
ABSTRACT
Expression of the miR-34 family (miR-34a, -34b, -34c) is elevated in settings of heart disease, and inhibition with antimiR-34a/antimiR-34 has emerged as a promising therapeutic strategy. Under chronic cardiac disease settings, targeting the entire miR-34 family is more effective than targeting miR-34a alone. The identification of transcription factor (TF)-miRNA regulatory networks has added complexity to understanding the therapeutic potential of miRNA-based therapies. Here, we sought to determine whether antimiR-34 targets secondary miRNAs via TFs which could contribute to antimiR-34-mediated protection. Using miRNA-Seq we identified differentially regulated miRNAs in hearts from mice with cardiac pathology due to transverse aortic constriction (TAC), and focused on miRNAs which were also regulated by antimiR-34. Two clusters of stress-responsive miRNAs were classified as “pathological” and “cardioprotective,” respectively. Using ChIPBase we identified 45 TF binding sites on the promoters of “pathological” and “cardioprotective” miRNAs, and 5 represented direct targets of miR-34, with the capacity to regulate other miRNAs. Knockdown studies in a cardiomyoblast cell line demonstrated that expression of 2 “pathological” miRNAs (let-7e, miR-31) was regulated by one of the identified TFs. Furthermore, by qPCR we confirmed that expression of let-7e and miR-31 was lower in hearts from antimiR-34 treated TAC mice; this may explain why targeting the entire miR-34 family is more effective than targeting miR-34a alone. Finally, we showed that Acsl4 (a common target of miR-34, let-7e and miR-31) was increased in hearts from TAC antimiR-34 treated mice. In summary, antimiR-34 regulates the expression of other miRNAs and this has implications for drug development.
Abbreviations
| Acsl4 | = | Acyl-CoA synthetase long-chain family member 4 |
| Akap6 | = | A-kinase anchor protein 6 |
| Alcam | = | Activated leukocyte cell adhesion molecule |
| ANOVA | = | Analysis of Varience |
| ChIP-Seq | = | Chromatin Immunoprecipitation-Sequencing |
| CTCF | = | CCCTC-binding factor |
| HF | = | Heart failure |
| HNF4A | = | Hepatocyte nuclear factor 4 alpha |
| HW/TL | = | Heart weight/tibia length |
| KEGG | = | Kyoto Encyclopedia of Genes and Genomes |
| LNA | = | Lock Nucleic Acid |
| miR | = | microRNA |
| qPCR | = | quantitative PCR |
| RPM | = | Reads per million |
| Sema4c,4f | = | Semaphorin 4c, Semaphorin 4f |
| Sirt1 | = | Sirtuin 1 |
| SOM | = | Self Organizing Maps |
| Sox2 | = | SRY-box 2 |
| TAC | = | Transverse Aortic Constriction |
| TF | = | Transcription factor |
| 3′-UTR | = | 3′-Untranslated Region |
| Vegf | = | Vascular endothelial growth factor |
Introduction
MicroRNAs (miRNAs) are a class of highly conserved, small (∼22 nucleotides in length) double-stranded non-coding RNAs that play a role in gene regulation. MiRNAs repress gene expression through base pairing to the 3′ untranslated region (3′UTR) of target mRNA and/or interfering with protein synthesis.Citation1 MiRNAs are involved in many biological processes and diseases. In the heart, miRNA expression is dysregulated during pathological processes (reviewed in Citation2-4) and miRNA-based therapies have shown success in attenuation of disease progression in preclinical animal models of heart failure (HF).Citation5-9
A single miRNA can target hundreds of mRNAs, and mRNAs contain binding sites for one or more miRNAs. Using genome-wide computational approaches, Hu et al demonstrated that over 1000 mRNAs were altered in the heart of the disease model of pressure overload, with only 5% of these genes directly regulated by miRNAs.Citation10 The non-direct mRNA targets typically encode for transcription factors (TFs), kinases, phosphatases and proteases, which cause up- and down-regulation of downstream genes, pointing to a complex regulatory network.Citation10 Besides mRNA, miRNAs have also been shown to regulate the expression of other secondary miRNAs in the heart.Citation11-13 MiRNA-mediated miRNA regulation adds an additional complexity to understanding the mechanism of miRNAs and should be taken into account when manipulating miRNAs.
MiR-34a and the entire miR-34 family (miR-34a, -34b, -34c) have received significant attention in the cardiac field as therapeutic targets.Citation5-7,14 Expression levels of the miR-34 family were elevated in hearts of mice with cardiac stressCitation5,6,15 and HF patients.Citation16,17 We have previously inhibited miR-34a and the miR-34 family using locked nucleic acid [LNA]-antimiR chemistry in 2 mouse models with failing hearts i.e. i) transverse aortic constriction (TAC) which induced pressure overload on the heart causing pathological hypertrophy or ii) myocardial infarction (MI, which mimics a heart attack). Inhibition of miR-34a and the miR-34 family attenuated TAC-induced cardiac remodeling and dysfunction,Citation6 however, only inhibition of the entire miR-34 family was effective in more severe cardiac disease settings (a more severe model of pressure overload which induces severe cardiac dysfunction and lung congestion, and the MI model).Citation5,6 Therefore, we propose that drugs which target the entire miR-34 family are likely to have more therapeutic benefit than inhibition of miR-34a alone.
In an increasingly competitive and financially restricted pharmaceutical environment, where potential new treatments must show a clear benefit over approved pharmacological agents, a comprehensive understanding of the complex regulatory networks of any potential treatment will be essential. The goal of this study was to understand the regulatory effects of miR-34 by assessing miRNA profiles in a diseased heart model where inhibition of miR-34 attenuated pathogenesis. We hypothesized that the efficacy of antimiR-34 may be due to its regulation of secondary miRNAs, producing direct and indirect mRNA target effects. The identification of other miRNAs regulated by miR-34 and the delineation of miR-34 interaction partners and signaling pathways will be essential for progressing miR-34 based therapies into the clinic.
Results
miRNA-Seq reveals distinctive miRNA gene expression patterns with cardiac pathology and antimiR-34 treatment
Massive parallel sequencing (miR-Seq) was performed on RNA isolated from heart tissue (ventricle) of 4 groups of mice (Sham control, Sham antimiR-34, TAC control, TAC antimiR-34). Heart tissue had been collected from a previous study in which adult male mice were subjected to either a sham or TAC surgery for 5 weeks, and subcutaneously administered either a LNA-control or LNA-antimiR-34 for 6 weeks.Citation6 Treatment of antimiR-34 in TAC animals for 6 weeks was able to improve cardiac function and attenuate pathological remodeling.Citation6 Thus, the experimental design compares 4 models with different morphological and molecular phenotypes: 1) Sham control: normal heart size and cardiac function, 2) Sham antimiR-34: normal heart size, normal cardiac function and reduced miR-34a, -34b, -34c expression by 80%, 3) TAC control: pathological cardiac hypertrophy, poor cardiac function, increased levels of miR-34a, -34b, -34c and 4) TAC antimiR-34: attenuated pathological hypertrophy and better cardiac function than TAC control, and reduced miR-34a, -34b, -34c expression.Citation6
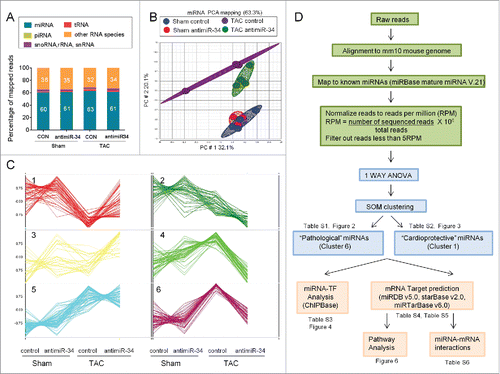
After miR-Seq, reads were mapped to the mouse genome (mm10 assembly). Approximately 62% of reads mapped to miRNA regions () and the remaining reads mapped to other non-coding RNAs such as transfer RNA (tRNA), ribosomal RNA (rRNA), small nucleolar RNA (snoRNA), small nuclear RNA (snRNA) and piwi-interacting RNA (piRNA) (). Two-dimensional Principal Component Analysis (PCA) highlighted differential expression profiles of miRNAs in response to TAC control and TAC antimiR-34 compared to Sham groups (). Following the normalization of reads and subsequent ANOVA analyses, 6 distinct patterns of miRNA expression were identified by Self-Organizing Map (SOM) clustering (). The experimental design, analysis pipeline and selection of miRNAs are outlined in .
Figure 1. MiR-Seq analysis of miRNA expression changes with pressure overload (TAC) and treatment with antimiR-34. (A) Percentage of total reads mapped to non-coding small RNAs and other RNA species identified by deep sequencing. (B) 2D Principal Component analysis plot of 313 miRNAs in Sham control, Sham antimiR-34, TAC control and TAC antimiR-34 hearts. (C) Results of SOM clustering analysis, showing 6 distinct patterns of miRNA expression in heart samples. Vertical scale indicates normalization of mean intensity reads. (D) Work flow of sequencing reads, processing, bioinformatic data analyses and selection of miRNAs of interest.
Classification of “pathological” and “cardioprotective” secondary miRNAs
A total of 60 miRNAs were identified in Cluster 6 () and have been referred to as “pathological” miRNAs (Table S1). These miRNAs showed similar expression patterns to the miR-34 family and represent miRNAs which may contribute to cardiac pathology, and could be therapeutically targeted. The top 9 “pathological” miRNAs in Cluster 6 are presented in and and include; let-7i-5p, miR-21a-5p, let-7e-5p, miR-423-5p, miR-27b-5p, miR-31-5p, miR-434-3p, miR-24-1-5p, and miR-300-3p. These 9 miRNAs were increased in the TAC control group, blunted in the TAC antimiR-34 group (Table S1, ), and positively correlated with pathological cardiac hypertrophy (i.e., HW/TL, ).
Figure 2. “Pathological” miRNAs are upregulated in TAC control vs. Sham control, and their upregulation blunted in TAC antimiR-34 versus TAC control. Left: Heat map of mean expression levels for 60 “pathological” miRNAs from Cluster 6, Right: MiRNA-Seq normalized read counts for the top 9 “pathological” miRNAs. *p < 0.05 vs. Sham of the same treatment, †p < 0.05 by 1 way ANOVA and Fisher post hoc test. n = 3–4/group. Data is presented as mean ± SEM.
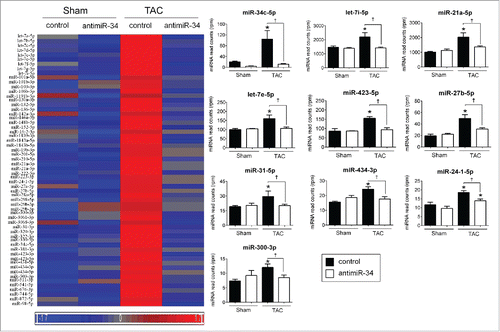
Table 1. “Pathological” and “cardioprotective” miRNAs significantly correlated with HW/TL ratio of Sham control, Sham antimiR-34, TAC control and TAC antimiR-34 mice.
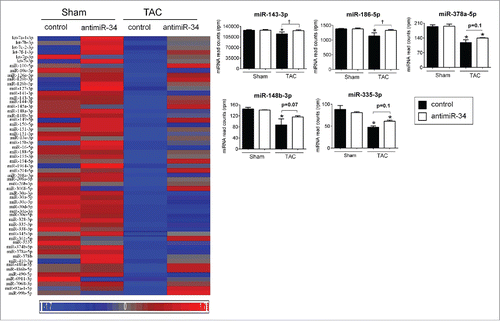
Cluster 1 which included 57 miRNAs () was also of interest, as these miRNAs showed downregulation in TAC control mice, and increased expression in TAC antimiR-34 treated animals (Table S2, ). Cluster 1 miRNAs have been defined as “cardioprotective” miRNAs because upregulation was associated with improved cardiac outcome (i.e. less pathological cardiac hypertrophy in TAC mice administered antimiR-34Citation6) (Table S2, ). The top 5 “cardioprotective” miRNAs in Cluster 1 include miR-143-3p, miR-186-5p, miR-378a-5p, miR-148b-3p, and miR-335-3p (, ). These 5 miRNAs were inversely correlated with heart weight/ tibia length (HW/TL) ().
Figure 3. “Cardioprotective” miRNAs are repressed in TAC control vs. Sham control, and restored, at least partially, with TAC antimiR-34 versus TAC control. Left: Heat map of mean expression levels for 57 “cardioprotective” miRNAs from Cluster 1, Right: MiRNA-Seq normalized read counts for the top 5 “cardioprotective” miRNAs. *p < 0.05 vs. Sham of the same treatment, †p < 0.05 by 1 way ANOVA and Fisher post hoc test. n = 3–4/group. Data is presented as mean ± SEM.
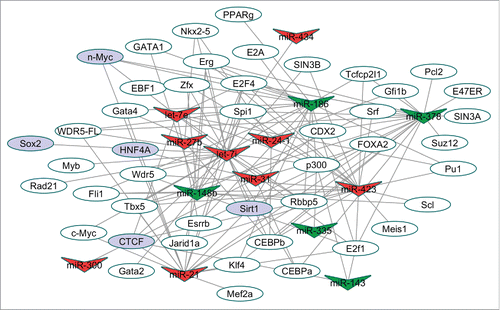
Modeling TF-miRNA regulatory networks revealed specific modulation by the miR-34 family
One mechanism by which miRNAs can regulate other (secondary) miRNAs is by binding to direct target genes which play roles as TFs.Citation10 To interrogate secondary miRNA interactions we devised a strategy to look for TF binding sites in the promoter region of the 9 “pathological” and 5 “cardioprotective” miRNAs which were regulated by antimiR-34 (). The TF-binding sites were determined based on miRNA promoters (5kb upstream and 1kb downstream of Transcription Start Site) within the TF ChIP-Seq datasets using ChIPBaseCitation18, and we constructed TF-miRNA regulatory networks. We identified a total of 134 TF-miRNA interactions involving 45 TFs and the 9 “pathological” and 5 “cardioprotective” miRNAs (). Detailed TF-miRNA interactions are listed in Table S3. illustrates the complexity of miRNA-TF interactions; one miRNA can be regulated by different TFs and one specific TF can modulate the expression of multiple target miRNAs. Five of the 45 TFs are verified as experimentally validated miR-34 target genes based on previous literature. Sirt1/SIRT1 has been validated as a target by qPCR and Western Blot in mouse hearts and human cancer cells, respectively.Citation6,19 Using luciferase assays, the promoters of HNF4α and SOX2 were negatively regulated by miR-34.Citation20,21 The TF CCCTC-binding factor (CTCF) is also a validated target of miR-34a based on reporter assays showing that miR-34a represses CTCF expression.Citation22 Finally, protein expression of N-Myc was inversely correlated to miR-34 in miR-34 knockout animals.Citation21 Collectively, these results implicate the miR-34 family in modulating the expression of other miRNAs via its target TFs.
Figure 4. TF-miRNA regulatory network. A total of 134 TF-miRNA interactions involving 45 TF and 14 miRNAs were collected for this network. V-shaped nodes correspond to the miRNAs, red V-shaped nodes are “pathological” miRNAs and green V-shaped nodes are “cardioprotective” miRNAs. The oval nodes represent TFs, while purple oval nodes represent validated miR-34 targets. TF binding sites were determined from ChIP-Seq data from miRNA promoters (defined as 5kb upstream and 1kb downstream) as defined from ChIPBase. Verified miR-34 targets are based on previous experimental reports.
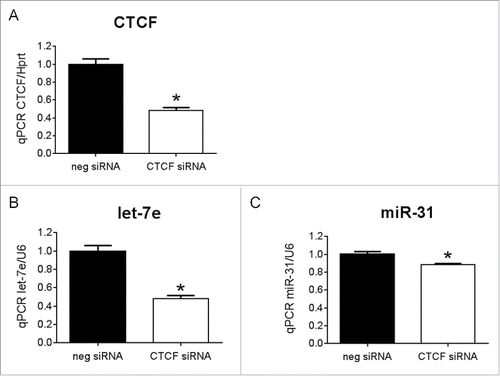
To provide further evidence that TF targets of miR-34 can regulate other miRNAs, we assessed a TF-miRNA interaction in vitro by transfecting CTCF siRNA into the cardiomyoblast H9c2 cell line. qPCR confirmed knockdown of CTCF by ∼50% compared to the negative control siRNA ( p < 0.05, ). Knockdown of CTCF was accompanied by a decrease in the expression levels of let-7e () and miR-31 ().
Figure 5. The transcription factor, CTCF, regulates let-7e and miR-31 (A) qPCR for CTCF in H9c2 cells transfected with CTCF siRNA or negative control siRNA (neg siRNA). N = 3/group *p < 0.05 vs. neg siRNA by unpaired t-test. qPCR for (B) let-7e and (C) miR-31 in H9c2 cells transfected with CTCF siRNA or neg siRNA, N = 3/group, *p < 0.05 vs. neg siRNA by unpaired t-test. Data is presented as mean ± SEM.
Pathways targeted by “pathological” and “cardioprotective” miRNAs
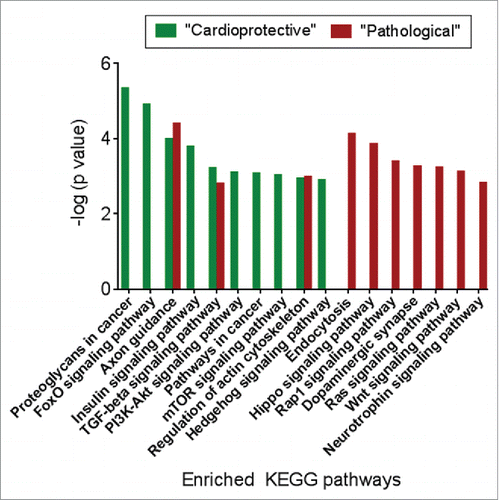
Next, we interrogated mRNA targets of the 9 “pathological” and 5 “cardioprotective” miRNAs (Table S4, Table S5) against KEGG pathways for enriched themes. These analyses highlighted distinct and differential enrichment patterns (). Pathways selectively identified for “cardioprotective” miRNAs included proteoglycans in cancer, FoxO signaling, insulin signaling, PI3K-Akt signaling, pathways in cancer, mTOR signaling, hedgehog signaling (all pathways, P < 0.01). In contrast, pathways associated with “pathological” miRNAs included endocytosis, Hippo signaling, Rap1 signaling, dopaminergic synapse, Ras signaling, Wnt signaling and Neutrophin signaling (all pathways, P<0.01) ().
Figure 6. The target genes of “pathological” and “cardioprotective” miRNAs are enriched for common and distinct functional pathways. Enriched pathways targeted by “pathological” miRNAs are shown in red, while pathways enriched for mRNA targets of “cardioprotective” miRNAs are shown in green.
Integrated analysis to identify miRNA-mRNA interactions
Previous studies have identified targets of miR-34a and the miR-34 family that have been implicated in providing cardiac protection with antimiR treatment by regulating processes including angiogenesis, telomere shortening, myocyte stability, cell survival, cardiac repair and regeneration.Citation6,7 Validated targets include vascular endothelial growth factors (VEGFs), vinculin, protein O-fucosyltranferase 1, Notch1, protein phosphatase 1, regulatory subunit 10 (PPP1R10; also known as PNUTS), sirtuin 1, Cyclin D1 and B-cell lymphoma 2.Citation6,7 Here, we were interested in assessing whether the cluster of “pathological” miRNAs () had common targets to the targets of the miR-34 family. We hypothesized that multiple miRNAs regulating the same target would have a larger effect than a single miRNA regulating the target, as previously reported.Citation23 The 9 “pathological” miRNAs shared a number of common predicted targets with the miR-34 family (Table S6). Akap6 contains binding sites for let-7i, let-7e, miR-21a and miR-34 (Table S6), while miR-423 is predicted to target Vegfa, a validated target of miR-34.
Let-7e and miR-31 were decreased in TAC mice treated with antimiR-34 but not in TAC mice treated with antimiR-34a
Previously, we used 2 antimiRs of different length (15-mer vs. 8- mer) targeting miR-34a/miR-34 in the pressure overload model (TAC). The 15-mer antimiR inhibits miR-34a alone whereas the 8-mer antimiR inhibits the entire miR-34 family (including miR-34a, -34b and -34c). We have previously demonstrated that the 8-mer antimiR was more effective at inhibiting pathological remodeling than the 15-mer antimiR.Citation5,6 One possible explanation for the reduced efficacy with the 15-mer antimiR is due to the increased expression of miR-34b and miR-34c. An additional explanation may be due to the regulation of other key miRNAs by antimiR-34 (but not antimiR-34a) important for cardiac protection. To assess this possibility, we selected 2 miRNAs from the “pathological” miRNA cluster (): miR-31 and let-7e. miR-31 was selected because: i) it was induced by pressure overload in the current study as well as a previous study, Citation24 and ii) miR-31 was upregulated in hearts of mice subjected to ischemia reperfusion, and an inhibitor of miR-31 (antagomiR-31) reduced infarct size in this model.Citation25 Let-7e was selected because i) the let-7 family represents a conserved and abundant miRNA in the heart, ii) let-7e was shown to be upregulated in patients with aortic stenosis and heart failure,Citation17,26 iii) let-7e was also found to be upregulated in mouse hearts subjected to MI,Citation27 and iv) there was a strong linear correlation between let-7e expression and a measure of pathological cardiac hypertrophy (HW/TL) and cardiac function (assessed by fractional shortening) ().
Figure 7. Secondary miRNAs regulated by inhibiting miR-34 have targets common to miR-34. (A) Linear correlation between let-7e expression and HW/TL ratios and fractional shortening, p < 0.05 for all correlations, (B) qPCR of let-7e and miR-31 in hearts of TAC control and TAC antimiR-34 treated mice. N = 6/group *p < 0.05 vs. TAC control by unpaired t-test. (C) qPCR of let-7e and miR-31 in hearts of TAC control and TAC antimiR-34 treated mice. N = 4–5/group. CON-control. (D) Venn diagram of common predicted targets for miR-34, let-7e and miR-31. (E) qPCR of Acsl4 in hearts of TAC control and TAC antimiR-34 treated mice. N = 6 = 8/group *p < 0.05 vs. TAC control by unpaired t-test. Data is presented as mean ± SEM.
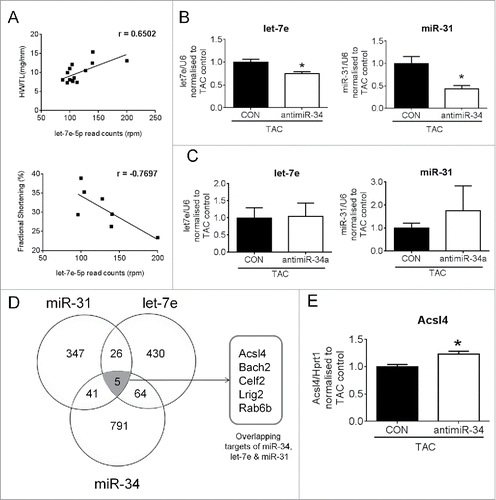
Next, we performed qPCR to examine whether miR-31 and let-7e were differentially regulated with the 15-mer antimiR-34a treatment versus the 8-mer antimiR-34 treatment in a setting of TAC. Consistent with the miRNA-Seq data (), let-7e and miR-31 expression levels were lower in antimiR-34 treated TAC mice vs. TAC control mice by qPCR (). By contrast, expression of let-7e and miR-31 was not different in TAC mice treated with 15-mer antimiR-34a compared to TAC control mice (), suggesting that the regulation of let-7e and miR-31 may be required by antimiR-34 to mediate cardiac protection. Using target prediction databases (miRDB, starBase or miRTarBase) we found miR-34, miR-31 and let-7e share 5 common targets (, Table S6). One of the 5 predicted targets was Acyl-CoA synthetase long-chain family member 4 (Acsl4) (, Table S6). Acsl4 was of interest to validate because it has relatively high expression in the heart and may regulate cardiac metabolism.Citation28,29 By qPCR, expression of Acsl4 was increased in hearts of TAC mice treated with antimiR-34 versus TAC control mice ().
Discussion
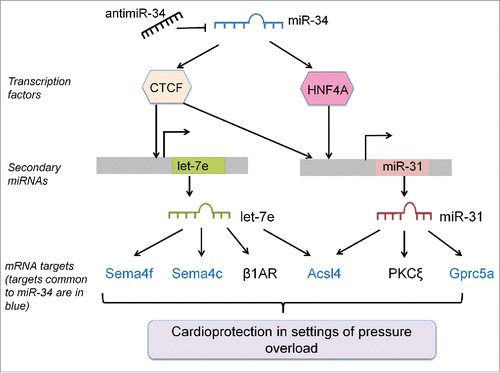
Our understanding of how miRNAs regulate other miRNAs in the heart is currently limited, Citation11-13 but potentially has significant implications for the therapeutic potential and possible side effects of any miRNA-based therapy. In the present study, we report that antimiR-34 therapy regulates the expression of numerous miRNAs in the heart, in addition to the direct targets i.e., miR-34a, -34b, -34c. Based on the evidence presented in this paper, we propose a mechanism showing how “secondary” miRNAs are regulated with antimiR-34 treatment (). miR-34 regulates numerous direct targets including TFs. TFs then have the potential to regulate the expression of other miRNAs. These miRNAs have both common and additional targets to miR-34. We have also identified 2 miRNAs within the pathological cluster (miR-31 and let-7e) which are likely to explain, at least in part, why antimiR-34 treatment was more effective than antimiR-34a in a cardiac disease setting.Citation5,6
Figure 8. Summary Figure- AntimiR-34 therapy regulates secondary miRNAs via interactions with TFs, and secondary miRNAs have effects via targets common and distinct to miR-34. MRNA targets of secondary miRNAs that are common to miR-34 are shown in blue, and mRNA targets specific to secondary miRNAs are in black.
MiRNA-mediated miRNA regulation in the heart has previously been reported between miRNAs that are encoded in the introns of myosin heavy chain genes.Citation12,13 In another study, the expression of 15–30 miRNAs were differentially regulated in hearts from mice overexpressing miR-378 or miR-499.Citation11 Regulation of the “secondary” miRNAs had a modulatory effect on cardiac gene expression, explaining direct and indirect mRNA targets for miR-378 and miR-499.Citation11 While these previous studies looked at miRNA-mediated regulation in a basal setting, to our knowledge, our study is the first to identify miRNA-mediated regulation in a cardiac stress model incorporating a therapeutic intervention targeting miR-34. Collectively, the current study, together with previous reports, highlights that miRNA signaling is non linear and more complex than originally anticipated.
We identified 2 clusters of “secondary” stress responsive miRNAs in the TAC model. “Pathological” miRNAs followed a very similar expression profile to members of the miR-34 family i.e. expression was elevated with TAC and attenuated with antimiR-34 treatment. In contrast, “cardioprotective” miRNAs were defined as those displaying an inverse relationship i.e., expression was decreased with TAC and completely or partially restored with antimiR-34 treatment. summarizes the role of “pathological” miRNAs in the heart based on previous reports. Of interest, a number of these miRNAs have also been found to be elevated in the human failing heart and other cardiac stress models in rodents (). Consistent with this cluster representing “pathological” miRNAs, overexpression of miR-27b was shown to augment pathological cardiac hypertrophy, and this was associated with cardiac dysfunction.Citation30 The role of other “pathological” miRNAs is more complex and may depend on the specific cardiac insult. For instance, inhibition of miR-21 in a pressure overload mouse model was beneficial,Citation31 but was also shown to be required for ischemic preconditioned-induced cardioprotection against ischemia/reperfusion.Citation32 In general, less is currently known about the “cardioprotective” miRNAs identified in Cluster 1 (). However, one example which is in alignment with this cluster representing “cardioprotective” miRNAs is miR-378. miR-378 was previously shown to be decreased in the heart in a setting of cardiac pathology due to TAC or MI, and restoration inhibited TAC-induced pathological cardiac hypertrophy.Citation33 Other miRNAs presented in have not been specifically assessed in the heart and may represent promising targets.
Table 2. List of “pathological” miRNAs (upregulated during TAC and decreased in TAC LNA-antimiR34 treatment) and their role in the heart based on previous reports.
Table 3. List of “cardioprotective” miRNAs (decreased during TAC and upregulated in TAC LNA-antimiR34 treatment) and their role in the heart based on previous reports.
As previously discussed, miRNAs are products of transcription and thus may contain binding sites for TFs to modulate their expression.Citation34 In our study, antimiR-34 treatment in a TAC setting regulated a number of other miRNAs. Since miR-34 targets mRNAs that encode for TFs, we propose that these TFs bind to the promoter and regulate the expression of other miRNAs. We bioinformatically identified putative TF binding sites on the promoters of miR-34 regulated miRNAs. A Sirt1 binding site was identified on the promoters of let-7i, miR-21a, miR-186 and miR-378. Therefore, the up- or downregulation of these miRNAs by miR-34 may be mediated via its direct targeting of Sirt1. Similarly, the expression of miR-423, let-7i, miR-21, let-7e, miR-31, miR-300 and miR-143 can be regulated by miR-34 via its direct target TF, CCCTC-binding factor (CTCF). In the current study, we examined the interaction of CTCF with miR-31 and let-7e by knocking down CTCF with siRNA in the cardiomyoblast H9c2 cell line (). Consistent with our working model (), decreased expression of CTCF was associated with changes in the expression of let-7e and miR-31. It is also noteworthy that some miRNAs (such as let-7i, miR-378 and others) have binding sites for several TFs. Similar to gene regulation, there is extra complexity as multiple TFs cooperate synergistically with one another to regulate miRNA activity.
Individually, miR-34 can exert subtle effects on its targets, but the functional networks of antimiR-34 therapy is likely due to the combinatorial effects of these stress responsive miRNAs working together. Table S6 shows that the mRNA targets of miR-34 are also targets for other “pathological” miRNAs. We have previously validated Semaphorin 4b as a validated target of miR-34, which was positively correlated with cardiac function in mice subjected to TAC.Citation6 In this study, we identified other semaphorins (Sema4f and Sema4c) to be targets of miR-34 and let-7e (Table S6, ). In another example of multiple miRNAs regulating the same targets, vinculin is not only suppressed by miR-34 but also by miR-21, and Alcam is a predicted target of miR-31, miR-434 and miR-34 (Table S6). These mRNA targets have known functional effects on pathways that could explain the cardioprotective properties of antimiR-34 therapy. The current results also suggest that the therapeutic benefit of antimiR-34 over antimiR-34a is in part due to the regulation of “secondary” miRNAs. Two “pathological” miRNAs (let-7e and miR-31) were attenuated in antimiR-34 treated TAC hearts but not antimiR-34a treated TAC hearts. Previous literature demonstrates that the expression of both miRNAs is elevated under cardiac stress settings, and silencing miR-31 was beneficial in an ischemia/reperfusion model (). Using prediction target databases we identified 5 mRNA targets which were common to miR-34a, let-7e and miR-31. One of these targets, Acsl4, was validated experimentally in cardiac tissue from the TAC model. A previous study also validated Acsl4 as a target of miR-34a by using a biotin pull down method which captures miRNA-bound mRNAs.Citation35 Acsl4 was of interest in the current study because it has relatively high abundance in the heart and has the potential to have an impact on energy supply to the heart.Citation28,29 The adult heart derives most of its energy from the oxidation of fatty acids, and a key process which allows fatty acids to be oxidised involves Acsl family members converting long chain fatty acids into long chain fatty acyl-CoAs.Citation36 The specific role of Acsl4 in the heart remains unclear.Citation28,29 However, interestingly, expression of Ascl4 was shown to be depressed in pig hearts which had undergone hypertrophy in response to a high-fat, high-sucrose diet which induced obesity.Citation37
A limitation of the present study is that mRNA-Seq data was not available. However, subsequent validation of differential expression of miRNAs in antimiR-treated mice (i.e. let-7e and miR-31) was based on interactions of miR-34 with experimentally validated TFs (CTCF and HNF4A), and we subsequently showed that CTCF was able to regulate the expression of let-7e and miR-31 (). Another limitation is the use of pharmacological approaches to inhibit miR-34a (15-mer antimiR) or the miR-34 family (8-mer antimiR). While the use of pharmacological tools is considered relevant for examining potential therapeutic effects, it is recognized that pharmacological approaches have the potential to have off-target effects. Furthermore, an 8-mer antimiR could potentially have more off-target effects than a 15-mer antimiR due to the shorter sequence. However, based on the original extensive characterization of the 8-mer LNA-antimiR chemistry, we consider off-target effects to be minimal. In this earlier report, a number of analyses were used to show that the 8-mer LNAs do not have significant off-target effects (e.g. Sylamer analysis of microarray, comprehensive proteomics including isobaric tag labeling of peptides combined with liquid chromatography and tandem mass spectrometry, and a Renilla luciferase reporter gene model).Citation38 In addition, we consider it unlikely that antimiR-34 had non-specific effects on other miRNAs (e.g., miR-31) because the antimiR sequence targeting the seed region of miR-34 is very specific. As an example, the alignment between the sequence for antimiR-34 and miR-31 or let-7e is very poor ().
Figure 9. Alignment of antimiR-34 with sequences of the miR-34 family (a,b,c) and “secondary” miRNAs (let-7e and miR-31). * highlight exact alignment between sequences.

Finally, the current study also has significant implications for other diseases in which members of the miR-34 family are dysregulated, and this aberrant expression is being actively targeted with miRNA therapeutics. For example, in contrast to cardiac pathology, the expression of miR-34 family members is down-regulated in a broad range of cancers.Citation39 A miR-34 mimic (MRX34, to increase miR-34 expression) is currently being evaluated by Mirna Therapeutics in a phase 1 clinical trial for patients with liver cancer.Citation40 As observed for antimiR-34 in the present study, a miR-34 mimic is also likely to have an effect on “secondary” miRNAs in addition to miR-34.
In summary, our results highlight that in addition to mRNA targets, miR-34 can also regulate other miRNAs, and this additional layer of complexity should be taken into account when understanding miRNA-mRNA interactomes and key networks in mediating cardiac stress and protection. Here, we demonstrate that therapeutic inhibition of the miR-34 family leads to the regulation of “secondary” miRNAs in the heart, and differential expression of these miRNAs is likely to contribute to the cardiac protection observed with antimiR-34 therapy (). With the successful completion of a clinical trial testing a miRNA-based therapy for the treatment of hepatitis C virus,Citation41 there has been an explosion of preclinical studies assessing the potential of miRNA-based therapies for multiple diseases. Great caution and care needs to be taken to understand the secondary effects and implications of targeting miRNAs because these “secondary” effects have the potential to significantly impact the desired therapeutic effects, as well as leading to potential side effects.
Materials and methods
Experimental design-animal models
Ventricles were collected from a previous study in which adult (12 weeks old) male FVB/N mice were subjected to either a sham or pressure overload surgery (TAC) for 5 weeks, and subcutaneously administered either a LNA-control (random sequence with no functional effect Citation38) or LNA-antimiR-34 for 6 weeks.Citation6
RNA extraction
Total RNA was extracted from ventricles of mice (n = 3–4) and H9c2 cells using TRIzol® Reagent (Thermo Fisher) according to the manufacturer's instructions. RNA concentration and purity was assessed by Nanodrop® ND-1000 UV-Vis Spectrophotometer (Nanodrop Technologies) and integrity (28S:18S ratio) by a total RNA kit using the Shimadzu MultiNA (Shimadzu). Samples with a RIN of >8 were used for experiments.
MicroRNA-Seq for miRNA expression analysis
Standard miRNA-Seq libraries were prepared according to the manufacturer's instructions (Illumina). Samples were multiplexed (24 samples per lane) and sequenced on 3 flowcell lanes on the Illumina Genome Analyzer HiSeq 2000 (read length: 50bp, Ramaciotti Center for Genomics, UNSW). MiRNA-Seq reads were aligned to the mouse reference genome (mm10) using Partek Flow®; version 3.0 (Partek Inc.) with default settings unless otherwise stated. After removing adaptor sequences, the bases of unaligned reads were trimmed for a minimum Phred quality score of 20 (default settings used except minimum read length = 17). Next, fastq files were aligned to the mm10 reference genome with Bowtie 1 (default parameters used except seed mismatch limit = 0, seed length = 22, alignments reported per read = 10) and Bowtie 2 (default parameters except interval between seeds S,1,1.25, ambiguous characters function L,O.15, default reporting mode = false, max alignments reported per read = 10). Bowtie aligner is an open-source software (http://bowtie-bio.sourceforge.net/index.shtml).Citation42 Prealignment and postalignment quality control was performed. Read counts were then filtered for reads with minimum mapping quality of 20 (99% of reads were of good quality).
Calculation of differential gene expression and bioinformatic analyses
The trimmed, aligned and filtered data were transferred to Partek Genomics Suite®, version 6.6 (Partek Inc.) and quantitated to known miRNAs using miRBase mature miRNA version 21 (released 3rd July, 2014). Reads were normalized to reads per million (RPM) and calculated as follows: (Number of sequenced reads / Total reads) X 1,000,000. MiRNA reads containing less than 5 RPM in all groups were subsequently removed. Next, normalized data were subjected to one-way analysis of variance (ANOVA) to examine differential gene expression (unadjusted p value <0.05). PCA, heat maps and SOM clustering Citation43 (map height = 3, map width = 2) were carried out based on default parameters specified in Partek Genomics Suite®, version 6.6 to visualize the data set. Candidate miRNAs were assigned “pathological” and “cardioprotective” based on 3 criteria: 1) p-value <0.05 for TAC control vs. Sham control, 2) fold change between TAC control versus Sham control > 1.5, and 3) significant correlation between miRNA expression with a measure of pathological cardiac hypertrophy (heart weight/tibia length, HW/TL). These analyses led to the identification of 9 “pathological” miRNAs and 5 “cardioprotective” miRNAs.
Transcription factor binding
TF-binding sites in the promoter region of the 9 “pathological” and 5 “cardioprotective” miRNAs were determined based on miRNA promoters (5kb upstream and 1kb downstream of Transcription Start Site) within the TF ChIP-Seq datasets using ChIPBase.Citation18 In addition, we utilized Cytoscape (version 3.2.1) to construct TF-miRNA regulatory networks for visualization.
Target gene prediction
Target genes (mRNAs) of “pathological” vs. “cardioprotective” miRNAs were extracted from miRDBCitation44 (Version 5.0, release date: August, 2014 ), starBase Version 2.0Citation45,46 and miRTarBaseCitation47 (Version 6.0, release date: September 2015). The parameters used for starBase is medium stringency (> = 2CLIP-Seq experiments supported the predicted miRNA target site).
Pathway enrichment
Pathway enrichment analysis was carried out based on Fisher's Exact test on the extracted target gene lists (described above) and built on annotated pathways from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Partek Genomics Suite®). Pathways were considered significantly enriched if they had a minimum count threshold of 2 genes and a modified Fischer's exact p-value for gene enrichment of less than 0.05.
Cell culture and transfection assays
H9c2 rat cardiomyoblast cells were maintained at 37°C and 5% CO2 in culture medium of Dulbecco's modified Eagle's medium, supplemented with 10% (v/v) fetal bovine serum, 4.5g/L D-glucose, 4mM L-glutamine and penicillin/streptomycin (100 units/ml). The Silencer-Select predesigned siRNA against rat CTCF were obtained from Ambion (Thermo Fischer Scientific). Nontargeting siRNA were used as a negative control. The siRNA was dissolved in nuclease-free water and transfected using Lipofectamine RNAi max (Thermo Fischer Scientific) according to the manufacturer's protocol. Lipofectamine RNAi max was diluted in Opti-MEM I (Thermo Fischer Scientific) and incubated for 10 minutes at room temperature. siRNA (100nM) were diluted in OptiMEM I medium, and then mixed with the diluted RNAi max. The mixture was incubated 20 minutes at room temperature, before addition to cells, incubated for 24 hours before changing to normal growth medium. RNA was harvested 48 hours after transfection. All transfection conditions were performed in triplicates.
Quantitative RT-PCR (qPCR)
To validate miRNA-Seq data, 50 ng of total RNA was reverse transcribed for each miRNA of interest using a TaqMan MicroRNA Reverse Transcription Kit (Life Technologies, Thermo Fisher Scientific) according to manufacturer's recommendations. qPCR was performed using Taqman MicroRNA Assays and amplified on an Applied Biosystems 7500 real-time PCR instrument according to the manufacturer's instructions. MiRNA expression was normalized against snoU6 and data analyzed using the 2−ΔΔCt algorithm.Citation48
To validate mRNA targets, 2μg of total RNA was reverse transcribed using the High Capacity RNA-to-cDNA kit (Life Technologies, Thermo Fischer Scientific) according to manufacturer's recommendations. qPCR was perfomed using SYBR ® Green PCR Master Mix and amplified on the Quant StudioTM 6 and 7 Flex real-time PCR instrument according to manufacturer's instructions. Hypoxanthine phosphoribosyltransferase 1 (Hprt1) was used to standardize for cDNA concentration and data was analyzed using the 2−ΔΔCt algorithm.Citation48 Primers for gene expression are mouse Acsl4, F: ctgaaagactggcaggaagg, R: aatatcgccagtgcaaaacc ; mouse Hprt1, F: tcctcctcagaccgctttt, R: cctggttcatcatcgctaatc; rat CTCF, F: gccagaaacagctccttgac, R: cgtctggaccagcacagtta and rat Hprt1, F: ggtccattcctatgactgtagatttt, R: caatcaagacgttctttccagtt.
Statistical analyses
Results are presented as mean ± SEM. Statistical significance and p-values were calculated using StatView 5.0.1. One-way ANOVA followed by Fisher's post hoc test was performed to identify differences between groups, unless otherwise stated. Student's unpaired t-test was used when comparing 2 groups. A p-value <0.05 was considered significant. Pearson's correlation was used to correlate miRNA expression with HW/TL ratio (Graphpad Prism 6).
Data access
MiRNA-seq data used for this research has been submitted to the NCBI Gene Expression Omnibus (GEO) under accession number GSE79050.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_Tables_1-6.zip
Download Zip (445.5 KB)Acknowledgement
The authors wish to thank P Kantharidis for technical advice.
Funding
This work was supported by a National Health and Medical Research Council (NHMRC) Project Grant (1045144 to JRM and RCYL) and supported in part by the Victorian Government's Operational Infrastructure Support Program. J.R.M. is an NHMRC Research Fellows (IDs 586604 & 1078985-SRF). J.R.M. was also supported by an Australia Research Council Future Fellowship (FT0001657). RCYL was supported by a University of New South Wales Vice-Chancellor Postdoctoral Fellowship.
Related Research Data
References
- Bernardo BC, Charchar FJ, Lin RC, McMullen JR. A microRNA guide for clinicians and basic scientists: background and experimental techniques. Heart Lung Circ 2012; 21:131-42; PMID:22154518; http://dx.doi.org/10.1016/j.hlc.2011.11.002
- Bernardo BC, Ooi JY, Lin RC, McMullen JR. miRNA therapeutics: a new class of drugs with potential therapeutic applications in the heart. Future Med Chem 2015; 7:1771-92; PMID:26399457; http://dx.doi.org/10.4155/fmc.15.107
- Olson EN. MicroRNAs as therapeutic targets and biomarkers of cardiovascular disease. Sci Transl Med 2014; 6:239ps3; PMID:24898744; http://dx.doi.org/10.1126/scitranslmed.300908
- Ooi JYY, Bernardo BC, McMullen JR. The therapeutic potential of microRNAs regulated in settings of physiological cardiac hypertrophy. Future Medicinal Chemistry 2014; 6:205-22; PMID:24467244; http://dx.doi.org/10.4155/fmc.13.196
- Bernardo BC, Gao XM, Tham YK, Kiriazis H, Winbanks CE, Ooi JY, Boey EJ, Obad S, Kauppinen S, Gregorevic P, et al. Silencing of miR-34a attenuates cardiac dysfunction in a setting of moderate, but not severe, hypertrophic cardiomyopathy. PLoS One 2014; 9:e90337; PMID:24587330; http://dx.doi.org/10.1371/journal.pone.0090337
- Bernardo BC, Gao XM, Winbanks CE, Boey EJ, Tham YK, Kiriazis H, Gregorevic P, Obad S, Kauppinen S, Du XJ, et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc Natl Acad Sci U S A 2012; 109:17615-20; PMID:23047694; http://dx.doi.org/10.1073/pnas.1206432109
- Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S, Kaluza D, Treguer K, Carmona G, Bonauer A, et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013; 495:107-10; PMID:23426265; http://dx.doi.org/10.1038/nature11919
- Hullinger TG, Montgomery RL, Seto AG, Dickinson BA, Semus HM, Lynch JM, Dalby CM, Robinson K, Stack C, Latimer PA, et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ Res 2012; 110:71-81; PMID:22052914; http://dx.doi.org/10.1161/CIRCRESAHA.111.244442
- Montgomery RL, Hullinger TG, Semus HM, Dickinson BA, Seto AG, Lynch JM, Stack C, Latimer PA, Olson EN, van Rooij E. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure / clinical perspective. Circulation 2011; 124:1537-47; PMID:21900086; http://dx.doi.org/10.1161/CIRCULATIONAHA.111.030932
- Hu Y, Matkovich SJ, Hecker PA, Zhang Y, Edwards JR, Dorn GW, 2nd. Epitranscriptional orchestration of genetic reprogramming is an emergent property of stress-regulated cardiac microRNAs. Proc Natl Acad Sci U S A 2012; 109:19864-9; PMID:23150554; http://dx.doi.org/10.1073/pnas.1214996109
- Matkovich SJ, Hu Y, Dorn GW, 2nd. Regulation of cardiac microRNAs by cardiac microRNAs. Circ Res 2013; 113:62-71; PMID:23625950; http://dx.doi.org/10.1161/CIRCRESAHA.113.300975
- van Rooij E, Quiat D, Johnson BA, Sutherland LB, Qi X, Richardson JA, Kelm RJ Jr, Olson EN. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev Cell 2009; 17:662-73; PMID:19922871; http://dx.doi.org/10.1016/j.devcel.2009.10.013
- van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007; 316:575-9; PMID:17379774; http://dx.doi.org/10.1126/science.1139089
- Yang Y, Cheng H, Qiu Y, Dupee DK, Noonan M, Lin YD, Fisch S, Unno K, Sereti KI, Liao R. MicroRNA-34a plays a key role in cardiac repair and regeneration following myocardial infarction. Circ Res 2015; 117:450-9; PMID:26082557; http://dx.doi.org/10.1161/CIRCRESAHA.117.305962
- Lin RC, Weeks KL, Gao XM, Williams RB, Bernardo BC, Kiriazis H, Matthews VB, Woodcock EA, Bouwman RD, Mollica JP, et al. PI3K(p110 alpha) protects against myocardial infarction-induced heart failure: identification of PI3K-regulated miRNA and mRNA. Arterioscler Thromb Vasc Biol 2010; 30:724-32; PMID:20237330; http://dx.doi.org/10.1161/ATVBAHA.109.201988
- Greco S, Fasanaro P, Castelvecchio S, D'Alessandra Y, Arcelli D, Di Donato M, Malavazos A, Capogrossi MC, Menicanti L, Martelli F. MicroRNA dysregulation in diabetic ischemic heart failure patients. Diabetes 2012; 61:1633-41; PMID:22427379; http://dx.doi.org/10.2337/db11-0952
- Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, et al. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation 2007; 116:258-67; PMID:17606841; http://dx.doi.org/10.1161/CIRCULATIONAHA.107.687947
- Yang JH, Li JH, Jiang S, Zhou H, Qu LH. ChIPBase: a database for decoding the transcriptional regulation of long non-coding RNA and microRNA genes from ChIP-Seq data. Nucleic Acids Res 2013; 41:D177-87; PMID:23161675; http://dx.doi.org/10.1093/nar/gks1060
- Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A 2008; 105:13421-6; PMID:18755897; http://dx.doi.org/10.1073/pnas.0801613105
- Takagi S, Nakajima M, Kida K, Yamaura Y, Fukami T, Yokoi T. MicroRNAs regulate human hepatocyte nuclear factor 4alpha, modulating the expression of metabolic enzymes and cell cycle. J Biol Chem 2010; 285:4415-22; PMID:20018894; http://dx.doi.org/10.1074/jbc.M109.085431
- Choi YJ, Lin CP, Ho JJ, He X, Okada N, Bu P, Zhong Y, Kim SY, Bennett MJ, Chen C, et al. miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat Cell Biol 2011; 13:1353-60; PMID:22020437; http://dx.doi.org/10.1038/ncb2366
- De Antonellis P, Carotenuto M, Vandenbussche J, De Vita G, Ferrucci V, Medaglia C, Boffa I, Galiero A, Di Somma S, Magliulo D, et al. Early targets of miR-34a in neuroblastoma. Mol Cell Proteomics 2014; 13:2114-31; PMID:24912852; http://dx.doi.org/10.1074/mcp.M113.035808
- Wirsing A, Senkel S, Klein-Hitpass L, Ryffel GU. A systematic analysis of the 3′UTR of HNF4A mRNA reveals an interplay of regulatory elements including miRNA target sites. PLoS One 2011; 6:e27438; PMID:22140441; http://dx.doi.org/10.1371/journal.pone.0027438
- El-Armouche A, Schwoerer AP, Neuber C, Emons J, Biermann D, Christalla T, Grundhoff A, Eschenhagen T, Zimmermann WH, Ehmke H. Common microRNA signatures in cardiac hypertrophic and atrophic remodeling induced by changes in hemodynamic load. PLoS One 2010; 5:e14263; PMID:21151612; http://dx.doi.org/10.1371/journal.pone.0014263
- Wang Y, Men M, Yang W, Zheng H, Xue S. MiR-31 downregulation protects against cardiac ischemia/reperfusion injury by targeting protein kinase C epsilon (PKCepsilon) directly. Cell Physiol Biochem 2015; 36:179-90; PMID:25925791; http://dx.doi.org/10.1159/000374062
- Ikeda S, Kong SW, Lu J, Bisping E, Zhang H, Allen PD, Golub TR, Pieske B, Pu WT. Altered microRNA expression in human heart disease. Physiol Genomics 2007; 31:367-73; PMID:17712037; http://dx.doi.org/10.1152/physiolgenomics.00144.2007
- van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A 2008; 105:13027-32; PMID:18723672; http://dx.doi.org/10.1073/pnas.0805038105
- de Jong H, Neal AC, Coleman RA, Lewin TM. Ontogeny of mRNA expression and activity of long-chain acyl-CoA synthetase (ACSL) isoforms in Mus musculus heart. Biochim Biophys Acta 2007; 1771:75-82; PMID:17197235; http://dx.doi.org/10.1016/j.bbalip.2006.11.007
- Durgan DJ, Smith JK, Hotze MA, Egbejimi O, Cuthbert KD, Zaha VG, Dyck JR, Abel ED, Young ME. Distinct transcriptional regulation of long-chain acyl-CoA synthetase isoforms and cytosolic thioesterase 1 in the rodent heart by fatty acids and insulin. Am J Physiol Heart Circ Physiol 2006; 290:H2480-97; PMID:16428347; http://dx.doi.org/10.1152/ajpheart.01344.2005
- Wang J, Song Y, Zhang Y, Xiao H, Sun Q, Hou N, Guo S, Wang Y, Fan K, Zhan D, et al. Cardiomyocyte overexpression of miR-27b induces cardiac hypertrophy and dysfunction in mice. Cell Res 2012; 22:516-27; PMID:21844895; http://dx.doi.org/10.1038/cr.2011.132
- Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008; 456:980-4; PMID:19043405; http://dx.doi.org/10.1038/nature07511
- Cheng Y, Zhu P, Yang J, Liu X, Dong S, Wang X, Chun B, Zhuang J, Zhang C. Ischaemic preconditioning-regulated miR-21 protects heart against ischaemia/reperfusion injury via anti-apoptosis through its target PDCD4. Cardiovasc Res 2010; 87:431-9; PMID:20219857; http://dx.doi.org/10.1093/cvr/cvq082
- Ganesan J, Ramanujam D, Sassi Y, Ahles A, Jentzsch C, Werfel S, Leierseder S, Loyer X, Giacca M, Zentilin L, et al. MiR-378 controls cardiac hypertrophy by combined repression of mitogen-activated protein kinase pathway factors. Circulation 2013; 127:2097-106; PMID:23625957; http://dx.doi.org/10.1161/CIRCULATIONAHA.112.000882
- Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 2014; 15:509-24; PMID:25027649; http://dx.doi.org/10.1038/nrm3838
- Lal A, Thomas MP, Altschuler G, Navarro F, O'Day E, Li XL, Concepcion C, Han YC, Thiery J, Rajani DK, et al. Capture of microRNA-bound mRNAs identifies the tumor suppressor miR-34a as a regulator of growth factor signaling. PLoS Genet 2011; 7:e1002363; PMID:22102825; http://dx.doi.org/10.1371/journal.pgen.1002363
- Tham YK, Bernardo BC, Ooi JY, Weeks KL, McMullen JR. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol 2015; 89:1401-38; PMID:25708889; http://dx.doi.org/10.1007//s00204-015-1477-x
- Xia J, Zhang Y, Xin L, Kong S, Chen Y, Yang S, Li K. Global transcriptomic profiling of cardiac hypertrophy and fatty heart induced by long-term high-energy diet in bama miniature pigs. PLoS One 2015; 10:e0132420; PMID:26161779; http://dx.doi.org/10.1371/journal.pone.0132420
- Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, Fu C, Lindow M, Stenvang J, Straarup EM, et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet 2011; 43:371-8; PMID:21423181; http://dx.doi.org/10.1038/ng.786
- Agostini M, Knight RA. miR-34: from bench to bedside. Oncotarget 2014; 5:872-81; PMID:24657911; http://dx.doi.org/10.18632/oncotarget.1825
- Bader AG. miR-34 - a microRNA replacement therapy is headed to the clinic. Front Genet 2012; 3:120; PMID:22783274; http://dx.doi.org/10.3389/fgene.2012.00120
- Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med 2013; 368:1685-94; PMID:23534542; http://dx.doi.org/10.1056/NEJMoa1209026
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25; PMID:19261174; http://dx.doi.org/10.1186/gb-2009-10-3-r25
- Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander ES, Golub TR. Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc Natl Acad Sci U S A 1999; 96:2907-12; PMID:10077610; http://dx.doi.org/10.1073/pnas.96.6.2907
- Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res 2015; 43:D146-52; PMID:25378301; http://dx.doi.org/10.1093/nar/gku1104
- Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res 2014; 42:D92-7; PMID:24297251; http://dx.doi.org/10.1093/nar/gkt1248
- Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res 2011; 39:D202-9; PMID:21037263; http://dx.doi.org/10.1093/nar/gkq1056
- Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ, et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res 2016; 44:D239-47; PMID:26590260; http://dx.doi.org/10.1093/nar/gkv1258
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001; 29:e45; PMID:11328886; http://dx.doi.org/10.1093/nar/29.9.e45
- Matkovich SJ, Van Booven DJ, Youker KA, Torre-Amione G, Diwan A, Eschenbacher WH, Dorn LE, Watson MA, Margulies KB, Dorn GW 2nd. Reciprocal regulation of myocardial microRNAs and messenger RNA in human cardiomyopathy and reversal of the microRNA signature by biomechanical support. Circulation 2009; 119:1263-71; PMID:19237659; http://dx.doi.org/10.1161/CIRCULATIONAHA.108.813576
- Wang X, Wang HX, Li YL, Zhang CC, Zhou CY, Wang L, Xia YL, Du J, Li HH. MicroRNA let-7i negatively regulates cardiac inflammation and fibrosis. Hypertension 2015; 66:776-85; PMID:26259595; http://dx.doi.org/10.1161/HYPERTENSIONAHA.115.05548
- Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, Rojas M, Hammond SM, Wang DZ. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol 2007; 42:1137-41; PMID:17498736; http://dx.doi.org/10.1016/j.yjmcc.2007.04.004
- Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res 2007; 100:416-24; PMID:17234972; http://dx.doi.org/10.1161/01.RES.0000257913.42552.23
- Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy?. Am J Pathol 2007; 170:1831-40; PMID:17525252; http://dx.doi.org/10.2353/ajpath.2007.061170
- van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A 2006; 103:18255-60; PMID:17108080; http://dx.doi.org/10.1073/pnas.0608791103
- Roy S, Khanna S, Hussain SR, Biswas S, Azad A, Rink C, Gnyawali S, Shilo S, Nuovo GJ, Sen CK. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res 2009; 82:21-9; PMID:19147652; http://dx.doi.org/10.1093/cvr/cvp015
- Muthusamy S, DeMartino AM, Watson LJ, Brittian KR, Zafir A, Dassanayaka S, Hong KU, Jones SP. MicroRNA-539 is up-regulated in failing heart, and suppresses O-GlcNAcase expression. J Biol Chem 2014; 289:29665-76; PMID:25183011; http://dx.doi.org/10.1074/jbc.M114.578682
- Urbich C, Kaluza D, Fromel T, Knau A, Bennewitz K, Boon RA, Bonauer A, Doebele C, Boeckel JN, Hergenreider E, et al. MicroRNA-27a/b controls endothelial cell repulsion and angiogenesis by targeting semaphorin 6A. Blood 2012; 119:1607-16; PMID:22184411; http://dx.doi.org/10.1182/blood-2011-08-373886
- Veliceasa D, Biyashev D, Qin G, Misener S, Mackie AR, Kishore R, Volpert OV. Therapeutic manipulation of angiogenesis with miR-27b. Vasc Cell 2015; 7:6; PMID:26161255; http://dx.doi.org/10.1186/s13221-015-0031-1
- Xu M, Wu HD, Li RC, Zhang HB, Wang M, Tao J, Feng XH, Guo YB, Li SF, Lai ST, et al. Mir-24 regulates junctophilin-2 expression in cardiomyocytes. Circ Res 2012; 111:837-41; PMID:22891046; http://dx.doi.org/10.1161/CIRCRESAHA.112.277418
- Fiedler J, Jazbutyte V, Kirchmaier BC, Gupta SK, Lorenzen J, Hartmann D, Galuppo P, Kneitz S, Pena JT, Sohn-Lee C, et al. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation 2011; 124:720-30; PMID:21788589; http://dx.doi.org/10.1161/CIRCULATIONAHA.111.039008
- Li RC, Tao J, Guo YB, Wu HD, Liu RF, Bai Y, Lv ZZ, Luo GZ, Li LL, Wang M, et al. In vivo suppression of microRNA-24 prevents the transition toward decompensated hypertrophy in aortic-constricted mice. Circ Res 2013; 112:601-5; PMID:23307820; http://dx.doi.org/10.1161/CIRCRESAHA.112.300806
- Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med 2011; 208:549-60; PMID:21383058; http://dx.doi.org/10.1084/jem.20101547
- Wang J, Huang W, Xu R, Nie Y, Cao X, Meng J, Xu X, Hu S, Zheng Z. MicroRNA-24 regulates cardiac fibrosis after myocardial infarction. J Cell Mol Med 2012; 16:2150-60; PMID:22260784; http://dx.doi.org/10.1111/j.1582-4934.2012.01523.x
- Deacon DC, Nevis KR, Cashman TJ, Zhou Y, Zhao L, Washko D, Guner-Ataman B, Burns CG, Burns CE. The miR-143-adducin3 pathway is essential for cardiac chamber morphogenesis. Development 2010; 137:1887-96; PMID:20460367; http://dx.doi.org/10.1242/dev.050526