
ABSTRACT
Mutations in the RNA-binding protein, RBM10, result in a human syndromic form of cleft palate, termed TARP syndrome. A role for RBM10 in alternative splicing regulation has been previously demonstrated in human cell lines. To uncover the cellular functions of RBM10 in a cell line that is relevant to the phenotype observed in TARP syndrome, we used iCLIP to identify its endogenous RNA targets in a mouse embryonic mandibular cell line. We observed that RBM10 binds to pre-mRNAs with significant enrichment in intronic regions, in agreement with a role for this protein in pre-mRNA splicing. In addition to protein-coding transcripts, RBM10 also binds to a variety of cellular RNAs, including non-coding RNAs, such as spliceosomal small nuclear RNAs, U2 and U12. RNA-seq was used to investigate changes in gene expression and alternative splicing in RBM10 KO mouse mandibular cells and also in mouse ES cells. We uncovered a role for RBM10 in the regulation of alternative splicing of common transcripts in both cell lines but also identified cell-type specific events. Importantly, those pre-mRNAs that display changes in alternative splicing also contain RBM10 iCLIP tags, suggesting a direct role of RBM10 in these events. Finally, we show that depletion of RBM10 in mouse ES cells leads to proliferation defects and to gross alterations in their differentiation potential. These results demonstrate a role for RBM10 in the regulation of alternative splicing in two cell models of mouse early development and suggests that mutations in RBM10 could lead to splicing changes that affect normal palate development and cause human disease.
Introduction
RNA-binding proteins (RBPs) bind cellular RNAs to form ribonucleoprotein (RNP) particles with unique specificity that profoundly influence RNA biogenesis and function,Citation1-3 affecting multiple steps in gene expression. The RBM10 (RNA binding motif 10) protein is a member of the RNA-binding motif (RBM) gene family, which includes the highly homologous RBM5 and RBM6 proteins. RBM10 (originally called S1-1), is alternatively spliced to produce two mRNA variants (v1 and v2), which differ in the inclusion/exclusion of exon 4, giving rise to two protein isoforms of 930 and 853 amino acids, respectively.Citation4,5 Both protein variants comprise several functional domains that bind to RNA, such as two RNA recognition motifs (RRM), a RanBP2-type zinc finger motif, a C2H2 Zn finger domain and a C-terminal G patch domain.Citation5 RBM5, also known as Luca-15/H37, is a gene frequently inactivated in lung cancers and overexpressed in breast tumors and regulates alternative splicing of apoptosis-related genes.Citation6,7
Mutations in RMB10 were originally found in two families manifesting an X-linked syndromic form of cleft palate, termed TARP (for talipes equinovarus, atrial septal defect, Robin sequence, and persistent left superior vena cava). This syndrome comprises Pierre–Robin sequence (micrognathia, glossoptosis, and cleft palate), talipes equinovarus, atrial septal defect (ASD), and persistence of the left superior vena cava and results in pre- or postnatal lethality in affected males.Citation8,9 The first two families were quite similar in phenotype, with uniform early lethality although a confirmatory case report showed survival into childhood. More recently, three families with mutations in RBM10, in which the affected males were more phenotypically diverse, were identified.Citation10 All six reported causative mutations in TARP syndrome are predicted to result in loss of function of RBM10 illustrating that this gene is critical for normal mammalian development. The orthologous murine Rbm10 isoform 1 gene product is 96% identical to the human protein. It is expressed in midgestation mouse embryos in the branchial arches and limbs, consistent with the human malformations observed in patients with TARP syndrome.Citation8
Identification of endogenous RNA targets for individual RBPs is crucial to understand their role in RNA biogenesis. A role for RBM10 in pre-mRNA splicing was initially suggested by the identification of RBM10 as a component of pre-spliceosomal A and B complexes,Citation11-13 and for its interaction with the alternative splicing regulator SRrp86.Citation14 Moreover, RBM10 was found to modulate alternative splicing (AS) of Fas and Bcl-x genes.Citation15 A variant of the CLIP (crosslinking and immunoprecipitation) protocol, termed PAR-CLIP was used to identify binding sites of RBM10 in HEK 293 cells. This led to the identification of thousands of binding sites of RBM10, many occurring in the vicinity of splice sites. This study also revealed an extensive role for RBM10 in splicing regulation, in particular in the regulation of the exon skipping-type of AS regulation.Citation16 A second study using other variant of CLIP (CLIP-Seq) and splicing-sensitive microarrays identified RBM10 targets in HeLa cells. As was the case in HEK 293 cells, RBM10 was also found to have a role in splicing regulation, in particular functioning in antagonistic manner to the related proteins, RBM5 and RBM6 in the regulation of alternative splicing of NUMB, a regulator of the Notch signaling pathway.Citation17
To understand how RBM10 loss of function leads to human disease, we aimed to identify endogenous RNA targets of RBM10 in a mouse mandibular embryonic cell line, which is relevant to the phenotype observed in TARP syndrome. For this, we used the iCLIP variant protocol that allows the mapping of protein-RNA interactions at an individual nucleotide resolution.Citation18,19 We observed that RBM10 binds preferentially to intronic regions of protein-coding genes. This was complemented by RNA-seq profiling of RBM10 knock-out mouse mandibular cells as well as mouse embryonic (ES) stem cells with disrupted RBM10 expression. This analysis revealed an extensive role for RBM10 in the regulation of alternative splicing, affecting the regulation of several alternative cassette exons in both cell types (mandibular and ES cells), but also displaying cell-type specific regulation of AS. In mandibular cells, the overlap of the iCLIP and the RNA-seq data revealed a direct role of RBM10 in the silencing of alternative cassette exons. Taken together, our analyses provide evidence for a widespread role for RBM10 in the regulation of alternative splicing in a mouse cell line, with relevance to the human disease. This strongly suggest that misregulation of alternative splicing upon loss of function of RBM10 could lead to the TARP syndrome.
Results
Genome-wide mapping of RBM10 binding sites using iCLIP
We used iCLIP (individual-nucleotide resolution UV crosslinking and immunoprecipitation) to determine the RNA-binding landscape of RBM10 in a mouse mandibular MEPA (Mouse Embryonic Pharyngeal Arch) cell line, since this is more relevant to the phenotype observed in TARP syndrome.Citation20 The RBM10 protein is highly conserved between man and mouse, and the spatiotemporal pattern of expression of the mouse gene during embryogenesis is very consistent with the phenotypes observed in TARP patients, which harbor loss of function mutations in RBM10.Citation8-10 We carried out four independent iCLIP experiments using an antibody that recognizes the two major isoforms of the mouse RBM10 protein (variants 1 and 2) in mouse mandibular MEPA cells. As a control, we performed an immunoprecipitation using rabbit IgG. The RNA-RBM10 protein complexes were extracted from the membrane (). Considering the small difference of size between the two RBM10 isoforms, the two different complexes cannot be analyzed separately. For each experiment, an extraction was also performed for the control lane (immunoprecipitation using IgG) at the same level in the membrane.
Figure 1. Genome-wide mapping of RBM10 RNA-binding sites in a mouse mandibular MEPA cell line using iCLIP. (A) Autoradiograph of cross-linked protein/RNA complexes after immunoprecipitation and 32P RNA labeling. Immunoprecipitations were performed with an antibody that recognizes both isoforms of RBM10 protein or with IgG, as a negative control. Three different dilutions of RNase I were used. Cells untreated with UV were also used as a control. Sections of the membrane cut for the library preparation are indicated by red boxes (B) Number of reads obtained for each iCLIP experiment after processing of the sequencing data (removal of PCR duplicates, trimming of adapter sequences, attribution of the read to one specific replicate using the barcode) (C) Number of cross-linked nucleotides obtained through all iCLIP experiments and the ones reproducibly found in several replicates of the iCLIP experiments. (D) Graph showing the proportion of cross-linked nucleotides supported by more than 10 reads in the list of binding sites present in all 4 iCLIP experiments (‘strong’ list) or in 3 out of 4 experiments (‘full’ list).
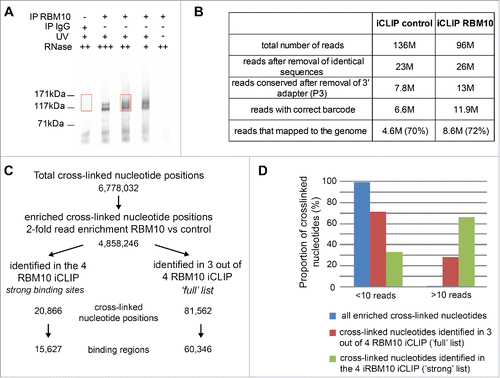
Libraries were prepared for each RBM10 iCLIP and its equivalent control and sequenced on an Illumina platform. Identical reads coming from the PCR amplification were removed and following removal of the adapter sequence, short reads were discarded (). 11.9 M reads were obtained for the four RBM10 iCLIP and 6.6 M for the control (). After attribution of the reads to each individual experiment using the barcode, reads were mapped to the mouse genome using Bowtie. Around 75% of the reads of the different experiments were mapped to the genome (). For the subsequent analysis, we decided to keep the reads that can be uniquely mapped to the genome. For each iCLIP replica experiment (RBM10 and control), the distinct cross-linked nucleotides were determined and associated to a number of reads (Fig. S1A). This analysis gave us a total of 6.8 M cross-linked nucleotides present in all four iCLIP experiments (). However, these positions can also be present in the equivalent control or be only supported by a small number of reads. Thus, these positions were filtered using different criteria. For each independent iCLIP experiment, we only use those cross-linked positions that had at least 2-fold more reads in the RBM10 iCLIP, as compared to the matching IgG iCLIP. The correlation between the 4 iCLIP experiments for these 4.9 M cross-linked positions was analyzed and confirmed a good reproducibility for all RBM10 iCLIP experiments (Fig. S1B). In comparison, we observed a strong reduction of the correlation coefficient when we compared each RBM10 iCLIP with its equivalent control (Fig. S1B).
Binding sites of RBM10 protein correspond to cross-linked nucleotides supported by a large number of reads that are enriched as compared to the control iCLIP. First, we decided, to obtain a list of ‘strong’ binding sites of RBM10 by keeping the cross-linked positions containing at least 1 read in each RBM10 iCLIP (if the control does not have any reads) or a 2-fold difference compared to the control. This stringent analysis gave us a total of 20,866 cross-linked nucleotides (‘strong’ list) that are common to the 4 RBM10 iCLIP experiments (, Table S1). We sometimes found adjacent cross-linked nucleotides and this observation probably reflects the fact that the binding of RBM10 is not limited to one individual nucleotide but probably spans several nucleotides. Taking this into account, we joined together the adjacent cross-linked nucleotides and estimated the number of ‘strong’ binding sites of RBM10 protein at 15,627 (). Then, in order to have a more complete list of RBM10 binding sites (‘full’ list), we focused our analysis to cross-linked nucleotides present in at least 3 out of 4 RBM10 iCLIP experiments. In this case, the number of cross-linked nucleotides was 81,562 and the number of RBM10 binding sites was estimated at 60,346 (, Table S1). To validate this analysis, we looked at the number of reads associated to each cross-linked nucleotide for the list of ‘strong’ and ‘full’ RBM10 binding sites. In comparison to the list of total cross-linked positions defined by all the iCLIP experiments, the proportion of cross-linked nucleotides supported by at least 10 reads increases drastically for the list of ‘full’ RBM10 binding sites and even more for the list of ‘strong’ RBM10 binding sites (). The number of RBM10 binding sites in mouse mandibular cells is in the same range as the one obtained using the PAR-CLIP method in human HEK 293 cells,Citation16 suggesting similar binding properties between mouse and human RBM10 protein and within different cell types.
iCLIP annotation and binding motifs
The annotation of the full list of RBM10 binding sites was performed using the cross-linked nucleotide positions () or the number of reads (Fig. S2). In both cases, this analysis showed a clear enrichment of RBM10 binding sites toward protein-coding transcripts ( and S2A) and more specifically toward introns of these protein coding genes ( and S2B). We also observed binding of RBM10 to non-coding (ncRNAs) RNAs ( and S2A), which are probably under evaluated as only uniquely mapped reads were kept. This analysis revealed that RBM10 binds transcripts of around 6,000 mouse protein coding genes (with a strong binding site for 3,000 genes), thus, associating with transcripts of around 30% of all annotated mouse protein coding genes.
Figure 2. RBM10 binding sites. (A) Distribution of RBM10 cross-linked nucleotide positions among protein coding transcripts, non-coding RNAs and intergenic regions. (B) Distribution of RBM10 cross-linked nucleotide positions among coding exons, 5′UTRs, 3′UTRs and introns. (C) Position of the RBM10 cross-linked positions within introns. The distribution of the number of intronic cross-linked nucleotide positions in regions upstream and downstream of the splice site is shown. (D) Nucleotide preference for RBM10 cross-linked positions (based on the top 1,000 RBM10 cross-linked nucleotides in introns). (E) In vivo RBM10 binding motif obtained with the MEME software using the 20 nt sequences surrounding the top 1,000 RBM10 cross-linked positions in introns.
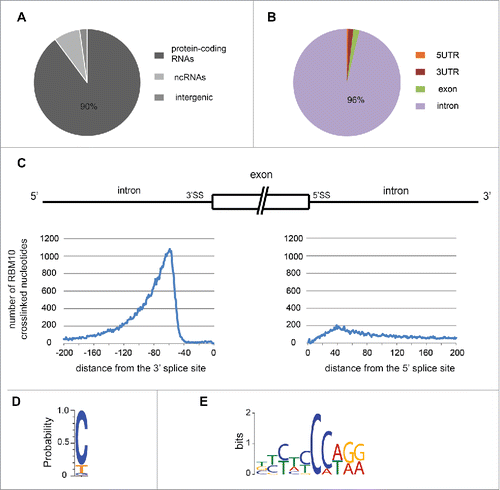
We observed that the distribution of RBM10 cross-linked nucleotides within the intron shows a clear enrichment on the 3′ side of the intron, with a small peak of RBM10 binding close to the 5′ splice site (). RBM10 preferentially binds to the 3′ side of the intron, from – 150 to –50 nucleotides upstream of the 3′ splice site. In humans, the branch point (BP) position has been well defined and is in average located between 18 and 35 nt upstream of the 3′ splice site.Citation21 However, BP recognition seems to be more relaxed than previously assumed and is highly dependent on the presence of downstream polypyrimidine tracts.Citation22 Mouse branch point positions display a similar pattern confirming that RBM10 binds preferentially upstream of the branch point.Citation23
A large number of RBPs recognize their RNA targets in a sequence-specific manner.Citation24,25 It has been shown that rat RBM10 protein binds in vitro to RNA homopolymers, with a preference for G and U polyribonucleotides.Citation4 The iCLIP data allowed us to search for RBM10 binding sites within the list of ‘strong’ RBM10 cross-linked nucleotides. Taking the top 1,000 cross-linked nucleotides in introns (ranked by the sum of reads in all 4 iCLIP experiments), we observed that cytosine represented more than 80% of the cross-linked nucleotides (). This C nucleotide bias could be directly related to RBM10 specificity as CLIP techniques are only subject to a modest uridine preference caused by UV-C cross-linking.Citation26 We then used the sequences surrounding the cross-linked nucleotides to obtain a consensus-binding motif, using the MEME software.Citation27 We obtained one significant motif enriched in C nucleotides (). We also look for enriched pentamers in the 20 nt sequences that surround the top 1,000 RBM10 cross-linked nucleotides. As control, we used 20 nt sequence chosen randomly within the same introns bound by RBM10, and we calculated a Z-score for each pentamer. The top 3 enriched pentamers are TCCAA, CCAAA and CCCCA. In fact, all top 15 enriched pentamers contains at least 2 consecutive C nucleotides (Fig. S3). These data show a strong binding preference for RBM10 for motifs containing at least 2 consecutive C nucleotides followed by a T or an A. This is consistent with one of the motifs identified by CLIP-seq for the human RBM10 protein in HeLa cells.Citation17
RBM10 binds to spliceosomal small nuclear RNAs
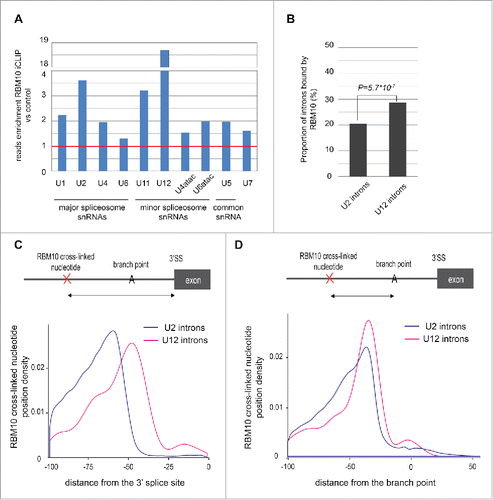
As described above, RBM10 also binds to non-coding RNAs ( and S2A) and we noticed a large number of reads corresponding to snRNAs. We decided to map reads directly to snRNA sequences to better characterize this binding (see details in methods). RBM10 preferentially binds to specific snRNAs, with a high enrichment of reads observed for the U12 minor spliceosomal snRNA, but we also observed significant binding to spliceosomal U2 snRNAs (), as was also described in human cells.Citation16 This data could suggest a role of RBM10 in the processing of snRNAs. Alternatively, and perhaps more likely taking into account the binding of RBM10 to introns in the vicinity of the branch point, we speculate that RBM10 interacts with the U2/U12 snRNP complex, as well as with the mRNA, during splicing and potentially regulates their association. In agreement with an enrichment of RBM10 binding to U12 snRNA, we observed that the proportion of U12 introns bound by RBM10 is higher compared to that of U2 introns (). RBM10 binding in U12 introns is closer to the 3′ splice site, as compared to U2 snRNAs (). This probably reflects the fact that most U12 introns do not contain a polypyrimidine tract and that RBM10 binding is relative to the branch point (BP), which is closer to the 3′ splice site for U12 introns.Citation28 To validate this hypothesis, we predicted the branch point location on U2 and U12 introns bound by RBM10 and analyzed the distance between RBM10 cross-linked nucleotides and the BP adenosine. We observed a strong enrichment of RBM10 binding upstream of the branch point (around −35nt) (). The profile is similar for U2 and U12-dependent introns suggesting that the distance between RBM10 binding and the branch point is important for splicing. In order to better characterize RBM10 binding to U2 and U12 snRNAs, we mapped the iCLIP reads to the consensus sequences of U2 and U12 and defined the cross-linked nucleotides. RBM10 binding was observed in different region of U12 snRNA (Fig. S4A and C) while RBM10 binding to U2 snRNA is preferentially localized to the first stem loop (Fig. S4B and D). RBM10 binds close to the branch point recognition region and this is probably important to regulate the interaction between U2/U12 and the pre-mRNA.
Figure 3. RBM10 binds small nuclear RNAs (snRNAs) of the major and minor spliceosome and their regulated introns. (A) Enrichment in the number of reads mapped to individual snRNAs for RBM10 iCLIP, as compared to the control. The red line corresponds to a ratio of 1 (i.e. no enrichment). (B) Proportion of introns spliced by the major or minor spliceosome that are bound by RBM10. Only introns of expressed genes (using RNA-seq data) have been analyzed (132,627 U2-introns and 606 U12-introns). U12 introns were retrieved from the U12DB database.Citation28 The comparison between the 2 samples was done using a z-test. (C) RBM10 binding profile upstream of the 3′ splice site for U2- (n = 27,123) and U12-introns (n = 174). The density of the cross-link nucleotides was plotted. (D) RBM10 binding profile upstream of the branch point (BP) adenosine for U2- (n = 26,234) and U12-introns (n = 133). The prediction of the BP was done using SVM-BPfinder.Citation22 The density of the cross-link nucleotides was plotted.
A role for RBM10 in alternative splicing in mandibular cells
In order to globally analyze the effects of RBM10 in alternative splicing, we performed RNA-Seq on polyA+ RNA isolated from cells in which RBM10 expression was disrupted. For this, we used the CRISPR/Cas9n system to generate a knockout of RBM10 in mouse mandibular cells. To avoid off-target effects, we used the double-nicking strategy using a Cas9 nickase mutant and we obtained two KO cell lines (KO1, KO2) containing a deletion in exon 3 and two additional KO cell lines (KO3 and KO4) harboring deletions in exon 9 (). The screening of RBM10 KO cells was carried out by Western blot analysis and showed the absence of both isoforms of RBM10 protein in the four selected RBM10 KO cells (). The RBM10 KO clones can contain one or two mutated copies of the RBM10 gene, which maps to the X chromosome, depending whether the clone is male or female. Mouse mandibular cell lines were obtained from dissection of a whole litter of embryos without sex assignment,Citation20 so we decided to genotype the WT population as well as some positive clones. While the WT population of mandibular (md) cells showed a mixed population of female and male cells, all the studied KO clones were female (Fig. S5A) and thus contained 2 copies of RBM10 genes. We sequenced RBM10 exon 3 and exon 9 from RBM10 KO cells to identify the specific deletions causing RBM10 KO (Fig. S5B). We identified two different alleles of KO3 containing two different deletions in exon 9. However, the sequencing of clones KO1, KO2 and KO4 showed only one version of the mutated exon suggesting that both copies of RBM10 gene are similar in each clone. The presence of identical deletion in both alleles of the targeted gene has been observed in other studies using CRISPR technology and is likely explained by an inter-allelic gene conversion event.Citation29
Figure 4. Alternative splicing changes in RBM10 KO mandibular cells. (A) Structure of the RBM10 gene indicating the position of the deletions in exon 3 and exon 9, respectively, generated using CRISPR technology. (B) Western blot showing the absence of RBM10 protein isoforms in 4 independent KO cell lines. (C) Heatmap showing the Euclidean distances between the RNA-seq samples as calculated from the variance stabilizing transformation of the count data using DESeq. Darker blue colors indicate more similar pattern of expression. The count data was obtained after mapping and transcript quantification using Sailfish.Citation30 (D) Summary of the splicing changes observed in the 4 RBM10 KO md cells compared to WT. Splicing analysis was carried out using SUPPA software.Citation32
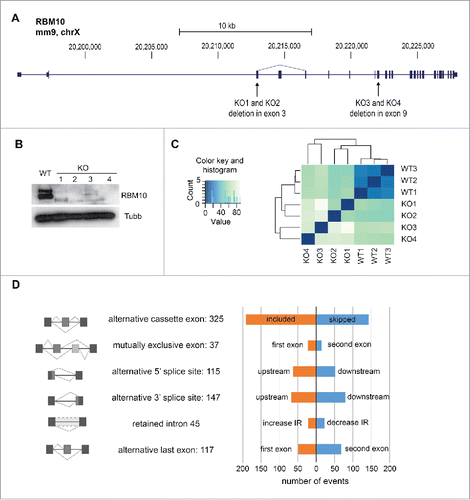
RNA-seq analysis was performed for each RBM10 KO cell line and compared to the WT mandibular MEPA cells that were analyzed in triplicate. Gene expression analysis was performed using Sailfish for mapping and transcript quantification,Citation30 followed by DESeq to assess differential expression.Citation31 As expected, the data obtained for the 3 WT md cells clustered strongly together while the 4 RBM10 KO samples formed a separate group with bigger distance between each other, as expected for 4 independent biological replicate KO cell lines (). Considering a 1.5-fold change and a <0.05 P-value, 288 genes displayed differential expression between WT and KO md cell lines, with 146 genes up-regulated, and 142 genes down-regulated (Table S2). We then assessed splicing changes in the RBM10 KO cell lines using the SUPPA tool,Citation32 which calculates the PSI (“Percentage Spliced In”) value for each splicing event extracted from the annotation of the mm10 mouse genome. We defined a list of differential splicing events based on the changes of PSI but also taking into consideration the standard deviation between replicates and the overlap between the 2 set of data (WT vs KO) (see details in Methods). As a result, we obtained a list of 786 splicing events that changed in the knockout cell lines (Table S3). These events fall into several categories but we identified a large number of alternative cassette exons, in particular inclusion of cassette exons upon abrogation of RBM10 expression ().
A role for RBM10 in alternative splicing in embryonic (ES) stem cells
Since RBM10 is ubiquitously expressed, its role in the regulation of housekeeping or ubiquitously expressed transcripts is probably conserved in different cell types or during development.Citation33 However, RBM10 might also play a role in the regulation of transcripts with specific expression profile. To assess how RBM10 affects splicing in a different mouse cell line, we took advantage of the availability of an RBM10 KO ES cell line. We chose the male gene trap cell line Rbm10Gt(CSI176)Byg. This cell line is hemizygous for the RBM10 mutated allele present in the single X chromosome, with an insertion on the fifth intron of this gene (Fig. S6A). We confirmed the presence of the insert at the genomic level (Fig. S6B). We could also detect a fusion transcript between the upstream part of RBM10 gene and the insertion by RT-PCR analysis. Furthermore, we could not detect any full-length RBM10 transcript (Fig. S6C) and we confirmed the absence of RBM10 protein by Western blot analysis (Fig. S6D).
RNA-seq analysis was performed for the RBM10 KO ES cell and compared to control cells (parental E14 cell line) that were analyzed in triplicate. The heatmap shows that the samples are strongly clustered, as expected for replicates of the same cell line (Fig. S7A). We also detected more significant changes in terms of gene expression and splicing in ES cells in comparison to mandibular cells probably because of the lower variability between replicates. Considering a 2-fold change and a P-value below 0.01, we found 653 genes upregulated and 772 genes downregulated in RBM10 KO ES cells (Table S2). The splicing analysis revealed a change in 2,967 splicing events (Table S3). Again, changes for all categories of events were reported but we observed a higher number of inclusion of alternative cassette exons upon RBM10 depletion (Fig. S7B).
Comparison between mandibular and ES RNA-seq
We compared the effect of abrogating RBM10 expression in md MEPA cells (CRISPR-mediated KOs) and ES cells (gene-trap) to assess a putative cell-type specific role for RBM10 in splicing regulation. In terms of gene expression, only 12 genes were shown to be upregulated in both cell lines, with 10 genes down-regulated in both cell types. In contrast, in terms of splicing, we found 111 event changes identified in both mandibular and ES cells (Fig S8A). The overlap between splicing events in both cell lines is significant for alternative cassette exons and also for intron retention events (Fig. S8A). Among the 60 shared alternative cassette exons, 44 correspond to included cassette exon and 16 are skipped cassette exons (). For validation, we selected a small subset of genes that showed large splicing changes in both mandibular and ES cells, as revealed by RNA-seq analysis, but also display a similar expression level in both cell lines. We confirmed by RT-PCR the cassette exon inclusion of 2 ubiquitously expressed genes, the protein kinase WNK1, a member of the WNK subfamily of serine/threonine protein kinasesCitation34 and also of the CDKN1A Interacting Zinc Finger Protein 1, CIZ1Citation35 (). We also validated splicing changes for AGFG2,Citation36 a co-factor of the Rev protein, and also for the oxidation resistance gene OXR1Citation37 (). We also identified cell line-enriched alternative splicing events. The inclusion of a cassette exon in the ST7 pre-mRNA was detected in both mandibular and ES cells; however, the effect is stronger in mandibular cells where ST7 expression is higher (). Its cellular function has not been precisely determined but ST7 has been linked with cancer, including mandibular and jaw cancer (Malacard Human Disease database). By contrast, splicing changes of NDRG2, a N-Myc downstream-regulated gene, were only observed in ES cells where the gene is expressed (). To understand whether the splicing differences observed between cell lines are linked to the expression level of the corresponding genes, we looked at the difference of expression between mandibular MEPA and ES cells for genes with mandibular-specific splicing changes, ES-specific splicing changes, or common events. Globally, genes with mandibular-specific splicing changes have a higher expression in mandibular cells (Fig S8B). Similarly, genes with ES-specific splicing changes have a higher expression in ES cells. Most of the genes that are spliced-regulated in both cell lines show similar level of expression. This data indicates that RBM10 regulates the splicing of ‘housekeeping’ genes but also of cell-specific expressed genes (Fig. S8B).
Figure 5. Comparison of Alternative splicing changes in mouse mandibular and ES cells depleted of RBM10. (A) Venn diagram showing the overlap of included or skipped cassette exons in RBM10 KO mandibular (md) and ES cells. (B) Validation of alternative cassette exon inclusion in RBM10 KO cells by RT-PCR. The size of the 2 PCR products (with or without the inclusion of the cassette exon), as well as the percentage of inclusion (estimated after quantification of the transcripts abundance by an Agilent Bioanalyzer) is indicated. We validated the splicing changes in 2 of the RBM10 KO md cells, as well as in the RBM10 KO ES cells. For AGFG2, OXR1, WNK1 and CIZ1, similar changes were observed in the 2 cells lines. ST7 splicing changes is more prominent in mandibular cells while NDRG2 splicing changes was only observed in ES cells. (C) RNA splicing map of RBM10 in mandibular cells. The count density of RBM10 cross-linked nucleotide positions was assessed for regulated alternative exons (100 silenced in red and 46 enhanced cassette exons in blue) in the introns upstream and downstream of the event.
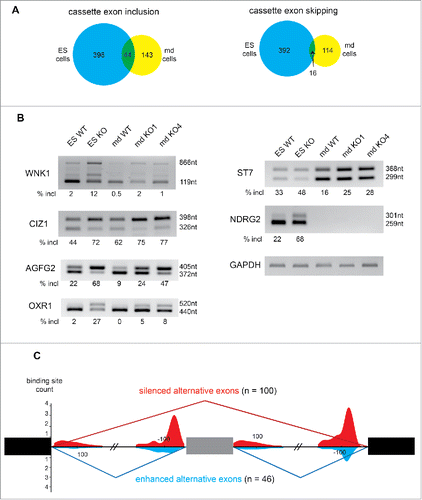
Next, we attempted to uncover whether the effect of RBM10 on the expression and/or splicing of target genes was direct. For this, we analyzed the overlap of bound and regulated RNA targets by comparing RMB10 binding as determined by iCLIP with RNA-seq data in mandibular mouse cells that were depleted of RBM10. While only 20% of the up- and downregulated genes in RBM10 KO md cells are bound by RBM10 (CLIP+), 60% of the spliced-regulated genes are CLIP+ (Fig. S9). To understand how RBM10 directly regulates splicing, and in particular alternative cassette exons, we used the iCLIP data to draw an RNA map in mouse mandibular cells ().Citation38 We observed binding of RBM10 to downstream introns for both enhanced and silenced alternative exons; however specific RBM10 binding to upstream introns was noticed only in the case of silenced alternative exons. This suggests, that RBM10 binding close to the 3′ splice site promotes the skipping of the downstream exon. We attempted to assess whether those changes in AS elicited by RBM10 were intrinsic to a particular exon-intron architecture. We only observed significant differences for exon length, as shown by included exons in RBM10 KO cells, which are shorter, whereas skipped exons are longer, as compared to cassette exons not affected by RBM10 in these cells. (Fig. S10).
RBM10 KO phenotype
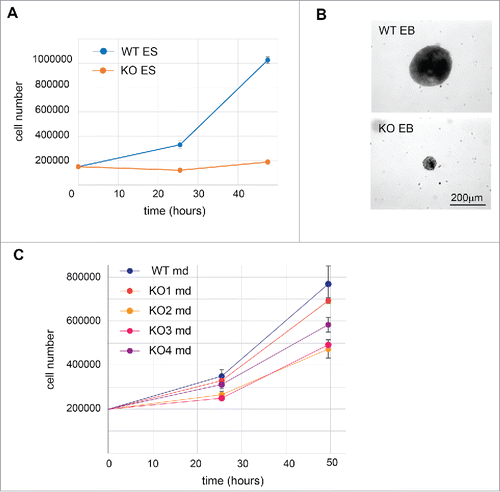
Next, we characterized the phenotypes of RBM10 KO cell lines. We observed a strong effect of abrogating RBM10 expression in ES cells that displayed a marked decrease in growth (). As the TARP syndrome affects early development, we tested whether RBM10 KO ES cells have conserved their differentiation potential. We induced embryonic body formation by culturing the cells in suspension without leukemia inhibitory factor (LIF). As expected, wild-type cells formed the characteristic three-dimensional multicellular aggregates called embryoid bodie (EBs) (). By contrast, there was drastic decrease in the size of the embryoid bodies in RBM10 KO cells (). The growth defect observed prior to differentiation probably contributes to this effect. It has been observed previously that lineage-specific differentiation could be affected by the size of the EBs during the differentiation process in vitro.Citation39 For this reason, we decided not to assess the expression of differentiation markers, as it would not be indicative of a biological effect of RBM10 knockout in vivo. In the case of mandibular cells, depletion of RBM10 seems to affect slightly their growth rate. It should be noted that mouse md (MEPA) cells were obtained from early embryos from CD1 females crossed with a male “Immortomouse’, which carries a constitutively expressed SV40 T (tumor) antigen (Tag) transgene, which could potentially affect its transformation potential.Citation20 Nonetheless, when comparing WT MEPA md cells with the RBM10 KO clones, 3 of the KO cell lines (KO 2, 3 and 4 lines) displayed a decrease of growth, as compared to the WT cells (). This effect in cell growth is different from the increase in proliferation observed following RBM10 knockdown in HeLa cells,Citation17 which was mainly attributed to the role of RBM10 in controlling the alternative splicing of the NUMB gene, an event that acts to regulate the proliferative capacity of cancer cells.Citation17 In these cells, direct binding of RBM10 upstream of the alternative exon 9 of NUMB gene leads to skipping of this exon and produces a NUMB isoform that acts as a negative regulator of the Notch signaling pathway. RBM10 was shown to antagonize the function of two other protein family members, RBM5 and RBM6 in the control of NUMB AS. By contrast, in mouse mandibular cells, we did not observe any binding of RBM10 upstream of the mouse alternative exon 9 of NUMB gene. Nonetheless, we analyzed NUMB splicing profile for WT and RBM10 KO in both ES cells and mandibular cells. In agreement with the observed lack of binding, there seems to be no effect of RBM10 in the splicing regulation of NUMB exon 9 (Fig. S11). This most likely reflects both differential roles for RBM10 in splicing regulation in cells derived from different origin and/or species-specific differences.
Figure 6. Phenotype of RBM10 KO in ES cells and mandibular cells. (A) Growth curve of WT and RBM10 KO ES cells. 150,000 cells were seeded in 6-well plates. The number of cells was analyzed after 25 h and 50 h. This analysis was done in triplicate. (B) Phase-contrast micrographs of embryoid bodies (EBs) obtained after differentiation of WT or RBM10 KO ES cells. Cells were cultured in suspension without LIF for 6 d. (C) Growth curve of WT and RBM10 KO mandibular cells. 200,000 cells were seeded in 6-well plates. The number of cells was analyzed after 25 h and 50 h. This analysis was done in triplicate.
Discussion
Mutations in RBM10 have been found to cause an X-linked human disorder, termed TARP syndrome. Here, we used the iCLIP protocol to identify endogenous RNA targets for RBM10 in a mouse mandibular cell line. Due to the very high conservation of the mouse and human RBM10 proteins and the study of a cell line model of early palate development, our analysis is relevant to the phenotypes observed in patients with loss-of-function RBM10 mutations. We observed a clear enrichment of RBM10 binding to introns of protein-coding genes and also to spliceosomal small nuclear RNAs of both canonical as well as minor spliceosomes. RBM10 preferentially binds in the proximity of the branch point (BP) and the 3′ splice site, which is in agreement with its specific association with the U2 and U12 snRNP complex. RBM10 iCLIP data revealed a strong enrichment for a cross-link C nucleotide and subsequently the identified enriched motifs contained at least 2 consecutive Cs, and resembled binding motifs identified in human cells.Citation17
We combined the iCLIP analysis with full transcriptome analysis of gene expression and splicing changes upon RBM10 inactivation in both mouse mandibular cells and also in ES cells. We could define a clear role for RBM10 in alternative splicing regulation. Thus, RBM10 has a role in cell house-keeping function by controlling the splicing of a set of genes expressed in early development, in stem cells as well as in embryonic differentiated mandibular cells. Combining iCLIP and RNA-seq data allowed us to draw an RBM10 RNA map, which suggested that the binding of RBM10 in the vicinity of 3′ splice sites leads to the silencing of the downstream exon. This is in agreement with the studies carried out in human cell lines.Citation16,17 The iCLIP data also shows that RBM10 binds U12 snRNA as well as U12 splicing-dependent introns. This suggests a role for RBM10 in splicing regulated by the minor spliceosome. However, we did not notice any defect in the splicing of U12 introns in the RNA-seq data of RBM10 KO cells. We do not know if RBM5 and RBM6 bind also to U12 introns but the absence of a defect in RBM10 KO cells could be explained by the redundant function of RBM5 and RBM6 proteins.
RBM10 was also found to be mutated in lung adenocarcinomasCitation40 and there is growing evidence for a role of RBM10 as a tumor suppressor that acts to repress the Notch signaling pathway and cell proliferation, by affecting the regulation of NUMB alternative splicing.Citation41 By contrast, we did not observe a role for RBM10 in the regulation of NUMB AS neither in mouse ES cells nor in embryonic mandibular cells. Moreover, RBM10 knockdown leads to slower growth rather than to proliferation, perhaps suggesting that a major role for RBM10 in cell growth is exerted earlier in development. Abrogating RBM10 expression in mouse ES cells led to a drastic decrease in cell growth and also dramatically affected the formation of embryoid bodies (). Among the list of splicing changes detected in RBM10 KO ES cells (Table S3), several genes could contribute to a proliferation phenotype, such as the pro-apoptotic Bcl-2 family member Bim (also known as Bcl2l11)Citation42 or the T-box transcription factor, TBX3, that promotes cell proliferation.Citation43
The TARP syndrome is a pleiotropic syndrome, affecting different aspects of development. Thus, it could be caused by many different alterations in gene expression and/or alternative splicing induced by RBM10 loss-of-function. As an example, depletion of RBM10 led to splicing changes in the EPN1 pre-mRNA, which is involved in the Notch signaling pathway.Citation44 Splicing changes were also observed in ES cells for the LEF1 gene that is required for the Wnt signaling pathway,Citation45 which is crucial for several steps of embryo development including palate morphogenesis.Citation46 Interestingly, we also detected splicing changes in the C2CD3 pre-mRNA, which is mutated in the oral facial digital syndromeCitation47 and in OPA1, whose mutations provoke an optic atrophy, a phenotype observed in TARP syndrome.Citation9 As was previously observed in human cells, RBM10 is also required for the splicing of CASK gene.Citation16
In summary, we have described the RNA-binding landscape of RBM10 in a mouse mandibular cell line and have clearly established a role for this RBP in alternative splicing regulation. The fact that RBM10 regulates AS in a tissue that is relevant to the malformations observed in the TARP syndrome and its crucial role during early development suggests that loss-of-function mutations of RBM10 may be compromising the AS regulation of selected genes leading to aberrant gene expression and causing human disease.
Materials and methods
Cell culture
Mouse mandibular MEPA (Mouse Embryonic Pharyngeal Arch) cell line was obtained, as previously described from 11.5 d.p.c. embryos from CD1 females crossed with a male 'Immortomouse'.Citation20 Cells were cultured in medium (DMEM, 10% FCS, 1% penicillin/streptomycin) containing 100 U ml−1 murine γ-interferon (Peprotech) at 33°C in an atmosphere containing 5% CO2. The hemizygote RBM10Y/− gene trap ES cells (CSI176), as well as the parental cell line E14Tg2a.4 were obtained from BayGenomics. Cells were cultured in gelatin-coated plates in GMEM medium supplemented with 15% FCS, 2 mM glutamine, 1 mM sodium pyruvate, 100 uM non-essential amino acids, 100 µM b-mercaptoethanol, antibiotics and LIF.
Generation of RBM10 knockout mandibular MEPA cells using CRISPR/Cas9 technology
To avoid off-target effects, we used the double nicking strategy with the Cas9 nickase mutant. We also conducted the targeting of two different exons and kept two cell lines for each exon targeted. Two single guide RNAs for each exon were cloned into pSpCas9(BB)-2A-GFP containing the nickase mutant of Cas9 (primer sequences in Table S4). The two plasmids were co-transfected into mandibular MEPA cells using Lipofectamine 2000 (Invitrogen). After 24h, single GFP positive cells were isolated by FACS and expanded in 96 well plates. Knockout cells were screened by western blot using anti-RBM10 antibody (ab72423, Abcam). Genotyping was carried out on gDNA extracted from cells using the Wizard® Genomic DNA Purification Kit (Promega).
Cell growth and differentiation
Cells were seeded at the indicated numbers (150,000 for ES cells and 200,000 for md cells) in 6-well plates keeping their respective culture condition. After 25 h and 50 hours, cells were collected and counted using a cell counter. This analysis was done in triplicate. Differentiation of ES cells into embryoid bodies (EBs) was done using the hanging drop method (drops of 20μL medium without LIF containing 600 ES cells), as previously described.Citation48 After two days, EBs were collected and grown in suspension for another 4 d.
iCLIP protocol
iCLIP experiments were performed following a published protocol,Citation49 with minor modifications.Citation50 The immunoprecipitation (IP) step was carried out using an anti-RBM10 antibody (ab72423, Abcam) that recognizes the 2 isoforms of the murine RBM10 proteins. Four independent experiments of the RBM10 protocol were performed. The four RBM10 iCLIP libraries with different barcodes were pooled together and sequenced on a single lane by single end sequencing 50 nt on an Illumina HiSeq 2000 system (BGI). Equivalent volume of the control libraries was sequenced on a different lane.
Read processing
Sequencing data from the iCLIP experiments were processed as followed using mostly the tools on Galaxy server https://usegalaxy.org/. Taking advantage of the iCLIP specific barcodes, identical reads, corresponding to PCR duplicates, were removed. Sequencing artifacts were discarded. Adapter sequences at the 3′ end were removed from the reads using CLIP tool. Barcode splitter tool attributed reads to each replicates. Reads were mapped to the Mouse July 2007 (NCBI37/mm9) genome data using Bowtie,Citation51 allowing only one mismatch in the seed. Only reads with only one genomic hit were kept. As reads come from cDNAs that have been truncated prematurely at the cross-linked nucleotide, we can deduce the cross-linked nucleotide positions that corresponds to the upstream nucleotide on the genome.
Annotation and motifs discovery
The cross-linked nucleotide position was used as the reference binding site. We annotated the cross-linked nucleotide positions based on ENSEMBL NCBI m37 and UCSC annotation mm9. We retrieved 20 nt sequences around the cross-linked nucleotides and searched for motifs on the top 1,000 sequences (ranked by total number of reads at the cross-linked nucleotide position). The search was done for a 6 to 10 nucleotide long motif using MEME (http://meme-suite.org/tools/meme)Citation27 allowing up to 20 motifs. We also looked for enriched pentamers and calculated their zscore. As a control set of sequences, we used, for each RBM10 binding site, a sequence randomly selected within the same intron. The occurrence (pentamer frequency) was calculated for each pentamer in each file. The z-score was calculated for each pentamer as: (occurrence in iCLIP sequences – average occurrence in control sequences) / standard deviation of occurrence in control sequences.
Analysis of snRNAs binding and U2/U12 intron binding
To study RBM10 binding to snRNAs, we mapped directly the iCLIP reads to the consensus sequence of the snRNAs. Only reads longer than 20 nt were used for this analysis. Then, we calculated the enrichment between the number of reads in RBM10 iCLIP and the control iCLIP.
To analyze how many U2- and U12-introns were bound by RBM10, we used the ‘full’ list of RBM10 binding sites. We only analyzed introns of expressed genes (using the RNA-seq data – see below). U12 introns were retrieved from the U12DB database.Citation28 The comparison between the proportion of U2- and U12-introns bound by RBM10 was done using a z-test. To analyze the distance between RBM10 binding site and the branch point, we predicted the branch point localization for U2 and U12 introns. For U2 introns we used SVM-BPfinder (http://regulatorygenomics.upf.edu/Software/SVM_BP/).Citation22 We only considered predictions with svm-score > 0 and one BP per intron. We first checked for BPs that that were at distance from the 3 ss within AGEZ (AG exclusion zone) length + 9 nt and had BP sequence score > 0. If there was one, we kept that one. If there was more than one, we took the one with the highest svm-score. If there were none, we dropped the condition on the BP sequence score and selected the best svm-score. If in this case there was still none, we dropped the AGEZ condition and simply reported the BP with the highest svm-score in the intron. To predict U12 branch points we used GeneIDCitation52 to score the splice-sites and locate BPs using the parameters for U12 intronsCitation28 and kept only those with positive scores for the branch-point and splice-sites.
RNA-seq
RNA extractions were performed using RNeasy Mini Kit (Qiagen) with DNAse treatment on column following manufacturer instructions. RNAs extracted from WT or RBM10 KO md cell lines were sent for sequencing to Beijing Genomics Institute (BGI). Three independent RNA extractions were carried out for the WT cell line while one RNA extraction was performed for each of the 4 KO cell lines, constituting 4 biological replicates. The libraries have been obtained using Truseq Transcriptome kit (polyA transcript enrichment) and sequenced on Illumina HiSeq providing a minimum of 10 Gb of data per sample (90 bp paired end reads) at the Beijing Genomics Institute (BGI). For ES cells, the same analysis was done on triplicate of WT and RBM10 KO RNA extractions.
RNA-seq bioinformatics analysis
Reads mapping and transcript quantification were done using Sailfish,Citation30 using the mm10 annotation of the mouse genome. To analyze the expression of a gene, we pooled the estimated read counts obtained by Sailfish for its different transcripts. We pre-filtered the data removing the bottom 25% of genes with low expression across all the cell lines. The differential expression between the WT and KO RBM10 cell lines was done using DESeq.Citation31 To generate heatmaps, the count values of each gene were first moderated by the variance stabilizing transformation (VST) method using DESeq. We calculated the Euclidean distances among samples and used heatmap2 function of the gplot package to visualize the sample clustering. The Genome-wide analyses of pre-mRNA splicing was done using SUPPA.Citation32 This method generated a list of splicing events from the mm10 annotation and then used the transcript quantification from Sailfish to calculate the PSI value for each splicing event (or transcript isoform) for each RNA-seq data. We only used transcripts with a level of expression (transcript per million TPM) above 1. We calculated the ΔPSI between KO and WT conditions (mean of PSI value for the 4 KO minus the mean of PSI value for the 3 WT). We also needed to take into consideration the variation within each cell lines WT or KO. So, we calculated the standard deviation but also the overlap between the 2 sets of data (overlap between the maximum (minimum) of the KO dataset and the minimum (maximum) of the WT data set for a negative (positive) ΔPSI. We filtered and ranked the list of splicing changes using these 3 parameters (ΔPSI, standard deviation and overlap).
RNA map
To investigate whether RBM10 directly regulates alternative exons, we looked for RBM10 ‘common’ cross-linked nucleotides in introns in proximity of the events. iCLIP cross-linked nucleotide positions based on the mm9 annotation were converted to the mm10 annotation using the Lift-Over tools to compare with the RNA-seq data.
We found 100 silenced exons and 46 enhances cassette exon with RBM10 binding in the neighboring introns. We analyzed the distribution of the relative distance in introns using a count density plot. We also analyzed features of those regulated events in terms of exon length, intron length, GC content and the strength of the splice site, which was evaluated on line using the maximum entropy score methodCitation53 (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html). For the comparison, we used 2 control sets: alternative cassette exons not regulated by RBM10 in genes expressed in md cells (DESeq_mean_normalized_count>1), as well as alternative cassette exons not regulated by RBM10 present in genes with an event regulated by RBM10.
Validation of splicing changes
RNA extractions were performed using RNeasy Mini Kit (Qiagen) following manufacturer instructions in WT or KO. RNAs were subsequently treated with DNase RQ1 (Promega) to remove any gDNA contamination. cDNA were obtained using Superscript III Reverse Transcriptase (Invitrogen). The PCRs to detect the splicing changes were done with GoTaq Hot Start Colorless Master Mix. The sequences of the used primers can be found in Supplemental Table S4. The quantification of each isoform abundance was done using an Agilent Bioanalyzer.
Data deposition
Raw sequencing data for both CLIP and RNA-seq experiments has been deposited in the Gene Expression Omnibus database (GSE89270).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KRNB_A_1247148_supplemental_material.zip
Download Zip (12.2 MB)Acknowledgments
We are grateful to Bob Hill and Ian Adams (MRC Human Genetics Unit, University of Edinburgh) for discussions and advice, and to Jacqueline Rainger (MRC HGU, Edinburgh) for providing the MEPA cell line. We are thankful to Dasa Longman (MRC HGU, Edinburgh) for critical reading of the manuscript. D.R.F. and J.F.C. were supported by Core funding from the Medical Research Council. J.F.C had also funding from the Wellcome Trust (Grant 095518/Z/11/Z). E.E. was supported by MINECO (Ministerio de Economía y Competitividad) and FEDER (Fondo Europeo de Desarrollo Regional) through grant BIO2014-52566-R, by Sandra Ibarra Foundation for Cancer and by AGAUR (Agència de Gestió d'Ajuts Universitaris i de Recerca) through grant 2014-SGR1121.
Related Research Data
References
- Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol 2002; 3:195-205; PMID:11994740; http://dx.doi.org/10.1038/nrm760
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BMM, Strein C, Davey NEE, Humphreys DTT, Preiss T, Steinmetz LMM, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012; 149:1393-406; PMID:22658674; http://dx.doi.org/10.1016/j.cell.2012.04.031
- Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 2012; 46:674-90; PMID:22681889; http://dx.doi.org/10.1016/j.molcel.2012.05.021
- Inoue A, Takahashi KP, Kimura M, Watanabe T, Morisawa S. Molecular cloning of a RNA binding protein, S1-1. Nucleic Acids Res 1996; 24:2990-7; PMID:8760884; http://dx.doi.org/10.1093/nar/24.15.2990
- Sutherland LC, Rintala-Maki ND, White RD, Morin CD. RNA binding motif (RBM) proteins: a novel family of apoptosis modulators? J Cell Biochem 2005; 94:5-24; PMID:15514923; http://dx.doi.org/10.1002/jcb.20204
- Bonnal S, Martínez C, Förch P, Bachi A, Wilm M, Valcárcel J. RBM5/Luca-15/H37 regulates Fas alternative splice site pairing after exon definition. Mol Cell 2008; 32:81-95; PMID:18851835; http://dx.doi.org/10.1016/j.molcel.2008.08.008
- Fushimi K, Ray P, Kar A, Wang L, Sutherland LC, Wu JY. Up-regulation of the proapoptotic caspase 2 splicing isoform by a candidate tumor suppressor, RBM5. Proc Natl Acad Sci U S A 2008; 105:15708-13; PMID:18840686; http://dx.doi.org/10.1073/pnas.0805569105
- Johnston JJ, Teer JK, Cherukuri PF, Hansen NF, Loftus SK, Chong K, Mullikin JC, Biesecker LG. Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am J Hum Genet 2010; 86:743-8; PMID:20451169; http://dx.doi.org/10.1016/j.ajhg.2010.04.007
- Gripp KW, Hopkins E, Johnston JJ, Krause C, Dobyns WB, Biesecker LG. Long-term survival in TARP syndrome and confirmation of RBM10 as the disease-causing gene. Am J Med Genet A 2011; 155A:2516-20; PMID:21910224; http://dx.doi.org/10.1002/ajmg.a.34190
- Johnston JJ, Sapp JC, Curry C, Horton M, Leon E, Cusmano-Ozog K, Dobyns WB, Hudgins L, Zackai E, Biesecker LG. Expansion of the TARP syndrome phenotype associated with de novo mutations and mosaicism. Am J Med Genet A 2014; 164A:120-8; PMID:24259342; http://dx.doi.org/10.1002/ajmg.a.36212
- Agafonov DE, Deckert J, Wolf E, Odenwälder P, Bessonov S, Will CL, Urlaub H, Lührmann R. Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol Cell Biol 2011; 31:2667-82; PMID:21536652; http://dx.doi.org/10.1128/MCB.05266-11
- Behzadnia N, Golas MM, Hartmuth K, Sander B, Kastner B, Deckert J, Dube P, Will CL, Urlaub H, Stark H, et al. Composition and three-dimensional EM structure of double affinity-purified, human prespliceosomal A complexes. EMBO J 2007; 26:1737-48; PMID:17332742; http://dx.doi.org/10.1038/sj.emboj.7601631
- Bessonov S, Anokhina M, Will CL, Urlaub H, Lührmann R. Isolation of an active step I spliceosome and composition of its RNP core. Nature 2008; 452:846-50; PMID:18322460; http://dx.doi.org/10.1038/nature06842
- Li J, Hawkins IC, Harvey CD, Jennings JL, Link AJ, Patton JG. Regulation of alternative splicing by SRrp86 and its interacting proteins. Mol Cell Biol 2003; 23:7437-47; PMID:14559993; http://dx.doi.org/10.1128/MCB.23.21.7437-7447.2003
- Inoue A, Yamamoto N, Kimura M, Nishio K, Yamane H, Nakajima K. RBM10 regulates alternative splicing. FEBS Lett 2014; 588:942-7; PMID:24530524; http://dx.doi.org/10.1016/j.febslet.2014.01.052
- Wang YY, Gogol-Döring A, Hu H, Fröhler S, Ma Y, Jens M, Maaskola J, Murakawa Y, Quedenau C, Landthaler M, et al. Integrative analysis revealed the molecular mechanism underlying RBM10-mediated splicing regulation. EMBO Mol Med 2013; 5:1431-42; PMID:24000153; http://dx.doi.org/10.1002/emmm.201302663
- Bechara EG, Sebestyén E, Bernardis I, Eyras E, Valcárcel J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol Cell 2013; 52:720-33; PMID:24332178; http://dx.doi.org/10.1016/j.molcel.2013.11.010
- König J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 2010; 17:909-15; PMID:20601959; http://dx.doi.org/10.1038/nsmb.1838
- Huppertz I, Attig J, D'Ambrogio A, Easton LE, Sibley CR, Sugimoto Y, Tajnik M, König J, Ule J. iCLIP: protein-RNA interactions at nucleotide resolution. Methods 2014; 65:274-87; PMID:24184352; http://dx.doi.org/10.1016/j.ymeth.2013.10.011
- Benko S, Fantes JA, Amiel J, Kleinjan DJ, Thomas S, Ramsay J, Jamshidi N, Essafi A, Heaney S, Gordon CT, et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat Genet 2009; 41:359-64; PMID:19234473; http://dx.doi.org/10.1038/ng.329
- Taggart AJ, DeSimone AM, Shih JS, Filloux ME, Fairbrother WG. Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat Struct Mol Biol 2012; 19:719-21; PMID:22705790; http://dx.doi.org/10.1038/nsmb.2327
- Corvelo A, Hallegger M, Smith CWJ, Eyras E. Genome-wide association between branch point properties and alternative splicing. PLoS Comput Biol 2010; 6:e1001016; PMID:21124863; http://dx.doi.org/10.1371/journal.pcbi.1001016
- Kol G, Lev-Maor G, Ast G. Human-mouse comparative analysis reveals that branch-site plasticity contributes to splicing regulation. Hum Mol Genet 2005; 14:1559-68; PMID:15857856; http://dx.doi.org/10.1093/hmg/ddi164
- Singh R, Valcárcel J. Building specificity with nonspecific RNA-binding proteins. Nat Struct Mol Biol 2005; 12:645-53; PMID:16077728; http://dx.doi.org/10.1038/nsmb961
- Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol 2007; 8:479-90; PMID:17473849; http://dx.doi.org/10.1038/nrm2178
- Sugimoto Y, König J, Hussain S, Zupan B, Curk T, Frye M, Ule J. Analysis of CLIP and iCLIP methods for nucleotide-resolution studies of protein-RNA interactions. Genome Biol 2012; 13:R67; PMID:22863408; http://dx.doi.org/10.1186/gb-2012-13-8-r67
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 2009; 37:W202-8; PMID:19458158; http://dx.doi.org/10.1093/nar/gkp335
- Alioto TS. U12DB: a database of orthologous U12-type spliceosomal introns. Nucleic Acids Res 2007; 35:D110-5; PMID:17082203; http://dx.doi.org/10.1093/nar/gkl796
- Li K, Wang G, Andersen T, Zhou P, Pu WT. Optimization of genome engineering approaches with the CRISPR/Cas9 system. PLoS One 2014; 9:e105779; PMID:25166277; http://dx.doi.org/10.1371/journal.pone.0105779
- Patro R, Mount SM, Kingsford C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat Biotechnol 2014; 32:462-4; PMID:24752080; http://dx.doi.org/10.1038/nbt.2862
- Anders S, Huber W. DESeq: Differential expression analysis for sequence count data. Genome Biol 2010; 11:R106; PMID:20979621; http://dx.doi.org/10.1186/gb-2010-11-10-r106
- Alamancos GP, Pagès A, Trincado JL, Bellora N, Eyras E. Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 2015; 21:1521-31; PMID:26179515; http://dx.doi.org/10.1261/rna.051557.115
- Ozuemba B, Masilamani TJ, Loiselle JJ, Koenderink B, Vanderbeck KA, Knee J, Larivière C, Sutherland LC, Ozuemba B, Masilamani TJ, et al. Co- and post-transcriptional regulation of Rbm5 and Rbm10 in mouse cells as evidenced by tissue-specific, developmental and disease-associated variation of splice variant and protein expression levels. Gene 2016; 580:26-36; PMID:26784654; http://dx.doi.org/10.1016/j.gene.2015.12.070
- Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem 2000; 275:16795-801; PMID:10828064; http://dx.doi.org/10.1074/jbc.275.22.16795
- Liu Q, Niu N, Wada Y, Liu J. The role of Cdkn1A-interacting zinc finger protein 1 (CIZ1) in DNA replication and pathophysiology. Int J Mol Sci 2016; 17:212; PMID:26861296; http://dx.doi.org/10.3390/ijms17020212
- Panaro MA, Acquafredda A, Calvello R, Lisi S, Dragone T, Cianciulli A. Organization patterns of the AGFG genes: an evolutionary study. Immunopharmacol Immunotoxicol 2011; 33:111-23; PMID:21284487; http://dx.doi.org/10.3109/08923973.2010.485617
- Volkert MR, Elliott NA, Housman DE. Functional genomics reveals a family of eukaryotic oxidation protection genes. Proc Natl Acad Sci U S A 2000; 97:14530-5; PMID:11114193; http://dx.doi.org/10.1073/pnas.260495897
- Witten JT, Ule J. Understanding splicing regulation through RNA splicing maps. Trends Genet. 2011; 27:89-97; PMID:21232811; http://dx.doi.org/10.1016/j.tig.2010.12.001
- Hwang Y-S, Chung BG, Ortmann D, Hattori N, Moeller HC, Khademhosseini A. Microwell-mediated control of embryoid body size regulates embryonic stem cell fate via differential expression of WNT5a and WNT11. Proc Natl Acad Sci U S A 2009; 106:16978-83; PMID:19805103; http://dx.doi.org/10.1073/pnas.0905550106
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012; 150:1107-20; PMID:22980975; http://dx.doi.org/10.1016/j.cell.2012.08.029
- Hernández J, Bechara E, Schlesinger D, Delgado J, Serrano L, Valcárcel J. Tumor suppressor properties of the splicing regulatory factor RBM10. RNA Biol 2016; 13:466-72; http://dx.doi.org/10.1080/15476286.2016.1144004
- Bouillet P, Purton JF, Godfrey DI, Zhang L-C, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM, Strasser A. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature 2002; 415:922-6; PMID:11859372; http://dx.doi.org/10.1038/415922a
- Willmer T, Hare S, Peres J, Prince S. The T-box transcription factor TBX3 drives proliferation by direct repression of the p21(WAF1) cyclin-dependent kinase inhibitor. Cell Div 2016; 11:6; PMID:27110270; http://dx.doi.org/10.1186/s13008-016-0019-0
- Chen H, Ko G, Zatti A, Di Giacomo G, Liu L, Raiteri E, Perucco E, Collesi C, Min W, Zeiss C, et al. Embryonic arrest at midgestation and disruption of Notch signaling produced by the absence of both epsin 1 and epsin 2 in mice. Proc Natl Acad Sci U S A 2009; 106:13838-43; PMID:19666558; http://dx.doi.org/10.1073/pnas.0907008106
- Hrckulak D, Kolar M, Strnad H, Korinek V. TCF/LEF transcription factors: An update from the internet resources. Cancers (Basel) 2016; 8:E70; PMID:27447672; http://dx.doi.org/10.3390/cancers8070070
- Greene RM, Pisano MM. Palate morphogenesis: current understanding and future directions. Birth Defects Res C Embryo Today 2010; 90:133-54; PMID:20544696; http://dx.doi.org/10.1002/bdrc.20180
- Thauvin-Robinet C, Lee JS, Lopez E, Herranz-Pérez V, Shida T, Franco B, Jego L, Ye F, Pasquier L, Loget P, et al. The oral-facial-digital syndrome gene C2CD3 encodes a positive regulator of centriole elongation. Nat Genet 2014; 46:905-11; PMID:24997988; http://dx.doi.org/10.1038/ng.3031
- Wang X, Yang P. In vitro differentiation of mouse embryonic stem (mES) cells using the hanging drop method. J Vis Exp. 2008; (17):825; PMID:19066514; http://dx.doi.org/10.3791/825
- Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. iCLIP–transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. J Vis Exp 2011:50:e2638; PMID: 21559008; http://dx.doi.org/10.3791/2638
- Rodor J, Pan Q, Blencowe BJ, Eyras E, Cáceres JF. The RNA-binding profile of Acinus, a peripheral component of the exon junction complex, reveals its role in splicing regulation. RNA 2016; 22:1411-26; PMID:27365209; http://dx.doi.org/10.1261/rna.057158.116
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25; PMID:19261174; http://dx.doi.org/10.1186/gb-2009-10-3-r25
- Blanco E, Parra G, Guigó R. Using geneid to identify genes. Curr Protoc Bioinformatics 2007; Chapter 4:Unit 4.3; PMID:18428791; http://dx.doi.org/10.1002/0471250953.bi0403s18
- Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 2004; 11:377-94; PMID:15285897; http://dx.doi.org/10.1089/1066527041410418