 This requires the ribosome to bind the RBS fast enough compared to the formation of the LDI,Citation8 which is demonstrated feasible.Citation61
Experimental evidence
Firstly, Groeneveld et al. indirectly tested the kinetic model by delineating the maturation protein synthesis contributed from the equilibrium model; the latter model states that RBS is occasionally freed from UCS during breathing if the fully-bound LDI forms faster than ribosome binding.Citation8 Through modulating the stability of the LDI via mutations, they eliminated the possibility of maturation protein being mostly synthesised by the equilibrated cloverleaf conformation.Citation7,Citation8 Secondly, they directly assessed the kinetic model via adjusting the co-transcriptional time delayed for the LDI formation, and concluded that this duration is positively correlated with the yield of maturation protein.Citation8 Moreover, computational simulation of the co-transcriptional folding trajectory using Kinwalker does predict both the transient kinetic trap and the cloverleaf structure.Citation51
Mutational analysis also provides evidence supporting the functional importance of this metastable hairpin - the kinetic trap for this kinetic model – for the translation of maturation protein. Mutants with the metastable hairpin disrupted produce no plaque whereas compensatory double mutation is able to rescue the fitness of the phage.Citation7 On the other hand, mutation stabilising the metastable structure via replacing the bulge by a base pair increases the translation of maturation.Citation7 Furthermore, bulk evolution of those mutants with no metastable hairpin eventually leads to the restoration of the metastable hairpin.Citation7 Taken together, they imply the necessity of the metastable hairpin for the synthesis of maturation protein and thus the infection fitness of the phage.
Half-life of the structures
In vitro, the cloverleaf structure requires several minutes to fully fold, whereas a tRNA with comparable size and conformation folds only on a millisecond time scale.Citation7,Citation60 This further suggests that the MS2 5′ UTR folding is delayed by being kinetically trapped in a non-native structure.Citation7 Given that coli-phage replicase proceeds at 30 nt per second (sec), the ribosome can stably bind the maturation protein start codon as long as the cloverleaf structure is postponed to form by about 1 sec.Citation7,Citation62 Hence, the proposed translational control is convincing in terms of time.
HDV Ribozyme
Regulation of the HDV ribozyme self-cleaving activity
HDV HDV is an RNA satellite virus depending on hepatitis B virus (HBV), and aggravates the virulency of HBV-causing hepatitis.Citation63 HDV has a circular and 1700 nt long ssRNA genome, encoding the delta antigen protein.Citation12,Citation64 The high self-complementarity of the genome enables it to assumes a rod-like structure.Citation65–68 A double-rolling-circle mechanism is exploited by HDV to replicate via the host RNA polymerase II, yielding linear multimers of both genomic and antigenomic sensesCitation64,Citation68.The multimers are subsequently self-cleaved into monomers via a trans-esterification reaction catalyzed by the HDV ribozyme in cis.Citation64,Citation68 The linear monomers are then ligated into a circular genome via a host factor, which harbours the ribozyme-targeting cleavage site again.Citation12,Citation13 The ribozyme activity is turned off in the ligated RNA by interacting a downstream attenuator in order to serve as a template during the upcoming replication cycles.Citation12,Citation69
HDV ribozyme catalyses the self-cleavage activity
This HDV ribozyme has a fast reaction rate which only depends on 85 nucleotides of either genomic or antigenomic RNAs requiring one nucleotide located 5′ of the cleavage site; limited variation is observed in this ribozyme.Citation64,Citation68,Citation70,Citation71 Both genomic and antigenomic ribozymes are enriched in G and fold into similar secondary structures, as concluded by aligning the genomic and antigenomic sequences in search of sequence and structural similarity, secondary-structure prediction via minimum-free-energy minimisation, and ribonuclease digestion.Citation72
Flanking sequence participates in the regulation of HDV ribozyme self-cleavage
Non-catalytic sequences neighbouring the group I intron of Tetrahymena thermophila can modulate the ribozyme self-processing activity through base-pairing a functional portion of the ribozyme sequence.Citation12,Citation73–75 Consistent with this regulatory model, flanking sequences originated from virus and vector have been shown to affect the HDV ribozyme self-cleaving activity.Citation71,Citation76 The upstream sequence 5′ to the self-cleavage site could thus be involved in regulating the self-cleavage activity of the HDV ribozyme when the self-cleavage activity is not desired, but the downstream attenuator not readily available. That is, alternative structures formed co-transcriptionally and being mutually exclusive to the active conformation could potentially temporarily adjust the HDV ribozyme activity.Citation12 This would require a thorough investigation of the co-transcriptional folding kinetics of the HDV ribozyme incorporating the 5′ upstream sequence.Citation13 Here, we summarise the findings in one inhibitory and one permissive structure for the HDV ribozyme self-cleaving activity.Citation12
Active structure
Active conformation consists of 5 stems: P1, P1.1, P2, P3, P4.Citation64()
The active double-pseudo-knotted structure assumes a nested structure folding the active site inside a catalytic cleft to shield it against the solvents.Citation64 Coaxial stacking occurs among P1, P1.1 and P4; P2 and P3 share another stack parallel to the aforementioned stack.Citation64 Both pseudo-knots are required for cleavage.Citation64 The composition of helix P1 can be modulated without affecting the activity as long as the length, base-pairing potential and the G1*U37 wobble base pair are intact.Citation64 Helix P1.1 consists of only two base pairs, but it is critical for rendering the ribozyme into its correct 3D conformation, especially the active site responsible for cleavage activity.Citation64 Mutations breaking the P1.1 stem result in a significantly reduced ribozyme activity.Citation64,Citation77,Citation78 The active conformation of the genomic HDV ribozyme has been examined by X-ray crystallography, ribonuclease probing and site-specific mutagenesis.Citation64
Figure 3. Schematic drawing for HDV ribozyme. The starting point of each structure on the viral genome in relation to the self-cleavage site (this site is annotated by an orange arrow) is labeled by the corresponding number. The 5 stems of the active structure are colored using similar color scheme employed by Chadalavada et al. (2000) [Citation12]: P1 in blue, P2 in green, P3 in yellow, P4 in dark red, P1.1 in purple. The stem P(-1) of the permissive alternative structure is colored in pink. In the inhibitory alternative structure, Alt 1, 2, and 3 disrupt the native stems of the active structure except P1 and P4.
![Figure 3. Schematic drawing for HDV ribozyme. The starting point of each structure on the viral genome in relation to the self-cleavage site (this site is annotated by an orange arrow) is labeled by the corresponding number. The 5 stems of the active structure are colored using similar color scheme employed by Chadalavada et al. (2000) [Citation12]: P1 in blue, P2 in green, P3 in yellow, P4 in dark red, P1.1 in purple. The stem P(-1) of the permissive alternative structure is colored in pink. In the inhibitory alternative structure, Alt 1, 2, and 3 disrupt the native stems of the active structure except P1 and P4.](/cms/asset/33c53266-3210-4af0-b3e9-297b67f38d64/krnb_a_1008373_f0003_oc.gif)
Structure preventing self-cleavage
The inhibitory structure consists of Alt1, Alt2 and Alt3
Due to the rapid self-cleavage of the HDV ribozyme, the ribonuclease experiments were performed on the 3′ self-cleavage product rather than the precursor.Citation72 An extended transcript extending from 30 nt upstream of the cleavage site to 15 nt downstream of the 3′-end, denoted as −30/99 RNA, was found to have extremely diminished activity.Citation12 The flanking sequence kinetically traps the ribozyme during transcription and results in a slow reaction rate, which can be improved by the addition of heat and denaturants to facilitate the formation of the active conformation.Citation12,Citation71,Citation72
Alt1, Alt2 and Alt3 disrupt the P1.1, P2 and P3 stems in the active conformation, as revealed by the biochemical, computational, and mutational studies conducted by Chadalavada et al. (2000); in contrast, P1 and P4 remain the native conformation.Citation12 P2 is proposed to form prior to the remaining HDV ribozyme and activate both genomic and antigenomic ribozyme, which may explain the resultant inactive conformation.Citation12,Citation79,Citation80 Alt1 is a 10-bp LDI formed between an upstream inhibitory stretch (nt −25/-15 related to the cleavage site) and the downstream stretch (nt 76/86).Citation12,Citation81,Citation82 Alt2 is an interaction between upstream flanking sequence and the ribozyme, and Alt3 is a non-native ribozyme-ribozyme interaction.Citation12
Experimental evidence
Three experimental approaches provide evidence for the inhibitory secondary structure.Citation12 Firstly, this extended transcript was directly probed via ribonucleases due to its slow self-cleaving rate, and the cleavage results were used to constrain the structural prediction by Mfold 3.0, yielding the structure shown in .Citation12 Secondly, a series of DNA oligomers were used to rescue the ribozyme activity of this inactive transcript.Citation12 Among the oligomers, AS1 anneals to the entire upstream inhibitory stretch of Alt1, raising the reactivity rate by 2700- to 20,000- fold; AS2 only partially disrupts Alt1, nevertheless, it still accelerates the reactivity by 14-fold.Citation12 These two oligomers have no additive effect since sequestering either portion of Alt1 is sufficient for disrupting this inhibitory LDI.Citation12 Thirdly, mutations were introduced outside the ribozyme to ensure that the observed ribozyme activity is caused by the stability of Alt1.Citation12 Single mutations, destabilising Alt1, exhibit 150-fold increase in the reactivity rate or even co-transcriptional self-cleavage; double compensatory mutation, restoring Alt1, has similar reactivity to the inactive RNA transcript.Citation12 Upon addition of AS1, these mutants show similar reactivity rate to the active ribozyme.Citation12 Lastly, Alt1 and Alt 3 are conserved among 21 genomic isolates.Citation12 These non-native structures are conserved perhaps due to their potential role in facilitating the formation of the genome rod-like structure which is required for replication and packaging. Citation12
Structure permitting self-cleavage
Self-cleavage-permissive structure is an upstream hairpin
The permissive structure for the self-cleavage of the HDV ribozyme is mapped to the nt −54/-18 of the RNA transcript using the secondary-structure prediction program Mfold.Citation12 This structure sequesters the inhibitory stretch of nt −24/-15 from Alt1 in a hairpin P(-1) located upstream of the cleavage site.Citation12,Citation81,Citation82 This extended RNA transcript is demonstrated experimentally to cleave co-transcriptionally.Citation12 P(-1) has bulges allowing G migrating, resulting in a more stable conformation due to the increased structural entropy.Citation12,Citation83 Hairpin P(-1) does not appear to interact with the ribozyme domain as the ribozyme has similar self-cleaving activity regardless of whether it is activated in trans (AS1 or AS2) or in cis.Citation83 However, the P(-1) motif is not found in the antigenomic sequences,Citation12 so only genomic sequences are assembled in our updated alignment for HDV ribozyme.
Experimental evidence
Firstly, structural mapping via ribonuclease was used to probe the nt −54/-1 fragment instead of the whole precursor transcript due to the fast-cleaving nature of this structure, which reveals a local hairpin P(-1) pairing nt −54/−40 with −18/-30.Citation12 This structure is consistent with the ss-count values calculated using Mfold which represent the propensity for a nucleotide position to be single-stranded.Citation12 Secondly, evolutionary conservation is found in hairpin P(-1) and the linking region between P(-1) and P1 among 21 genomic HDV RNA isolates.Citation12 Minimal sequence variation is also observed in this linking region (nt −17/−1) which is pyrimidine-rich and suspected to melt the annealing between the nascent transcript and the template, facilitating the subsequent ribozyme folding.Citation12
Half-life of the structures
No direct data exists regarding the half-life, but the mechanism proposed for the HDV ribozyme resembles the one utilised by the group I intron.Citation12 Moreover, in the human HDV-like CPEB3 ribozyme, a similar regulatory mechanism involving the flanking sequence was discovered.Citation13 As a follow-up study, an equilibrium model is proposed comprising two intermediate and the native fold, which is confirmed by mutagenesis and kinetic characterization.Citation84 In this model, the 5′ portion of P2 can base-pair with either the native 3′ portion or non-native ribozyme sequence made of nucleotides from P1, P3 and single-stranded regions.Citation84 Given that P2 has a driving role in the correct folding of the HDV ribozyme, the direction of the shifting of this equilibrium may explain the resultant ribozyme activity.Citation84
SAM-responsive riboswitch
Regulation of gene expression via a riboswitch
Riboswitches are proposed to be regulatory mechanisms that derive from the RNA world.Citation85,Citation86 They correspond to non-coding RNA structure elements located in the leader sequence of mRNA strands, which selectively bind certain metabolites to regulate the synthesis of downstream products relevant to this metabolite.Citation2-4,Citation11,Citation87 This regulatory response is achieved by the coordination between the embedded metabolite-binding aptamer domain and the expression platform which switches between alternative structures upon sensing a change in the aptamer domain when a metabolite binds.Citation3,Citation4,Citation11 The riboswitch can generally assume two mutually-exclusive structures – one metabolite-bound, and one metabolite-unbound. This ligand recognition via aptamer can sequentially affect the interaction between the mRNA and the translational or transcriptional apparatus.Citation88 The structure of the aptamer is typically evolutionarily conserved; for instance, the S-adenosylmethionine(SAM) riboswitch discussed in this paper is well conserved among bacteria species.Citation11 In SAM riboswitch, S box is the aptamer where the coenzyme SAM binds with high affinity, which often precedes a putative transcription terminator hairpin; the binding of SAM triggers an allosteric change that subsequently terminates the transcription.Citation11,Citation89 Overall, a riboswitch assigns the same mRNA both a sensory and an action role without requiring an intermediate.Citation90 This amounts to a speedy and sensitive responsive mechanism that is able to sense the conditions of the cellular environment with an accuracy comparable to mechanisms involving protein factors.Citation11,Citation90
Sam-unbound structure
If the SAM is unbound, the anti-terminator sequence can sequester the terminator sequence to prevent the formation of terminator and the polymerase can progress through the downstream geneCitation11 (). The structure without binding of SAM was derived from in vitro transcription and in-line probing using the first 251 nt of yitJ gene in B. subtilis as the model.Citation11,Citation89
Figure 4. Schematic drawing for SAM riboswitch. For both RNA structures, the stem of the terminator is colored in red, and the stem of the anti-anti-terminator is colored in blue. In the SAM-unbound structure, the anti-terminator is formed by the 3' portion of the anti-anti-terminator and the 5' portion of the terminator hairpin; thus, the terminator hair can no longer form. The pseudoknot is not shown in this drawing.
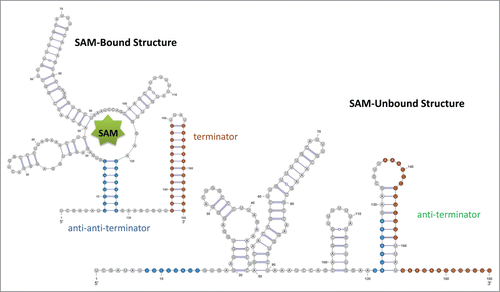
Sam-bound structure
Once the SAM is bound to the conserved core of the aptamer, the anti-terminator is sequestered by an anti-anti-terminator; the terminator sequence is then freed to assume a terminator hairpin to end the transcription.Citation11,Citation91
Experimental evidence
The SAM-bound structure was originally derived from the phylogenetic conservation observed in an alignment,Citation11,Citation89 and subsequently verified with disruptive and compensatory mutations using a 124-nt long construct (from leader mRNA of yitJ gene in B. subtilis) fused with a reporter gene,Citation11 and later determined by X-ray crystallography.Citation97
The proposed transcriptional termination mechanism upon SAM addition was tested in vitro using the transcription of 11 DNA templates harbouring the S box.Citation11,Citation86 The percentage of transcription termination was compared between presence and absence of SAM, which demonstrated increasing transcription termination upon addition of SAM.Citation11 Moreover, all structural features involved in the transcription termination mechanism (i.e., the anti-terminator, terminator, and anti-anti-terminator) were directly confirmed using disruptive and compensatory mutations.Citation11 Such disruptive mutations destabilise terminator and anti-terminator individually or both; in comparison, the compensatory mutations restore terminator and anti-terminator individually or simultaneously.Citation11 The corresponding percentage of SAM-induced transcription termination was compared among these mutants.Citation11 The results show that terminator is required for the mechanism to respond to SAM and anti-terminator is critical for relieving the transcriptional termination.Citation11 Hence, such mutational analysis supports the functional roles of those proposed structural features participating in the SAM-induced transcriptional termination mechanism.Citation11
Results
Alignments
We summarize the key features of our alignments in and . A small multiple sequence alignment (MSA) from a previous project published by usCitation14 was used to build a primary covariance model (CM) using the Infernal programCitation92 to search the full alignments in RfamCitation1 and the NCBI Taxonomy Browser.Citation93 In our previous research,Citation14 these small MSA were compiled using evolutionarily related sequences for these four RNA families, respectively. These small MSAs are of a high-quality with positive covariation and few gaps, making them convenient starting points to build the corresponding primary CMs. For each of our four alignments, we aligned the sequences of the seed alignment in Rfam with the primary CM first and then curated this against all experimentally verified structures which were first mapped to a reference sequence. We then build a secondary CM incorporating sequences from the Rfam seed alignment which we used to align the hits returned by the search with the primary CM. These hits were clustered to reduce primary sequence redundancy, choosing a representative sequence for each cluster based on structural fit and covariation. Hits passing this selection strategy were retained only if they contained all alternative structures. We then calculated quality measures for the resulting hits to determine which ones to add to the existing small MSA. The resulting alignment was then curated recursively according to the aligning results returned by Infernal using the secondary CM for each of the alternative structures.
Table 1. The basic alignment statistics for our new alignments and the corresponding seed alignments in Rfam, if it exists. The structural quality measures allow a swift comparison between the structural annotation of our new alignments and that of the corresponding seed alignments in Rfam.
Table 2. The alignment quality measures for the transient structural features for our alignments. For Trp, the numbers refer to the anti-terminator structure.
Mapping structures
Each of the alternative structures is mapped to the curated alignments via the respective reference sequence, see and for the structure-specific quality measures. All quality measures for all new alignments show an improvement with respect to the corresponding Rfam alignments, if they exist, see . As the numbers in show, the evolutionary evidence supporting the alternative structures is strong and comparable to those supporting the nominal structural features, see . This is also illustrated in the arc-plots shown in .
Figure 5. Arc-plot of Tryptophan operon leader made using the visualisation program R-chie [Citation96]. The left legend specifies the percentage of canonical base-pairs in the paired alignment columns, i.e. those connected by an arc. The right legend specifies the evolutionary support (e.g., covariation, etc.) for each position in the alignment. The alignment and arcs at the top show the information for the terminator structure, whereas the bottom ones correspond to the anti-terminator structure for the same underlying alignment. The lines in each alignment correspond to the respective sequences with every box representing either a nucleotide or a gap in the respective sequence. Every arc represents a base-pair involving the respective two alignment columns. The arcs are colour-coded according to their percentage of canonical base pairs, whereas the evolutionary information supporting each base pair is encoded in the colouring of the underlying nucleotides in the base paired alignment columns, see the right legend for details. Two green blocks connected by an arc implies that this is a canonical base pair (i.e., GC, AU or GU) which corresponds to the most abundant type of base pair for this pair of alignment columns. Cyan means this is a canonical base pair but that it has an one-sided mutation with respect to the most abundant (green) base-pair. Blue refers to a canonical base-pair which differs on both sides from the most abundant (green) base-pair. Red means this is a non-canonical base pair. Unpaired nucleotides are shown in black and gaps in grey.
![Figure 5. Arc-plot of Tryptophan operon leader made using the visualisation program R-chie [Citation96]. The left legend specifies the percentage of canonical base-pairs in the paired alignment columns, i.e. those connected by an arc. The right legend specifies the evolutionary support (e.g., covariation, etc.) for each position in the alignment. The alignment and arcs at the top show the information for the terminator structure, whereas the bottom ones correspond to the anti-terminator structure for the same underlying alignment. The lines in each alignment correspond to the respective sequences with every box representing either a nucleotide or a gap in the respective sequence. Every arc represents a base-pair involving the respective two alignment columns. The arcs are colour-coded according to their percentage of canonical base pairs, whereas the evolutionary information supporting each base pair is encoded in the colouring of the underlying nucleotides in the base paired alignment columns, see the right legend for details. Two green blocks connected by an arc implies that this is a canonical base pair (i.e., GC, AU or GU) which corresponds to the most abundant type of base pair for this pair of alignment columns. Cyan means this is a canonical base pair but that it has an one-sided mutation with respect to the most abundant (green) base-pair. Blue refers to a canonical base-pair which differs on both sides from the most abundant (green) base-pair. Red means this is a non-canonical base pair. Unpaired nucleotides are shown in black and gaps in grey.](/cms/asset/ddabdbbd-a9cc-4517-aefe-db703ca77f40/krnb_a_1008373_f0005_oc.gif)
Figure 6. Arc-plot for the Levivirus alignment. The alignment and arcs at the top correspond to the final inhibitory structure, whereas the bottom ones correspond to the transient structure permissive for maturation protein translation. See the caption of for more information on arc-plots and two legends.
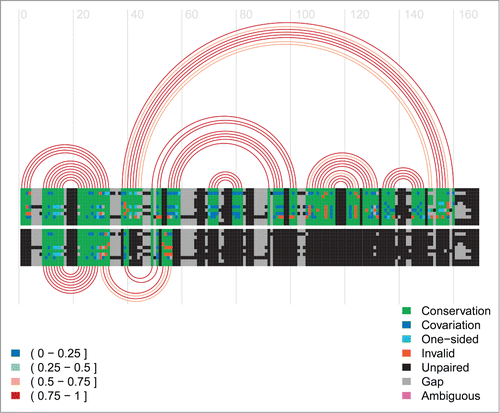
Figure 7. Arc-plot for the HDV ribozyme. The alignment and arcs at the top show the permissive alternative structure 2 and the active structure. The alternative structure 2, P(-1), is the 13-bp hairpin on the left side on top of the arc diagram. The bottom arcs correspond to the inhibitory alternative structure 1. See the caption of for more information on arc-plots and two legends.
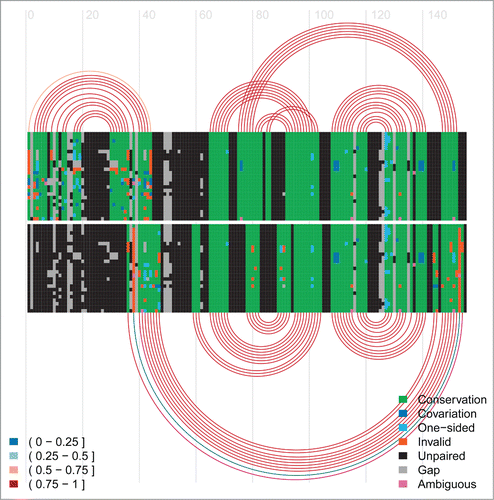
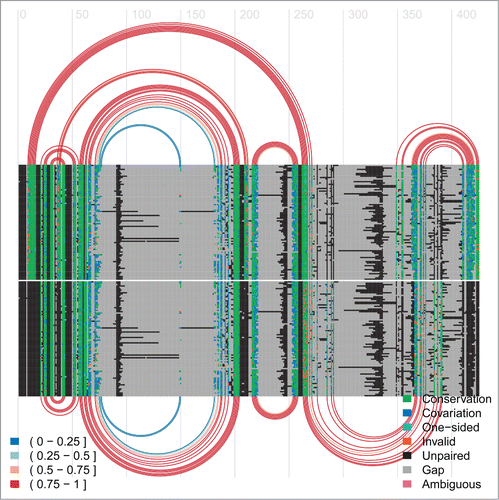
Figure 8. Arc-plot of SAM riboswitch. The top covariation and arcs correspond to the SAM-bound conformation and the bottom ones refer to the SAM-unbound conformation.
Conclusions
We introduce four RNA families, the Trp operon leader, the Levivirus 5′ UTR, the HDV ribozyme, and the Sam riboswitch, with transient structures that convey important functions critical to the regulation of their gene expression. The functional roles of these transient structures are diverse and comprise transcription regulation (Trp operon and SAM riboswitch), self-cleavage (HDV ribozyme) and translation regulation (Levivirus 5′UTR). Moreover, we show for all four alignments that both the dominant and transient structures are evolutionarily conserved. Our four alignments and structural annotations either significantly update and extend the existing Rfam family or introduce a new RNA family.
Overall, we hope to make that the case that the structural annotation of any RNA family ought to naturally also comprise functional transient RNA structures. This will not only require further experimental research regarding their structure determination and the study of their functional roles, but also some adjustments in Rfam. It should be fairly easy to provide a dedicated structural annotation for each functional role for a given RNA family. It will be more difficult to extend seed alignments in Rfam into full alignments using the current computational analysis pipeline of Rfam as more than one covariance model may be required to capture both the dominant and transient RNA structures of a given RNA family. Finally, translating experimental evidence into a structural annotation of an RNA family, as shown here for four families, is not always a straightforward task for transient structures and may require more manual curation and expertise. Ribonuclease probing, for example, of regions embedded in alternative structures may display only minor hits, rendering the interpretation ambiguous.Citation72 A concerted effort using a variety of experimental techniques as well as more sophisticated computational analysis tools will thus be required to arrive at complete structural annotations that also comprise functional transient structures.
Materials and Methods
Primary covariation model: Initial Small Alignment
A primary covariation model (cm) is constructed using a small good-quality alignment assembled previously by our group;Citation14 repeat the CM compilation (i.e., build and calibrate in Infernal-1.1rc2) for each of the alternative structures. If the structure contains pseudo-knots, it must be split into non-pseudo-knotted substructures in order to process it in Infernal; if possible, compatible helices of such non-pseudo-knotted substructures and the remaining alternative structures are combined into one structure so as to simplify the subsequent curation (e.g., in HDV, a non-pseudo-knotted substructure of the active conformation is added to the alt2 structure). The aforementioned pre-assembled alignments of Trp operon leader, Levivirus, HDV and SAM riboswitch have 10, 7, 10 and 15 sequences, respectively.
Secondary covariation model: pre-assembled alignment + unique Rfam seed alignment sequences
Firstly, the primary CM is used to align the sequences from the Rfam seed alignment (or only part of it, e.g., the alternative structures of HDV are only valid for the genomic sequences, so only genomic sequences are aligned here). This aligning procedure is repeated for the primary CM built from each of the alternative structures. Secondly, redundant sequences are removed from this expanded alignment (i.e., sequence from pre-assembled alignment + Rfam seed alignment), which is subsequently manually curated to optimise the covariation for all alternative structures. Curation is initiated on the alignment generated by the CM with the structure with the largest number of base pairs, and the other alternative structural features are mapped onto the same alignment. During curation, RaleeCitation94 is used to visually compare the resultant alignments generated by the covariation models pertaining to each alternative structure, and structural overlapping regions are identified and then manually curated (section 4.1 (iv) of Supplementary Material). Thirdly, a secondary CM is thus built and calibrated based on this expanded alignment.
Expanded Alignment: select sequences to add into the original small MSA
Overlapped hits
Firstly, the primary CM for each alternative structure is used to search the Rfam full alignment via Infernal (alternatively, for Levivirus, NCBI Taxonomy browser is used by downloading the branch of the tree of life in which this non-coding RNA resides; for SAM riboswitch, we start our curation from an alignment provided by Winkler et al. (2003)Citation11 and the SAM-bound structure is also annotated by them). A sequence is retained only if the searches using the CM of each individual alternative structure all return this sequence as a hit, and if this sequence spans the full length of all the CM without truncated ends. A hit is defined as a candidate sequence that yields log-odds score greater than 0 in the search result. Secondly, the secondary CM is used to align the overlapped hits; repeat the aligning step for all secondary CM corresponding to each of the alternative structures. Since the alignment constructing the secondary CM is optimised for all alternative structures, aligning the hits using the secondary CM facilitates the following curation among all alternative structures.
The search for homologs to add is only conducted on the Rfam full alignment rather than searching the NCBI from scratch, which is due to the fact that the qualified sequence to add must satisfy the requirement for all alternative structures. Thus, it would be more efficient but without loss of generality to start from the candidates fitting well with at least one of the alternative structures, i.e., the Rfam full alignment.
Cluster the aligned overlapped hits
In order to reduce the sequence redundancy and increase the diversity, the alignment sequences comprised of the overlapped hits are clustered based on primary sequence conservation via USEARCH;Citation95 different percentage identity cutoffs are tested to obtain around 50 to 100 clusters. For each alternative structure, a best-fit sequence is chosen for each cluster using home-made scripts scoring the covariation. Best-fit sequences overlapped among all of the structures are identified as common hits. These common hits are then ranked and filtered based on the fit of the sequence to a structure and the extent of insertions relative to the reference sequence, which gives rise to a MSA for each structure.
Criteria for selecting sequences to add
The structural measures (e.g., covariation, gappiness) of these ranked common hit sequences in the alignment are then incrementally calculated starting from the top sequence for each alternative structure, respectively. Thus, a set of sequences could be chosen systematically to be added to the original small alignment to enhance the alignment quality. Sequences added should maintain the positive overall covariation score of this alignemnt, have few invalid base-pairs, the least number of insertions introducing gaps, and no significant redundancy in terms of primary sequence identity among them.
Curation
Visual inspection and manual comparison are finally conducted to improve the alignment quality in terms of structural fitting. Homologous regions must be aligned based on their primary sequence: they cannot be shuffled around merely to satisfy the reference structure if they are obvious to be homologous to a neighbouring region and not to the reference helix region. Ensure any region (including non-structural) with known binding site, or other knowledge concerning the conservation in primary sequence is properly aligned. The detailed curation steps are described in section 4 of Supplementary Material.
Reference sequence and structure
The calculation of covariation and structure mapping involved in the aforementioned work-flow use the following reference sequence: (1) Trp operon leader: AE005174.2/2263095-2263188, from E.coli O157:H7 strain EDL933. (2) Levivirus: GQ153927.1/1-132, from Enterobacterio phage MS2. (3) HDV ribozyme: M28267.1/635-775, isolated from patient with acute delta-hepatitis. (4) Sam: AL009126.3/1258276-1258464, from Bacillus subtilis subspecies. subtilis strain 168.
References for the identification of the alternative/dominant structures can be found in section 2 of Supplementary Material.
1008373_suppl_material.pdf
Download PDF (134.8 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This project was supported by grants to I.M.M. from the Natural Sciences and Engineering Research Council (NSERC) of Canada and from the Canada Foundation for Innovation (CFI).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Burge, S. W. et al. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 2013; 41: D226-32.
- Nahvi, A. et al. Genetic control by a metabolite binding mRNA. Chem. Biol. 2002; 9: 1043-1049.
- Winkler, W., Nahvi, A. & Breaker, R. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature 2002; 419: 952-956.
- Winkler, W., Cohen-Chalamish, S. & Breaker, R. An mRNA structure that controls gene expression by binding FMN. PNAS 2002; 99: 15908-15913.
- Lai, D., Proctor, J. & Meyer, I. On the importance of cotranscriptional RNA structure formation. RNA 2013; 19: 1461-1473.
- Wong, T., Sosnick, T. & Pan, T. Folding of noncoding RNAs during transcription facilitated by pausing-induced nonnative structures. PNAS 2007; 104: 17995-18000.
- Van Meerten, D., Girard, G. & Van Duin, J. Translational control by delayed RNA folding: Identification of the kinetic trap. RNA 2001; 7: 483-494.
- Groeneveld, H., Thimon, K. & van Duin, J. Translational control of maturation-protein synthesis in phage MS2: a role for the kinetics of RNA folding? RNA 1995; 1: 79-88.
- Kolter, R. & Yanofsky, C. Attenuation in amino acid biosynthetic operons. Annu Rev Genet 1982; 16: 113-34.
- Yanofsky, C. Attenuation in the control of expression of bacterial operons. Nature 1981; 289: 751-758.
- Winkler, W. C., Nahvi, A., Sudarsan, N., Barrick, J. E. & Breaker, R. R. An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nature Structural Biology 2003; 10: 701-707.
- Chadalavada, D. M., Knudsen, S. M., Nakano, S. & Bevilacqua, P. C. A role for upstream RNA structure in facilitating the catalytic fold of the genomic hepatitis delta virus ribozyme. J Mol Biol 2000; 301: 349-367.
- Chadalavada, D. M., Gratton, E. A. & Bevilacqua, P. C. The human HDV-like CPEB3 ribozyme is intrinsically fast-reacting. Biochemistry 2010; 49: 5321-30.
- Zhu, J., Steif, A., Proctor, J. & Meyer, I. Transient RNA structure features are evolutionarily conserved and can be computationally predicted. Nucleic Acids Res 2013; 41: 6273-6285.
- Rose, J. K. & Yanofsky, C. Interaction of the operator of the tryptophan operon with repressor. PNAS 1974; 71: 3134-38.
- Bennett, G. N. & Yanofsky, C. Sequence analysis of operator constitutive mutants of the tryptophan operon of Escherichia coli. J Mol Biol 1978; 121: 179-192.
- Yanofsky, C. Tryptophan biosynthesis in Escherichia coli. genetic determination of the proteins involved. JAMA 1971; 218: 1026-1035.
- Stroynowski, I., van Cleemput, M. & Yanofsky, C. Superattenuation in the tryptophan operon of Serratia marcescens. Nature 1982; 298: 38-41.
- Squires, C. et al. Nucleotide sequence of the 5′ end of tryptophan messenger RNA of Escherichia coli. J Mol Biol 1976; 103: 351-81.
- Bertrand, K., Korn, L. J., Lee, F. & Yanofsky, C. The attenuator of the tryptophan operon of Escherichia coli: Heterogeneous 3′-OH termini in vivo and deletion mapping of functions. J Mol Biol 1977; 117: 227-47.
- Oppenheim, D., Bennett, G. & Yanofsky, C. Escherichia coli RNA polymerase and trp repressor interaction with the promoter-operator region of the tryptophan operon of Salmonella typhimurium. J Mol Biol 1980; 144: 133-42.
- Rose, J. K., Squires, C. L., Yanofsky, C., Yang, H. L. & Zubay, G. Regulation of in vitro transcription of the tryptophan operon by purified RNA polymerase in the presence of partially purified repressor and tryptophan. Nature new Biol 1973; 245: 133-37.
- Imamoto, F. Immediate cessation of transcription of the operator-proximal region of the tryptophan operon in Escherichia coli after repression of the operon. Nature 1968; 220: 31-34.
- Hiraga, S. & Yanofsky, C. Inhibition of the progress of transcription on the tryptophan operon of Escherichia coli. J Mol Biol 1973; 79: 339-49.
- Jackson, E. & Yanofsky, C. Region between the operator and first structural gene of the tryptophan operon of Escherichia coli may have a regulatory function. J Mol Biol 1973; 76: 89-101.
- Bertrand, K., Squires, C. & Yanofsky, C. Transcription termination in vivo in the leader region of the tryptophan operon of Escherichia coli. J Mol Biol 1976; 103: 319-37.
- Lee, F., Squires, C. L., Squires, C. & Yanofsky, C. Termination of transcription in vitro in the Escherichia coli tryptophan operon leader region. J Mol Biol 1976; 103: 383-393.
- Bronson, M. J., Squires, C. & Yanofsky, C. Nucleotide sequences from tryptophan messenger RNA of Escherichia coli: the sequence corresponding to the amino-terminal region of the first polypeptide specified by the operon. PNAS 1973; 70: 2335-9.
- Miozzari, G. F. & Yanofsky, C. Translation of the leader region of the Escherichia coli tryptophan operon. J.Bact 1978; 133: 1457-1466.
- Yanofsky, C. & Soil, L. Mutations affecting tRNATrp and its charging and their effect on regulation of transcription termination at the attenuator of the tryptophan operon. J Mol Biol 1977; 113: 663-677.
- Zurawski, G., Elseviers, D., Stauffer, G. V. & Yanofsky, C. Translational control of transcription termination at the attenuator of the Escherichia coli tryptophan operon. PNAS 1978; 75: 5988-92.
- Lee, F. & Yanofsky, C. Transcription termination at the trp operon attenuators of Escherichia coli and Salmonella typhimurium: RNA secondary structure and regulation of termination. PNAS 1977; 74: 4365-9.
- Oxender, D., Zurawski, G. & Yanofsky, C. Attenuation in the Escherichia coli tryptophan operon: role of RNA secondary structure involving the tryptophan codon region. PNAS 1979; 76: 5524-5528.
- Bertrand, K. & Yanofsky, C. Regulation of transcription termination in the leader region of the tryptophan operon of Escherichia coli involves tryptophan or its metabolic product. J Mol Biol 1976; 103: 339-49.
- Lee, F., Bertrand, K., Bennett, G. & Yanofsky, C. Comparison of the nucleotide sequences of the initial transcribed regions of the tryptophan operons of Escherichia coli and Salmonella typhimurium. J Mol Biol 1978; 121: 193-217.
- Miozzari, G. F. & Yanofsky, C. The regulatory region of the trp operon of Serratia marcescens. Nature 1978; 276: 684-9.
- Miozzari, G F. & Yanofsky, C. Naturally occurring promoter down mutation: Nucleotide sequence of the trp promoter/operator/leader region of Shigella dysenteriae 16. PNAS 1978; 75: 5580-4.
- Farnham, P. J. & Platt, T. A model for transcription termination suggested by studies on the trp attenuator in vitro using base analogs. Cell 1980; 20: 739-48.
- Farnham, P. J. & Platt, T. Effects of DNA base analogs on transcription termination at the tryptophan operon attenuator of Escherichia coli. PNAS 1982; 79: 998-1002.
- Zurawski, G. & Yanofsky, C. Escherichia coli tryptophan operon leader mutations, which relieve transcription termination, are cis-dominant to trp leader mutations, which increase transcription termination. J Mol Biol 1980; 142: 123-9.
- Stauffer, G. V., Zurawski, G. & Yanofsky, C. Single base-pair alterations in the Escherichia coli trp operon leader region that relieve transcription termination at the trp attenuator. PNAS 1978; 75: 4833-7.
- Stroynowski, I. & Yanofsky, C. Transcript secondary structures regulate transcription termination at the attenuator of S. marcescens tryptophan operon. Nature 1982; 298: 34-38.
- Winkler, M. E., Mullis, K., Barnett, J., Stroynowski, I. & Yanofsky, C. Transcription termination at the tryptophan operon attenuator is decreased in vitro by an oligomer complementary to a segment of the leader transcript. PNAS 1982; 79: 2181-5.
- Bollback, J. P. & Huelsenbeck, Phylogeny J. P., genome evolution, and host specificity of single-stranded RNA bacteriophage (family Leviviridae). J Mol Evol 2001; 52: 117-128.
- Furuse, K. Distribution of coliphages in the environment: general considerations. In: Goyal SM (ed) Phage ecology (Wiley, New York, 1987).
- Crawford, E. M. & Gesteland, R. F. The adsorption of bacteriophage R17. Virology 1964; 22: 165-167.
- Bradley, D. E. Shortening of Pseudomonas aeruginosa pili after RNA-phage adsorption. J Gen Microbiol 1972; 72: 303-319.
- Shapiro, L. & Bendis, I. RNA phages of bacteria other than E. coli. In: N Zinder (ed) RNA phages (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, 1975).
- Miyake, T. et al. Grouping of RNA phages based on template specificity of their RNA replicases. PNAS 1971; 68: 2022-2024.
- Murphy, F. A. et al. Virus taxonomy: The classification and nomenclature of viruses. The sixth report of the International Committee on Taxonomy of Viruses (Springer-Verlag, Vienna, 1995).
- Geis, M. et al. Folding kinetics of large RNAs. J Mol Biol 2008; 379: 160-173.
- van Himbergen, J., van Geffen, B. & van Duin, J. Translational control by a long range RNA-RNA interaction; a basepair substitution analysis. Nucleic Acids Res 1993; 21: 1713-7.
- Kolakofsky, D. & Weissmann, C. Possible mechanism for transition of viral RNA from polysome to replication complex. Nature (London) New Biol 1971; 231: 42-46.
- Klovins, J., Tsareva, N. A., de Smit, M. H., Berzins, V. & van Duin, J. Rapid evolution of translational control mechanisms in RNA genomes. J Mol Biol 1997; 265: 372-84.
- Robertson, H. D. & Lodish, H. F. Messenger characteristics of nascent bacteriophage RNA. PNAS 1970; 67: 710-716.
- van Duin, J. Single-stranded RNA bacteriophage. In: Calendar R, ed. The bacteriophages, vol 1 (Plenum Press, New York, 1988).
- Beekwilder, J., Nieuwenhuizen, R., Poot, R. & van Duin, J. Secondary structure model for the first three domains of Qbeta RNA. Control of A-protein synthesis. J Mol Biol 1996; 256: 8-19.
- Skripkin, E. A., Adhin, M. R., de Smit, M. H. & van Duin, J. Secondary structure of the central region of bacteriophage MS2 RNA. Conservation and biological significance. J Mol Biol 1990; 211: 447-63.
- Olsthoorn, R. C., Licis, N. & van Duin, J. Leeway and constraints in the forced evolution of a regulatory RNA helix. EMBO J 1994; 13: 2660-8.
- Poot, R. A., Tsareva, N. V., Boni, I. V. & van Duin, J. RNA folding kinetics regulates translation of phage MS2 maturation gene. PNAS 1997; 94: 10110-5.
- de Smit, M. H. & van Duin, J. Secondary structure of the ribosome binding site determines translational efficiency: A quantitative analysis. PNAS 1990; 87: 7668-7672.
- Kondo, M. Structure and function of RNA replicase of bacteriophage Qβ. Arch Int Physiol Biochim 1975; 83: 909-948.
- Lai, M. M. The molecular biology of hepatitis delta virus. Annu Rev Biochem 1995; 64: 259-286.
- Ferre-D′Amare, A. R., Zhou, K. & Doudna, J. A. Crystal structure of a hepatitis delta virus ribozyme. Nature 1998; 395: 567-74.
- Wang, K. S. et al. Structure, sequence and expression of the hepatitis delta (delta) viral genome. Nature 1986; 323: 508-514.
- Kos, A., Dijkema, R., Arnberg, A. C., van der Meide, P. H. & Schellekens, H. The hepatitis delta (delta) virus possesses a circular RNA. Nature 1986; 323: 558-560.
- Makino, S. et al. Molecular cloning and sequencing of a human hepatitis delta (delta) virus RNA. Nature 1987; 329: 343-346.
- Kuo, M. Y. et al. Molecular cloning of hepatitis delta virus RNA from an infected woodchuck liver: sequence, structure, and applications. J Virol 1988; 62: 1855-1861.
- Lazinski, D. & Taylor, J. Regulation of the hepatitis delta virus ribozymes: to cleave or not to cleave? RNA 1995; 1: 225-233.
- Been, M. D. & Wickham, G. S. Self-cleaving ribozymes of hepatitis delta virus RNA. Eur. J.Biochem 1997; 247, 741--753.
- Perrotta, A. T. & Been, M. D. The self-cleaving domain from the genomic RNA of hepatitis delta virus: sequence requirements and the effects of denaturant. Nucleic Acids Res 1990; 18: 6821-6827.
- Rosenstein, S. P. & Been, M. D. Evidence that genomic and antigenomic RNA self-cleaving elements from hepatitis delta virus have similar secondary structures. Nucleic Acids Res 1991; 19: 5409-16.
- Woodson, S. & Cech, T. Alternative secondary structures in the 5′ exon affect both forward and reverse self-splicing of the tetrahymena intervening sequence RNA. Biochemistry 1991; 30: 2042-2050.
- Woodson, S. Exon sequences distant from the splice junction are required for efficient self-splicing of the tetrahymena IVS. Nucleic Acids Res 1992; 20: 4027-4032.
- Nikolcheva, T. & Woodson, S. Facilitation of group 1 splicing in vivo: Misfolding of the Tetrahymena IVS and the role of ribosomal RNA exons. J Mol Biol 1999; 292: 557-567.
- Perrotta, A. & Been, M. A pseudoknot-like structure required for efficient self-cleavage of hepatitis delta-virus RNA. Nature 1991; 350: 434-436.
- Tanner, N. K. et al. A three-dimensional model of hepatitis delta virus ribozyme based on biochemical and mutational analyses. Curr Biol 1994; 4: 488-498.
- Perrotta, A. T. & Been, M. D. Core sequences and a cleavage site wobble pair required for HDV antigenomic ribozyme self-cleavage. Nucleic Acids Res 1996; 24: 1314-1321.
- Matysiak, M., Wrzesinski, J. & Ciesiolka, J. Sequential folding of the genomic ribozyme of the hepatitis delta virus: structural analysis of RNA transcription intermediates. J Mol Biol 1999; 291: 283-294.
- Perrotta, A., Nikiforova, O. & Been, M. A conserved bulged adenosine in a peripheral duplex of the antigenomic HDV self-cleaving RNA reduces kinetic trapping of inactive conformations. Nucleic Acids Res 1999; 27: 795-802.
- Mathews, D., Sabina, J., Zuker, M. & Turner, D. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol 1999; 288: 911-940.
- Zuker, M., Mathews, D. & Turner, D. Algorithms and thermodynamics for RNA secondary structure prediction practical guide In RNA Biochemistry and Biotechnology, J.B.B.R.C. Clark (Ed.), NATO ASI Series (Kluwer Academic Publishers, Dordrecht, the Netherlands, 1999).
- Zhu, J. & Wartell, R. The effect of base sequence on the stability of RNA and DNA single base bulges. Biochemistry 1999; 38: 15986-15993.
- Chadalavada, D. M., Senchak, S. E. & Bevilacqua, P. C. The folding pathway of the genomic hepatitis delta virus ribozyme is dominated by slow folding of pseudoknots. J Mol Biol 2002; 317: 559-575.
- Benner, S., Ellington, A. & Tauer, A. Modern metabolism as a palimpsest of the RNA world. PNAS 1989; 86: 7054-7058.
- Joyce, G. The antiquity of RNA-based evolution. Nature 2002; 418: 214-221.
- Mironov, A. et al. Sensing small molecules by nascent RNA: a mechanism to control transcription in bacteria. Cell 2002; 111: 747-756.
- Haller, A., Rieder, U., Aigner, M., Blanchard, S. C. & Micura, R. Conformational capture of the SAM-II riboswitch. Nat Chem Biol 2011; 7: 393-400.
- Grundy, F. & Henkin, T. The s box regulon: a new global transcription termination control system for methionine and cysteine biosynthesis genes in gram-positive bacteria. Mol Microbiol 1998; 30: 737-749.
- Nudler, E. & Mironov, A. The riboswitch control of bacterial metabolism. Trends Biochem Sci 2004; 29: 11-17.
- Epshtein, V., Mironov, A. & Nudler, E. The riboswitch-mediated control of sulfur metabolism in bacteria. PNAS 2003; 100: 5052-5056.
- Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013; 29: 2933-2935.
- Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J. & Sayers, E. W. Genbank. Nucleic Acids Res 2009; 37: D26-31.
- Griffiths-Jones, S. RALEE--RNA ALignment editor in Emacs. Bioinformatics 2005; 21: 257-259.
- Edgar, R. C. Search and clustering orders of magnitude faster than blast. Bioinformatics 2010; 26: 2460-2461.
- Lai, D., Proctor, J., Zhu, J. & Meyer, I. R-chie: a web server and r package for visualizing RNA secondary structures. Nucleic Acids Res 2012; 40:e95.
- Montange, R. K & Batey, R. T. Structure of the S-adenosylmethionine riboswitch regulatory mRNA element. Nature 2006; 441: 1172-1175.
- Darty, K., Denise, A., & Ponty, Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 2009; 25 (15):1974-1975.