
ABSTRACT
MicroRNAs (miRNAs) are small non-coding RNAs that function as negative gene expression regulators. Emerging evidence shows that, except for function in the cytoplasm, miRNAs are also present in the nucleus. However, the functional significance of nuclear miRNAs remains largely undetermined. By screening miRNA database, we have identified a subset of miRNA that functions as enhancer regulators. Here, we found a set of miRNAs show gene-activation function. We focused on miR-24-1 and found that this miRNA unconventionally activates gene transcription by targeting enhancers. Consistently, the activation was completely abolished when the enhancer sequence was deleted by TALEN. Furthermore, we found that miR-24-1 activates enhancer RNA (eRNA) expression, alters histone modification, and increases the enrichment of p300 and RNA Pol II at the enhancer locus. Our results demonstrate a novel mechanism of miRNA as an enhancer trigger.
Introduction
MicroRNAs (miRNAs) are small non-coding RNAs (ncRNAs) that primarily function through the destabilization or translational repressionCitation1 by targeting the 3′ untranslated region (3′UTR) of mRNA transcripts in the cytoplasm.Citation2,3 However, a number of recent studies indicate that miRNAs have also been implicated to positively-regulate gene transcription by targeting promoter elements,Citation4,5 a phenomenon known as RNA activation (RNAa).Citation6-9 With the exception of altering chromatin states,Citation5,6,10 the other fundamental intermediate steps that permits miRNA transcriptional activation is still a mystery. Thus, due to much uncertainty surrounding the intrinsic mechanisms of positive miRNA regulation, this unconventional yet biologically significant function by miRNAs is generally unappreciated.
Enhancers are traditionally known as cis-acting DNA elements that can increase gene transcription.Citation11 It is well established that precise regulation of gene expression in specific tissue and cell type is inseparably linked with enhancers.Citation12 Hence, enhancers are able to enable their genomic function to determine exactly when, where and at what levels is each gene is expressed to adapt to specific physiological, pathological or environmental conditions.Citation13 For instance, the super enhancer, a particular group of enhancers, is newly recognized to be essential for maintaining cell identity and the network of cell function.Citation14 Furthermore, markers for enhancers include binding of p300, histone H3K27 acetylation/H3K4 monomethylation, overlapped with DNase I hypersensitive sites and enhancer RNAs (eRNAs) expression.Citation12 Primary research showed that eRNAs were strong indicators of enhancer activity,Citation12,15,16 but recent evidence suggests eRNAs directly contributing to enhancer functions.Citation17,18 In fact, it is not only eRNA, but also enhancer-like long non-coding RNA (lncRNA)Citation19,20 and the DNMT1 interacting ncRNA,Citation21 play a crucial role in gene activation. However, unlike the lncRNA, miRNA has limited anecdotal evidenceCitation22 for contributing to enhancer induced gene activation currently.
Knowingly, miRNA is identified as a negative regulating ncRNA that functions in the cytoplasm. Yet unexpectedly, small RNA deep sequencing shows that some miRNAs are also overwhelmingly present in the nucleus,Citation23 and certain evidence indicates that some miRNAs exert biological function in the nucleus.Citation24 Besides, emerging data suggests that miRNA and protein-coding genes undergoes coordinated expression through their chromosomal loci interactions.Citation25 Recently, analysis of a large number of breast cancer samples has indicated that to some extent, miRNAs and their neighboring genes may have a positive correlative expression.Citation26 Thus, these results drive us to explore whether miRNAs also function as positive regulators in gene expression.
In this study, miRNAs overexpression in HEK293T revealed their positive influence on the neighboring protein-coding genes. Furthermore, detailed analysis of miR-24-1 demonstrates that miR-24-1 can also function as an enhancer trigger by modifying chromatin status that are favorable for transcriptional gene activation. Thus, we anticipate that miRNA mediated transcriptional gene activation may represent a novel mechanism of miRNAs.
Results
Characterization of miRNAs in enhancer loci from human annotated miRBase
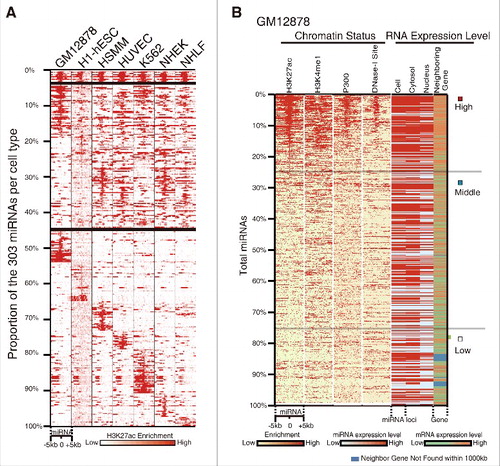
We compared the 1594 annotated human miRNA precursor loci and the regions with enrichment of the active enhancer marker H3K27ac among commonly used cell types, a total of 303 miRNAs loci were identified within the peaks of H3K27ac modification ( and Table S1). These above selected miRNAs are significantly differential conserved in sequence (p < 0.0001) (Fig. S1A). We then took one of the cell lines as examples, and found that the enrichment of H3K4me1, p300/CBP, and DNase-I-hypersensitive sites on miRNAs loci, all exhibited a similar pattern to H3K27ac ( and Fig. S1E), these evidence showed that these miRNAs are related to the enhancer. Interestingly, when we integrated miRNAs expression (), protein-coding gene expression (), and histone modification (), we found that our collection of H3K27ac-enriched miRNA tend to be present in the nuclear subcompartment (p = 2.533 × 10−10, one-side Wilcoxon rank sum test) (, Fig. S1B, C); in addition, we also found that there is a positive correlation between expression of nuclear miRNAs and their neighboring genes (p < 2.2 × 10−16, one-sided Wilcoxon rank-sum test) (, Fig. S1C, D).
Figure 1. Classification and characterization of enhancer-associated miRNAs. (A) Classification of enhancer-associated miRNAs based on H3K27ac enrichment of miRNA loci across 7 cell lines. miRNAs are organized by the different cell types, starting with the H3K27ac-enriched among all 7 cell types at the top and proceeding down to specifically enriched in one cell type. Each column represents a different cell type and each row shows relative H3K27ac enrichment at one miRNA genomic locus, with signal around ±5 kb region plotted. (B) Relationship between miRNA expression, enhancer activity and transcription of neighboring genes in GM12878 (UCSC). miRNAs are ranked from high to low based on H3K27ac density. The left 4 columns depict the patterns of enhancer markers within ±5 kb of miRNA loci. The right 4 columns are the transcription level of miRNAs in different cellular compartments and the neighboring genes. Correlation coefficiency between H3K27ac and others are shown in Fig. S1E. Color bars at the bottom indicate the range for each depth-normalized data set. See also Fig. S1.
MicroRNAs in enhancer loci display a transcriptional activator function
We reasoned that similar to miRNAs function at the promoter loci, our collection of miRNAs may act to regulate their neighboring genes. To test this hypothesis, we initially chose to investigate the well studied miR-26a-1, which is surrounded by protein-coding genes ITGA9, CTDSPL, VILL and PLCD1 in a 400kb window (, upper panel) and the miR-26a-1 gene derived DNA region was experimentally validated its enhancer activity by luciferase (Fig. S2A). We then constructed a miR-26a-1 plasmid and obtained a reproducible ectopic high-level expression of miRNA. Contrary to the repressive action of miRNA in the cytoplasm, overexpression of miR-26a-1 in HEK293T cells resulted in a concurrent increase in the transcription of neighboring ITGA9 and VILL genes (, upper panel). To further confirm the activation potential of our miRNAs, we next investigated miR-339 (Fig. S2A), and found its expression also activated the neighboring gene GPER by 4-fold (, lower panel).
Figure 2. Enhancer associated miRNA mediates transcriptional gene activation. (A) miR-26a-1 (upper panel), miR-339 (lower panel) up-regulate their respective neighboring genes expression. (B) UCSC genome browser: miR-3179-2 and miR-3180-2 accompanied by the same neighboring genes sit in the peak and valley of H3K4me1/H3K27ac derived from 7 cell lines in ENCODE UCSC, respectively (upper panel). Overexpression of these two miRNAs, the neighboring gene transcripts are quantified by qPCR (lower panel). (C) Western blot analysis of FBP1 and FANCC levels. (D) miRNA mimic up-regulates the neighbor genes expression. (E) Depletion of miR-24-3p using antagomir reduced its neighboring genes mRNA expression. All values are normalized to GAPDH and error bars show mean ± SEM for 3 biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001 by 2-tailed student's t-test. See also Fig. S2.
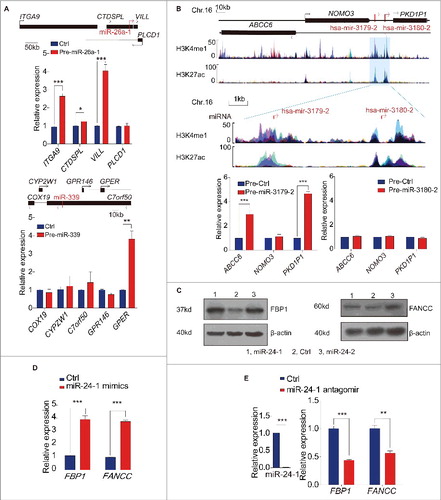
Next we analyzed miR-3179 and miR-3180, two miRNAs with obviously different enrichment levels of H3K27ac. These two miRNAs are located at the same intergenic region between NOMO and PKD1P1 gene on chromosome 16, which are ideal pairs of miRNA to investigate regulatory capacity by simultaneously detecting the expression of both neighboring genes (, upper panel). Firstly, we confirmed that the DNA fragments harboring miR-3179 possess enhancer activity in luciferase reporter assays (bar 4) (Fig. S2A), while miR-3180 itself does not possess any enhancer activating capacity (bar 3) (Fig. S2A), indicating that the H3K27ac enrichment is a reliable marker for potential enhancer elements. Secondly, upon transient transfection of miR-3179 into HEK293T cells, we found that the neighboring genes ABCC6 and PKD1P1 were up-regulated by 3-fold and 5-fold, respectively (, lower panel, left). In contrast, miR-3180 did not activate either local gene (, lower panel, right), implying that miRNA associated with enhancer features may be a prerequisite for enhancer-associated miRNA function.
To further investigate the function of those miRNAs, we focused on miR-24-1 and miR-24-2. Coincidentally, the mature sequences of them are identical, yet their pre-miRNAs are respectively derived from chromosome 9 and chromosome 19 with different neighboring genes (Fig. S2B, C, upper panels). As predicted, miR-24-1 and miR-24-2 precursors can indeed simultaneously activate their neighboring genes from both loci (Fig. S2B–E). In addition, in these experiments, a nuclear fraction with high purity was isolated from HEK293T cells (Fig. S2H) and Northern blotting showed that mature miR-24 can be detected both in cytoplasm and nucleus in the pre-miR-24 overexpressed HEK293T cells (Fig. S2I). As shown in Fig. S2J-O, pre-miRNA transfection can not only increase the miRNAs expression in the cytoplasm but also increase the expression in the nucleus.
Next, we decided to choose miR-24-1 as an example to do further research, and firstly we observed that the protein levels of miR-24-1s neighboring genes, FBP1 and FANCC, consequently increased as well (). Meanwhile, as expected, genes whose 3′UTRs are targeted by miR-24-1 in the cytoplasm showed a down-regulation pattern (Fig. S2F), implying that this miRNA can also function as a canonical miRNA in the cytoplasm.
To validate that mature miRNA participated in triggering activation, we conducted transfection of miR-24 mimics and found that mature miRNA can elevate its respective neighboring gene's mRNA level like precursor miRNAs, as did the miR-3179 mimics ( and Fig. S2G). To further evaluate the miRNA function in vivo, we used the antisense oligonucleotide (antagomiR) to exclusively and effectively knock down the endogenous mature miR-24. As shown in , qPCR revealed that miR-24 antagomiRs led to significant mRNA reduction of FBP1 and FANCC in HEK293T cells.
miR-24-1 facilitated gene transcription dependent on the presence of the intact enhancer and miRNA
To explore the importance of base-pairing rule for miRNA targeting to enhancer, we either deleted or mutated the seed region of miR-24-1 () and found that both the deletion and mutations abolished miRNA-induced transcriptional activation ().
Figure 3. miRNA mediated transcriptional gene activation requires the intact miRNA and its targeted enhancer. (A) Sequences of miR-24-1 and its mutants. (B) Analysis of HEK293T transfected with wild-type and mutant miR-24-1 by real-time RT-qPCR. GFP is used to normalize the transfection efficiency. (C) Schematic representation of the TALEN constructs targeting to the miRNA site in the enhancer locus (red, ChIP-seq track). TALENs were designed for pairwise heterodimeric binding to indicated target sequences (blue box) and the sketch map shows the reference sequence and miR-24-1 deletion#1 sequence. (D-E) miR-24-1 deletion#1 abolished neighboring gene activation. Different expression level of miR-24-1 did not up-regulate FBP1 expression in miR-24-1 deletion#1. The trigon represents the miRNA expression level from low to high. All values are normalized to GAPDH and error bars show mean ± SEM for 3 technical replicates. *P < 0.05, **P < 0.01, ***P < 0.001 by 2-tailed student's t-test. See also Fig. S3.
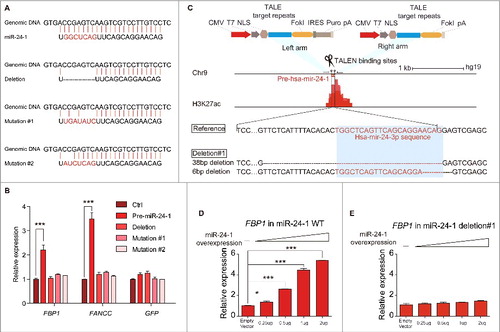
It is important to explore whether the miRNA targeted enhancer genome locus is essential for activating gene expression. We then undertook a TALE nuclease genome-editing approach Citation28 to delete the miR-24-1 enhancer locus in the HEK293T cell line ( and Fig. S3A, B). Specific primers designed around the mutation sites allowed detection of the allelic mutable editing events (Table S3). Two clones (miR-24-1 deletion1# and deletion2#) were identified in the miR-24-1 enhancer locus (, and Fig. S3C, D), in which the enhancer region were disrupted and the miRNA expression was significantly reduced (Fig. S3E). In these 2 TALEN-deletion cell strains, miR-24-1 neighboring genes were not able to be activated any further (), even by high levels of miR-24-1 ectopic overexpression (Fig. S3E, F), whereas the expression level of the neighboring gene of miR-24-2, which was the homologous originating locus of miR-24-1 mentioned before, was upregulated (Fig. S3G). The above results indicate that the transcriptional activation of miR-24-1 neighboring genes is dependent on the presence of the intact enhancer.
miR-24-1 functions through increased endogenous miRNA expression, chromatin state alteration of the enhancer
To explore the underlying mechanism by which our miRNA modulates the activity of the enhancer, we first ascertained that miR-24-1 can in fact enter the nucleus by observing the nuclear translocation of Cy5 labeled miR-24-1 (Fig. S4A, B). In order to determine whether endogenous miRNA is involved in miRNA function, we quantified the endogenous miRNA after ectopic expression of miR-24-1. To this end, we transfected the miR-24-1 mimics and then specifically detected endogenous miR-24-1 precursor expression (, upper panel). Surprisingly, we found that the exogenous miRNA can increase endogenous miR-24 expression in the activated enhancer region (, lower panel).
Figure 4. Epigenetic events involved in miRNAs' transcriptional activation. (A) miRNA mimic (green lines) increases the endogenous miRNA expression (red rectangle, mature miRNA; blue rectangle, pre-miRNA). Arrows indicate primer locations for qPCR analysis of endogenous miRNA. (B) Depletion of AGO2 blocks transcriptional activation of neighboring genes. (C) Pol II, p300 and H3K27ac ChIP assays on the miR-24-1 locus. (D) The schematic diagram (upper panel) represents the position of eRNA primers. qPCR data from the lower panel shows that eRNAs are produced from the miR-24-1 locus upon miRNA overexpression. Fig. A-D, data shown are mean ± SEM from 3 independent transfections. **P < 0.01, ***P < 0.001 by 2-tailed Student's t-test. (E) The occupancy pattern of chromatin status of miR-24-1 enhancer locus by NOMe-seq. Nuclei were extracted from HEK293T cells and treated with M.CviPI GpC methyltransferase and subjected to bisulfite conversion and cloning. Circles represent GpC dinucleotides (Blue: the reference GpC sites; Gray: occupied, unmethylated and inaccessible to M.CviPI; Orange: unoccupied, methylated and accessible to M.CviPI). Horizontal lines represent randomly selected clones. Y-axis indicates the clone number and X-axis marks the sequence length of DNA. The changed occupancy in the enhancer and promoter region is around 70bp, unable to accommodate a singular nucleosome, hinting that those regions may be occupied by regulation factors. Both regions gained occupancy by miR-24-1 overexpression as compared with the control.
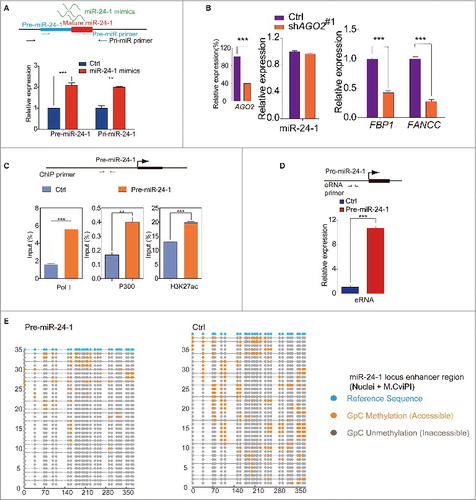
Growing evidence suggests that AGO2 can also exist and functions in the nucleus.Citation30-33 To inquire whether AGO2 is involved in our miRNA function, pre-hsa-mir-24-1 was transfected, and then AGO2 was knocked down. As shown in , the expression levels of miR-24-1 neighboring genes were significantly decreased in cells transfected with AGO2 shRNA (Fig. S4C, D). This observation implies that AGO2 is important for miR-24 activity.
It is known that enhancer activation is associated with the initiation of RNA polymerase II (RNAPII) transcription,Citation15,16 producing functional transcripts from the enhancer.Citation15,16,34 As shown in (left panel), RNAPII was enriched in the miR-24-1 enhancer region. Furthermore, we can detect increased eRNA transcripts from the active enhancers provoked by the exogenous expression of miR-24-1, miR-26a-1, miR-339, or miR-3179, respectively ( and Fig. S4E-G). Consistently, in response to miR-24-1 overexpression, ChIP-qPCR showed that the AGO2 and p300/CBP binding was enriched (, middle panel, Fig. S4H, left panel). Meanwhile, the active enhancer marker H3K27ac was increased and the repressive marker H3K9me3 was depleted (, right panel, and Fig. S4H, middle panel), while the poised enhancer Citation35 marker H3K4me1 also exhibited a slight increase (Fig. S4H, right panel). Taking advantage of NOMe-Seq,Citation36 which can monitor in vivo protein occupancy on the DNA sequence, we found significant changes of protein occupancy in the miR-24-1 enhancer locus and promoter region of FBP1 (, Fig. S4I-K). These results suggest that miR-24-1 overexpression can adjust the chromatin occupancy of the enhancer locus and its corresponding gene promoter. Overall, our data demonstrate that miRNA overexpression leads to an increased endogenous miR-24 expression and direct chromatin state alteration of the enhancer, which are all involved in gene activation (Fig. S4L).
miR-24-1 promotes global gene transcription through the binding and activating of its targeted enhancers
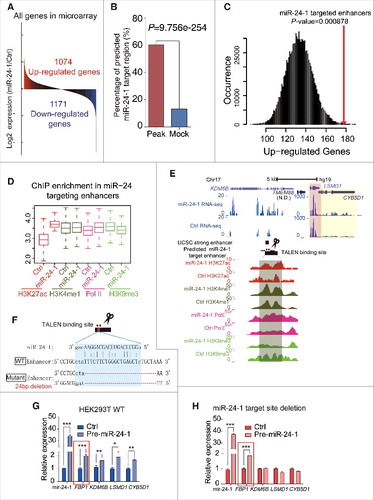
miRNA as a small molecule can target the 3′UTR of many genes.Citation3 As miR-24-1 and miR-24-2 can activate each of their neighboring genes, we wondered whether miRNA may also be able to activate global gene expression. From microarray data, we noticed that miR-24-1 ectopic expression could increase as well as decrease gene expression ( and Fig. S5A). Bioinformatics analysis by miRanda showed that down-regulated genes caused by miR-24-1 overexpression has more potential binding sites in their 3′UTR (Fig. S5B), suggesting that miRNAs generally function in the cytoplasm. Meanwhile, we propose that some miRNAs may activate gene expression through the enhancer-associated miRNA mechanism. To further investigate whether there are some genes activation caused by miR-24-1, we performed ChIP-seq assay of active enhancer markers H3K27ac and identified a prominent enrichment of H3K27ac in 3282 enhancer regions responding to miR-24-1 overexpression. In addition, a motif similar to the seed sequence of miR-24-3p was found in the elevated H3K27ac enhancer region (Fig. S5C), where a statistically significant high frequency of DNA sequence that perfectly matches the miR-24-1 seed sequence was also discovered (Fig. S5D). Importantly, miR-24-1 targets predicted by the miRanda algorithm were significantly more prevalent in the activated enhancer regions than randomly selected sequences (). Moreover, the up-regulated genes revealed by microarray assay tend to flank the activated enhancers (), and taking one as an example, we can see that in the KDM6B gene locus, the enrichment of enhancer related markers Citation37 changed accordingly (). Thus, we speculate that miR-24-1 can bind to loci beyond their original sequence and activate genes transcription, which is additionally supported by the findings that the long-range target genes DUSP16 and ENO3 mRNA levels were activated by miR-24-1 (Fig. S5E-G), but conversely decreased in miR-24-1 antagomiR transfection (Fig. S5H). To further confirm the importance of the integrity of the miRNA targeted enhancer, we disrupted the miRanda predicted enhancer in the KDM6B gene locus using FastTALE™ TALEN Assembly Kit (SIDANSAI) ( and Fig. S5I-K) and found that this deletion can abolish the neighboring genes up-regulation of KDM6B, LSMD1 and CYB5D1 when we transfected miR-24-1 ().
Figure 5. miRNAs activate genome-wide gene transcription. (A) Microarray shows that 1074 genes were upregulated after miR-24-1 transfection to HEK293T cells. Up- and downregulated genes are shaded in red and blue, respectively. (B) The miRanda algorithm analysis shows that 60.42% of total 3282 H3K27ac enriched regions were predicted as miR-24-1 targeted sites, while only 13.24% of total 2070 mock regions were predicted (P = 9.756e-254, χ2 Test). (C) Correlation between miR-24-1 target enhancers and adjacent up-regulated gene number calculated in ±100k of miR-24-1 targeted H3K27ac peaks. Difference in upregulated gene number is significant (P = 0.000878) relative to mock. The x-axis is the up-regulated genes number, and y-axis is the occurrence of every number in total 1 million times. In detail, we identified 1984 potential miR-24-1 targeted enhancers through H3K27ac histone ChiP-seq data and miRanda prediction. A total number of 179 upregulated genes revealed by microarray assay are located within ±100Kb of these enhancers. As the control, we created a background by randomly sampling sets of mock regions which has same length with targeted enhancers for 1 million times and computing the up-regulated genes number within ±100Kb of every mock region. Distribution of the upregulated genes number in the background is plotted as the black curve. Up-regulated genes number for potential miR-24-1 targeted enhancers is significant (P-value = 0.000878, red line) relative to our background model. (D) As shown by ChIP-seq boxplots, H3K27ac, H3K4me1 and Pol II was enriched while H3K9me3 decreased in the predicted miR-24-1 target region when miR-24 overepressed. (E) An example of . Genomic locus of the predicted miR-24-1 target region overlapped with the strong enhancer (blue, left). After miR-24-1 overexpressed, H3K27ac, H3K4me1 and Pol II was enriched while H3K9me3 decreased. LSMD1 (pink shadow) and CYB5D1 (yellow shadow) became upregulated as RNA-seq analysis shown. Also by microarray data, LSMD1 appears to be up-regulated. (F) Schematic of TALENs design for miR-24-1 targeted locus predicted by miRanda overlapped with an enhancer (blue). (G–H) miR-24-1 target site deletion abolished neighboring gene activation. FBP1 in red box as a positive control. All values are normalized to GAPDH and error bars show mean ± SEM for 3 biological replicates *P < 0.05, **P < 0.01, ***P < 0.001 by 2-tailed student's t-test.
Discussion
MicroRNA induced translation repression results in gene silencing, and mainly occurs in the cytoplasm. However, miRNAs also exist in the nucleus, leaving us to question the precise function of nuclear miRNAs. Recent reports have demonstrated that miRNA or small RNACitation38,39 may be able to up-regulate transcriptionCitation6,7 through the interaction between miRNAs and promoters, as well as enhance translation.Citation40 Our work on the function of enhancer-associated miRNA presents a somewhat novel concept.
In our research, we found that a subset of miRNAs deriving from the enhancer loci is capable of activating transcription, and these miRNAs are associated with active enhancers. Choosing miR-24-1 for further investigation, we found it function as unconventional mediators for transcriptional gene activation through chromatin remodeling at enhancer regions. Meanwhile, when it is within the cytoplasm, miR-24-1 would still be able to function canonically as a repressor for its target genes (Fig. S2F and Fig. S5B). In knowing this, one would visualize that miRNA carries a dual function ability, activation in the nucleus as well as repression in the cytoplasm (Fig. S6). The first miRNA, lin-4, was discovered 30 years ago and is an inhibitor of lin-14 by targeting its 3′UTR in the cytoplasm.Citation41,42 This model, was later predominantly applied to the understanding of the miRNA-mRNA regulation network among diverse organismal systems.Citation2,43,44 Our work has identified that miRNAs can function as an unconventional mediator for transcriptional gene activation through chromatin remodeling at enhancer regions. The positive and negative modulating effects of miRNAs in gene regulation may both contribute to gene expression, our findings demonstrate that miRNAs can function as an activator. Thus, we expect that miRNA activated gene transcription may represent a novel mechanism.
Author contributions
W.Y. and M.X. conceived the project and planned experiments; M.X. performed most of them; J.L. W.L and Y.X performed computational analyses of ChIP-seq, RNA-seq and miRNA target prediction. F.W. assisted in computational analyses; Plasmid construction, Transfection and qPCR were contributed by C.D. and Y.W.; L.Z. and Y.X. performed western blot; H.L. assisted in NOMe-seq; Y.L. assisted in ChIP-seq library preparations and sequencing; L.P. assisted in RNA-seq library preparations; the project was supervised by W.Y.; W.Y. and M.X. wrote the manuscript with contributions from other authors.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_Datas.zip
Download Zip (37.2 MB)Acknowledgments
We thank Yang Shi for valuable suggestions and results interpretation. We thank colleagues, Alastair Murchie, Hengmi Cui and Emily Niemitz, have reviewed the final version of the manuscript. We also would like to thank Yue Yu for critical reading and revising of the manuscript and Shanhe Yu for assistance with figure preparation. We also want to thank Zhentian Wang, Zhihu Zhao, Kankan Wang, Xianwen Yang, Qijun Qian for assistance of experiments; Gang Wei and Ruitu Lv's help for statistical analyses; Jia Guo assistance in Confocal Microscope; Lu Chen, Bo Wen for providing the pLenti-EF1a-IRES-EGFP construct and assistance with lentivirus package; Gangning Liang for suggestions on NOMe-seq; Jin Li for Confocal Microscope immaging and providing MEF cells. We also need to thank all the members in Yang Shi group in Fudan University for support and discussions. This work was supported by the grant from National Natural Science Foundation of China (31271355), National Basic Research Program of China 973 Program (2012CB517606), National Natural Science Foundation of China (81272392), the Major Project of Basic Research of Technology Committee in Shanghai of China (12DJ1400200), National Basic Research Program of China 973 Program (2009CB825603).
Related Research Data
References
- Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem 2010; 79:351-79; PMID:20533884; https://doi.org/10.1146/annurev-biochem-060308-103103
- Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet 2012; 13:271-82; PMID:22411466; https://doi.org/10.1038/nrg3162
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116:281-97; PMID:14744438; https://doi.org/10.1016/S0092-8674(04)00045-5
- Matsui M, Chu Y, Zhang H, Gagnon KT, Shaikh S, Kuchimanchi S, Manoharan M, Corey DR, Janowski BA. Promoter RNA links transcriptional regulation of inflammatory pathway genes. Nucleic Acids Res 2013; 41:10086-109; PMID:23999091; https://doi.org/10.1093/nar/gkt777
- Majid S, Dar AA, Saini S, Yamamura S, Hirata H, Tanaka Y, Deng G, Dahiya R. MicroRNA-205-directed transcriptional activation of tumor suppressor genes in prostate cancer. Cancer 2010; 116:5637-49; PMID:20737563; https://doi.org/10.1002/cncr.25488
- Huang V, Place RF, Portnoy V, Wang J, Qi Z, Jia Z, Yu A, Shuman M, Yu J, Li LC. Upregulation of Cyclin B1 by miRNA and its implications in cancer. Nucleic Acids Res 2012; 40:1695-707; PMID:22053081; https://doi.org/10.1093/nar/gkr934
- Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci U S A 2008; 105:1608-13; PMID:18227514; https://doi.org/10.1073/pnas.0707594105
- Guo D, Barry L, Lin SS, Huang V, Li LC. RNAa in action: from the exception to the norm. RNA Biol 2014; 11:1221-5; PMID:25602906; https://doi.org/10.4161/15476286.2014.972853
- Turner MJ, Jiao AL, Slack FJ. Autoregulation of lin-4 microRNA transcription by RNA activation (RNAa) in C. elegans. Cell Cycle 2014; 13:772-81; PMID:24398561; https://doi.org/10.4161/cc.27679
- Huang V, Zheng J, Qi Z, Wang J, Place RF, Yu J, Li H, Li LC. Ago1 Interacts with RNA polymerase II and binds to the promoters of actively transcribed genes in human cancer cells. PLoS Genet 2013; 9:e1003821; PMID:24086155; https://doi.org/10.1371/journal.pgen.1003821
- Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA. Interchromosomal associations between alternatively expressed loci. Nature 2005; 435:637-45; PMID:15880101; https://doi.org/10.1038/nature03574
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. An atlas of active enhancers across human cell types and tissues. Nature 2014; 507:455-61; PMID:24670763; https://doi.org/10.1038/nature12787
- Nord AS, Blow MJ, Attanasio C, Akiyama JA, Holt A, Hosseini R, Phouanenavong S, Plajzer-Frick I, Shoukry M, Afzal V, et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 2013; 155:1521-31; PMID:24360275; https://doi.org/10.1016/j.cell.2013.11.033
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013; 153:307-19; PMID:23582322; https://doi.org/10.1016/j.cell.2013.03.035
- De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei CL, Natoli G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol 2010; 8:e1000384; PMID:20485488; https://doi.org/10.1371/journal.pbio.1000384
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010; 465:182-7; PMID:20393465; https://doi.org/10.1038/nature09033
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 2013; 498:516-20; PMID:23728302; https://doi.org/10.1038/nature12210
- Lam MT, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, Benner C, Kaikkonen MU, Kim AS, Kosaka M, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 2013; 498:511-5; PMID:23728303; https://doi.org/10.1038/nature12209
- Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010; 143:46-58; PMID:20887892; https://doi.org/10.1016/j.cell.2010.09.001
- Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, Shiekhattar R. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 2013; 494:497-501; PMID:23417068; https://doi.org/10.1038/nature11884
- Di Ruscio A, Ebralidze AK, Benoukraf T, Amabile G, Goff LA, Terragni J, Figueroa ME, De Figueiredo PL, Alberich-Jorda M, Zhang P, et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 2013; 503:371-6; PMID:24107992; https://doi.org/10.1038/nature12598
- Garcia PB, Cai A, Bates JG, Nolla H, Schlissel MS. miR290-5p/292-5p activate the immunoglobulin kappa locus in B cell development. PLoS One 2012; 7:e43805; PMID:22928038; https://doi.org/10.1371/journal.pone.0043805
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature 2012; 489:101-8; PMID:22955620; https://doi.org/10.1038/nature11233
- Roberts TC. The MicroRNA biology of the mammalian nucleus. Mol Ther Nucleic Acids 2014; 3:e188; PMID:25137140; https://doi.org/10.1038/mtna.2014.40
- Chen D, Fu LY, Zhang Z, Li G, Zhang H, Jiang L, Harrison AP, Shanahan HP, Klukas C, Zhang HY, et al. Dissecting the chromatin interactome of microRNA genes. Nucleic Acids Res 2014; 42:3028-43; PMID:24357409; https://doi.org/10.1093/nar/gkt1294
- Dvinge H, Git A, Graf S, Salmon-Divon M, Curtis C, Sottoriva A, Zhao Y, Hirst M, Armisen J, Miska EA, et al. The shaping and functional consequences of the microRNA landscape in breast cancer. Nature 2013; 497:378-82; PMID:23644459; https://doi.org/10.1038/nature12108
- Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 2012; 148:84-98; PMID:22265404; https://doi.org/10.1016/j.cell.2011.12.014
- Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, Zhang F. A transcription activator-like effector toolbox for genome engineering. Nat Protoc 2012; 7:171-92; PMID:22222791; https://doi.org/10.1038/nprot.2011.431
- Mulepati S, Heroux A, Bailey S. Structural biology. Crystal structure of a CRISPR RNA-guided surveillance complex bound to a ssDNA target. Science 2014; 345:1479-84; PMID:25123481; https://doi.org/10.1126/science.1256996
- Castanotto D, Lingeman R, Riggs AD, Rossi JJ. CRM1 mediates nuclear-cytoplasmic shuttling of mature microRNAs. Proc Natl Acad Sci U S A 2009; 106:21655-9; PMID:19955415; https://doi.org/10.1073/pnas.0912384106
- Nishi K, Nishi A, Nagasawa T, Ui-Tei K. Human TNRC6A is an Argonaute-navigator protein for microRNA-mediated gene silencing in the nucleus. Rna 2013; 19:17-35; PMID:23150874; https://doi.org/10.1261/rna.034769.112
- Tang R, Li L, Zhu D, Hou D, Cao T, Gu H, Zhang J, Chen J, Zhang CY, Zen K. Mouse miRNA-709 directly regulates miRNA-15a/16-1 biogenesis at the posttranscriptional level in the nucleus: evidence for a microRNA hierarchy system. Cell Res 2012; 22:504-15; PMID:21862971; https://doi.org/10.1038/cr.2011.137
- Zisoulis DG, Kai ZS, Chang RK, Pasquinelli AE. Autoregulation of microRNA biogenesis by let-7 and Argonaute. Nature 2012; 486:541-4; PMID:22722835; https://doi.org/10.1038/nature11134
- Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 2011; 474:390-4; PMID:21572438; https://doi.org/10.1038/nature10006
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 2010; 107:21931-6; PMID:21106759; https://doi.org/10.1073/pnas.1016071107
- Kelly TK, Liu Y, Lay FD, Liang G, Berman BP, Jones PA. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res 2012; 22:2497-506; PMID:22960375; https://doi.org/10.1101/gr.143008.112
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009; 459:108-12; PMID:19295514; https://doi.org/10.1038/nature07829
- Janowski BA, Younger ST, Hardy DB, Ram R, Huffman KE, Corey DR. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat Chem Biol 2007; 3:166-73; PMID:17259978; https://doi.org/10.1038/nchembio860
- Yue X, Schwartz JC, Chu Y, Younger ST, Gagnon KT, Elbashir S, Janowski BA, Corey DR. Transcriptional regulation by small RNAs at sequences downstream from 3′ gene termini. Nat Chem Biol 2010; 6:621-9; PMID:20581822; https://doi.org/10.1038/nchembio.400
- Zhang X, Zuo X, Yang B, Li Z, Xue Y, Zhou Y, Huang J, Zhao X, Zhou J, Yan Y, et al. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell 2014; 158:607-19; PMID:25083871; https://doi.org/10.1016/j.cell.2014.05.047
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993; 75:843-54; PMID:8252621; https://doi.org/10.1016/0092-8674(93)90529-Y
- Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993; 75:855-62; PMID:8252622; https://doi.org/10.1016/0092-8674(93)90530-4
- Ambros V. The functions of animal microRNAs. Nature 2004; 431:350-5; PMID:15372042; https://doi.org/10.1038/nature02871
- Shenoy A, Blelloch RH. Regulation of microRNA function in somatic stem cell proliferation and differentiation. Nat Rev Mol Cell Biol 2014; 15:565-76; PMID:25118717; https://doi.org/10.1038/nrm3854